
Abstract
Cholesterol oxidase, steroid C27 monooxygenase and 3-ketosteroid-Δ1-dehydrogenase are key enzymes involved in microbial catabolism of sterols. Here, three isoenzymes of steroid C27 monooxygenase were firstly characterized from Mycobacterium neoaurum as the key enzyme in sterol C27-hydroxylation. Among these three isoenzymes, steroid C27 monooxygenase 2 exhibits the strongest function in sterol catabolism. To improve androst-1,4-diene-3,17-dione production, cholesterol oxidase, steroid C27 monooxygenase 2 and 3-ketosteroid-Δ1-dehydrogenase were coexpressed to strengthen the metabolic flux to androst-1,4-diene-3,17-dione, and 3-ketosteroid 9α-hydroxylase, which catalyzes the androst-1,4-diene-3,17-dione catabolism, was disrupted to block the androst-1,4-diene-3,17-dione degradation pathway in M. neoaurum JC-12. Finally, the recombinant strain JC-12S2-choM-ksdd/ΔkshA produced 20.1 g/L androst-1,4-diene-3,17-dione, which is the highest reported production with sterols as substrate. Therefore, this work is hopes to pave the way for efficient androst-1,4-diene-3,17-dione production through metabolic engineering.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Androst-1,4-diene-3,17-dione (ADD), an important pharmaceutical androgen steroid, is widely used as an important precursor in the synthesis of steroid hormone medicines [25, 29]. Traditionally, in the pharmaceutical industry, ADD was synthesized by multistep chemical methods from natural steroid sapogenin, diosgenin [18]. One of the well-established route in its commercial usage is chemical modification of diosgenin to ADD followed by further synthesis to pharmaceutical steroids [14]. However, the chemical conversion of diosgenin to ADD and other valuable steroids has many shortcomings such as high-cost processes, relatively low yields, waste of land resources, and high pollution [35]. Therefore, with the constant improvement of the environmental protection consciousness and the further development of green technology, non-pollution and non-toxic technology have inevitably become the main direction of industrial development [1, 30].
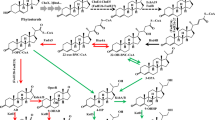
Microbial conversion of the widely spread natural sterols (phytosterol, cholesterol, ergosterol, etc.) to produce valuable steroid precursors has been an important alternative way in the pharmaceutical industry [11]. Since the initial discovery of the gene cluster encoding sterol catabolism, many key enzymes involved in this process have been well identified and characterized in recent years [22, 36]. So far, the main key intermediates produced from sterol microbial metabolism were the C19 steroids such as 4-androstene-3,17-dione (AD) and androst-1,4-diene-3,17-dione (ADD) [11]. Since these products are widely used as important precursors in the synthesis of steroid hormone medicines, it has great significance and value to realize the biotransformation of the low-cost sterols into these high-value products. Generally, sterols were firstly bio-converted to AD [48], AD was then transformed to other intermediates that were catalyzed by the corresponding enzymes [7]. The biotransformation of sterols to AD involves two processes: the modification of 3β-ol-5-ene to 3-oxo-4-ene moiety on steroid nucleus catalyzed by 3β-hydroxysteroid dehydrogenase (3β-HSD) or cholesterol oxidase [8, 9, 44], and the side-chain degradation of sterols [34]. Then AD is catalyzed to ADD by 3-ketosteroid-Δ1-dehydrogenase (KSDD) [39]. Besides, AD and ADD are easily catalyzed to 9α-hydroxy-AD (9α-OH-AD) and 9α-hydroxy-ADD (9α-OH-ADD) by 3-ketosteroid-9α-hydroxylase (KSH), respectively [38]. Moreover, 9α-OH-ADD further degrades to carbon dioxide and water by spontaneous ring B destruction [41, 42], which results in the degradation of the key intermediates (Fig. 1).

Microbial metabolic pathway of sterols in Mycobacterium neoaurum. The depicted metabolites are: (I) cholesterol, (II) 4-cholesten-3-one, (III) cholest-4-en-3-one-27-oic acid, (IV) 4-androstene-3,17-dione (AD), (V) androst-1,4-diene-3,17-dione (ADD), (VI) 9α-hydroxy-4-androstene-3,17-dione (9α-OH-AD), (VII) 9α-hydroxy-androst-1,4-diene-3,17-dione (9α-OH-ADD). ChoM cholesterol oxidase, SMO steroid C27 monooxygenase, KSDD 3-ketosteroid-Δ1-dehydrogenase, KSH 3-ketosteroid 9α-hydroxylase
In the biotransformation process of sterols’ to AD, the initial step of sterols side-chain oxidation is hydroxylation catalyzed by steroid C27 monooxygenase at C27 [11]. CYP125, a well-studied steroid C27 monooxygenase belongs to cytochrome P450 family and catalyzes three successive oxidations of the sterol terminal carbon to an acid as shown for Rhodococcus jostii RHA1, Mycobacterium bovis BCG and M. tuberculosis H37Rv [5, 24, 40]. A crucial role of CYP125A1 in hydroxylating cholest-4-en-3-one at C27 and oxidizing to cholest-4-en-3-one-27-oic acid was demonstrated for M. tuberculosis H37Rv [21]. However, there are no literatures report about the effect of steroid C27 monooxygenase on AD/ADD production and any other steroid C27 monooxygenase isoenzymes involved in sterol catabolism in mycobacteria. Thus, it is necessary to characterize steroid C27 monooxygenase isoenzymes in Mycobacterium and identify their functions in AD/ADD production.
It is well known that the genus Mycobacterium is the most efficient AD/ADD producer [11, 19, 33]. Many efforts have been devoted to increasing the AD/ADD yield by mutation breeding and genetic engineering. For instance, nitrosoguanidine mutagenesis and the combination of mitomycin C and UV treatments were used to achieve the high sterol transformation efficiency and AD/ADD yield [13, 15]. By disrupting ksddM gene in Mycobacterium, AD production was improved from sterol bioconversion, while by overexpressing ksddM gene, ADD production was improved [3, 39]. In a recent study, the cholesterol oxidases in M. neoaurum were identified and applied by increasing the AD and ADD production [45]. However, so far, there is still no report concerning the improvement of AD/ADD yield through metabolic engineering techniques. Thus, it is imperative to tune sterols’ metabolic flux to improve the AD/ADD yield and block their degradation pathway.
In our previous studies, strain M. neoaurum JC-12, capable of transforming phytosterol into ADD as the main product, was obtained [28]. Enzymes cholesterol oxidase (ChoM) and 3-ketosteroid-Δ1-dehydrogenase (KSDD) were identified as the key enzymes playing important roles in bio-converting sterols into AD/ADD in M. neoaurum [27, 48]. In this study, we firstly discovered three steroid C27 monooxygenase isoenzymes (SMO1, SMO2 and SMO3) involved in the C27-hydroxylation in M. neoaurum. By gene knockout and complementation experiment, SMO2 was confirmed to possess the strongest function in C27-hydroxylation. To enhance ADD production, metabolic engineering strategy was carried out by disrupting KSH to block the ADD degradation pathway and coexpressing ChoM, SMO2 and KSDD to strengthen the metabolic flux toward ADD in M. neoaurum JC-12. Finally, the recombinant strain JC-12S2-choM-ksdd/ΔkshA produced ADD of 20.1 g/L, which is the highest production ever reported. In this work, we firstly reported the optional regulation of the sterol metabolism to drive increased metabolic flux toward the freewheeling products by metabolic engineering strategy, which supplies a new insight into the redesigned metabolic pathway for improving the steroid intermediates production from sterols.
Experimental
Strains, plasmids, primers and culture conditions
All strains, plasmids and primers used in this work are listed in Table 1 and Table S1. E. coli strains JM109 and BL21 (DE3), cultured in Luria–Bertain (LB) medium, were used for plasmid construction and heterologous expression, respectively. Strain M. neoaurum JC-12 was stored in our laboratory and used for constructing engineering strains. For sterol biotransformation, M. neoaurum strains were inoculated in 10 ml seed medium (10 g/L glucose, 10 g/L peptone, 6 g/L beef extract, 10 g/L NaCl, pH 7.5) and cultivated at 30 °C and 160 rpm for 48 h. Then, 5 mL culture was transferred into 100 mL fermentation medium containing 20 g/L glucose, 10 g/L peptone, 6 g/L beef extract, 3 g/L K2HPO4, 0.5 g/L MgSO4·7H2O and 5 × 10−4 g/L MnCl2·4H2O, pH 7.5. To improve the sterol biotransformation, hydroxymethyl-β-cyclodextrin (HP-β-CD) was used to enhance the solubility of sterols [32, 47]. Sterols and HP-β-CD with a ratio of 1:3 (w/w) were added into the fermentation medium to conduct the transformation. Without special conditions, the fermentation was carried out with 20 g/L phytosterol (added with 60 g/L HP-β-CD). A 5-L fermentor (Biotech Co., Shanghai, China) was used to scale up the flask cultures with agitation speed of 400 rpm and 1 volume of air per unit of medium per minute (vvm) at 30 °C and pH 7.5. In addition, to identify the function of SMO in M. neoaurum, 4-cholesten-3-one agar medium containing 5 g/L 4-cholesten-3-one based on the minimal medium (g per liter of distilled water, (NH4)2HPO4 1.5, MgSO4·7H2O 0.2, K2HPO4 0.4, FeSO4·7H2O 5 × 10−4, ZnSO4·7H2O 2 × 10−4) and added with 20 g/L agar was used in this study. The corresponding antibiotics and inducers were added when needed.
Heterologous expression and purification of SMO isoenzymes in E. coli
The genome of M. neoaurum JC-12 was sequenced and three putative steroid C27 monooxygenase genes designated as Smo1 (gene ID: MH881437), Smo2 (gene ID: MH881438) and Smo3 (gene ID: MH881439) were selected for further research in this study. The genes Smo1, Smo2 and Smo3 were amplified by PCR techniques using primers listed in Table S1. The amplified fragments were inserted into the Sac I/Hind III or BamH I/EcoR I sites of pET28a vector to create pET28a-Smo1, pET28a-Smo2 and pET28a-Smo3, respectively. Then these recombinant plasmids were transformed into E. coli BL21 to construct recombinant strains BL21/pET28a-Smo1, BL21/pET28a-Smo2 and BL21/pET28a-Smo3, which were confirmed by DNA sequencing. These recombinant strains were cultured in 50 mL LB medium with 50 mg/L kanamycin at 37 °C. The protein expression was induced by 0.05 mM IPTG when the OD600 value of the culture reached 0.6–0.8. After continuous cultivation for 12 h at 16 °C, the cells were harvested by centrifugation at 10,000×g for 10 min and washed with 50 mM phosphate buffer (pH 7.0). Then the pellets were suspended in phosphate buffer for sonication. The cell-free extracts were obtained by centrifugation at 10,000×g for 40 min and used for further protein sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis, protein purification and enzyme activity assay. Purification was performed as the instructions of HisTrap™ HP column [43]. The method of Bradford was used to determine the protein concentration and bovine serum albumin was used as the standard [2].
Activity assay and protein properties of the SMO isoenzymes
The SMO activity was determined by monitoring substrate concentrations using HPLC as described previously with minor modification [21]. One unit of SMO activity is defined as the amount of enzyme required to convert 1 µmol of 4-cholesten-3-one to 4-cholesten-3-one-27-oic acid at 30 °C and pH 7.0 per minute. The optimum pH for SMO activity was determined by SMO enzyme activity assay at 30 °C in several buffers (50 mM citrate–sodium citrate buffer, pH 3.0–6.0; 50 mM phosphate buffer, pH 6.0–8.0; 50 mM glycine–sodium hydroxide buffer, pH 8.0–10.0). The optimum temperature was examined in 50 mM phosphate buffer (pH 7.0) using the standard reaction mixture with temperatures ranging from 10 to 60 °C. The kinetic parameters (KM, Vmax and kcat) of these three isoenzymes were determined by fitting a plot of rate versus substrate concentration to the Michaelis–Menten equation using nonlinear regression in the software GraphPad Prism 5.0 (GraphPad Software, Inc. La Jolla, CA, USA).
Functional analysis of SMO1, SMO2 and SMO3 in sterols metabolism
To identify the function of SMO isoenzymes, the disruption and complementation of their corresponding genes were carried out and the engineered strains used were constructed as given in the Supplementary Methods. By measuring the viability of the Smo-deleted strains grown on 4-cholesten-3-one agar medium plates with 4-cholesten-3-one as the sole energy and carbon source, the roles of SMO1, SMO2 and SMO3 on the growth of M. neoaurum with sterol as the sole energy and carbon source were identified. The different Smo-deleted strains were firstly inoculated into 10 mL seed medium and cultivated on a rotary shaker at 30 °C and 160 rpm for 24 h. Then, 1 mL of these cultures were diluted appropriately with distilled physiological saline and then plated on the 4-cholesten-3-one agar medium plates. CFU (colony forming units) of these strains were calculated at 3 and 7 days, respectively. Besides, the SMO enzyme activity of these Smo-deleted strains in the seed medium was also detected at 3 and 7 days, respectively. The 4-cholesten-3-one degradation properties of the different Smo-deleted strains were analyzed by shake-flask fermentation. After cultivation on a rotary shaker at 30 °C and 160 rpm for 24 h, 2 mL of the seed medium cultures were inoculated into 50 mL fermentation medium added with 1 g/L 4-cholesten-3-one and 3 g/L HP-β-CD. Then, the difference in the behavior among the Smo-deleted mutants in 4-cholesten-3-one degradation was clarified with the cultivation period elongated to 250 h. The residue of substrate was analyzed by HPLC.
Overexpression of SMO isoenzymes in M. neoaurum JC-12
To analyze the effects of SMOs on sterol transformation, plasmid pMV261 was used as an overexpression vector to augment the expression of SMO1, SMO2 and SMO3 in M. neoaurum JC-12. Genes Smo1, Smo2 and Smo3 were amplified using primers Smo1-f & r, Smo2-f & r and Smo3-f & r (Table S1). The Sac I/Hind III fragment of Smo1, BamH I/EcoR I fragment of Smo2, and BamH I/EcoR I fragment of Smo3 genes were inserted into the corresponding sites of pMV261 to construct the recombinant plasmids p261-Smo1, p261-Smo2 and p261-Smo3. Then these plasmids were transferred into M. neoaurum JC-12 to generate the recombinant strains JC-12S1, JC-12S2 and JC-12S3 (Table 1).
Metabolic engineering of M. neoaurum JC-12 for the production of ADD
A metabolic engineering strategy was carried out by disrupting KSH and coexpressing ChoM, SMO2 and KSDD. To delete kshA gene in M. neoaurum JC-12, recombinant strain JC-12ΔkshA with p2N-ΔkshA was constructed in accordance with MutS1 as described in the Supplementary Methods. To coexpress ChoM, SMO2 and KSDD in the kshA-deleted mutant JC-12ΔkshA, the primers choM-SD-f (containing an SD sequence for ribosome binding) and choM-r were used to amplify the choM gene. The choM fragment was solely digested by EcoR I and inserted into the EcoR I site of plasmid p261-Smo2 to create the p261-Smo2-choM with SMO2 and ChoM. Then, the ksdd gene was amplified using primers ksdd-SD-f and ksdd-r, and the solely Hind III digested fragment was inserted into the Hind III site of plasmid p261-Smo2-choM to create p261-Smo2-choM-ksdd with SMO2, ChoM and KSDD. The plasmid p261-Smo2-choM-ksdd was transformed into JC-12ΔkshA to construct the recombinant strain JC-12S2-choM-ksdd/ΔkshA, in which the kshA gene was knocked out and Smo2, choM and ksdd genes were augmented. The KSH, ChoM and KSDD enzyme activities were assayed according to previous studies [27, 28, 46].
Analytical methods
A 1 mL sample was taken from culture broth and extracted with 4 mL ethyl acetate. After centrifugation, 2 mL of the supernatant was analyzed by Shimadzu HPLC equipped with C18 column (Diamonsil ®C18, 5 µm particles, 250 mm × 4.6 mm) and UV/visible detector. 4-Cholesten-3-one was detected at 240 nm and the mobile phase composed of acetonitrile and isopropanol (85:15, v/v) [26]. ADD was detected at 254 nm and the mobile phase composed of methanol and water (70:30, v/v). The flow rate was 1 mL/min and the column temperature was 30 °C [48]. Biomass accumulation was determined as the number of CFU per mL of cultural liquid during fermentation [20]. Residual glucose detection was done by a biological sensing analyzer (SBA, China) [49].
Results and discussion
Characterization of SMO isoenzymes from M. neoaurum
Mycobacterium neoaurum JC-12 is a good producer of ADD with few by-products of AD using sterols as substrate [28]. By analyzing the genome sequence results of M. neoaurum JC-12, three putative steroid C27-monooxygenases encoded by the genes Smo1 (gene ID: MH881437), Smo2 (gene ID: MH881438) and Smo3 (gene ID: MH881439) were selected for further research. The amino acid identity between SMO1 and SMO2 was about 54%, and the identity between SMO1 and SMO3 was 65.6%. The phylogenetic tree is shown in Fig. S1 and SMO1, SMO2 and SMO3 share high similarity with the cytochrome P450 of Mycobacteriaceae (WP_019510071.1, 99%), cytochrome P450 of Mycobacteriaceae (WP_023986299.1, 99%) and cytochrome P450 of Mycobacteriaceae (WP_019512517.1, 99%), respectively.
To characterize the C27-hydroxylation catalytic activity, these three SMO isoenzymes were purified (Fig. S2) and the enzyme activities were analyzed (Table S2). Besides, the steady-state kinetic constants (KM and kcat) for the oxidation of 4-cholesten-3-one by SMO1, SMO2 and SMO3 are shown in Table 2. The KM of SMO2 was lower than that of SMO1 and SMO3, and the kcat and kcat/KM of SMO2 was higher than that of SMO1 and SMO3. These results indicate that the catalytic activity of SMO2 is higher than SMO1 and SMO3. Since the enzymatic properties of SMOs from M. neoaurum ATCC 25795 and M. neoaurum JC-12 have no differences, we designated SMO1, SMO2 and SMO3 as the same.
SMO2 exhibits stronger functions in sterol catabolism than SMO1 and SMO3
Since sterol catabolic pathway in M. neoaurum ATCC 25795 is intact, and there are no intermediates (AD/ADD) accumulated to inhibit cell growth and respiration [10]. Thus, we selected it as an experimental strain rather than M. neoaurum JC-12 to determine the specific functions of the enzymes SMO1, SMO2 and SMO3 in sterol catabolism. On the bases of M. neoaurum ATCC 25795, seven Smo-deleted mutants (MutS1, MutS2, MutS3, Mut(S1&S2), Mut(S1&S3), Mut(S2&S3) and Mut(S1&S2&S3)) and their corresponding complemented strains were constructed as described in Supplemental Methods. As shown in Table S3, all Smo-deleted mutants exhibited low enzyme activities and Smo2-deleted mutant showed lower activity than Smo1 or Smo3-deleted mutants. Compared with wild-type strain ATCC 25795, all Smo-deleted mutants grew poorly on 4-cholesten-3-one agar plates and Smo2-deleted mutant grew poorer than Smo1 or Smo3-deleted mutants (Fig. S3). The cell growth of mutant strain Mut(S1&S2) was inhibited the most among two-gene deleted mutants. When Smo1, Smo2 and Smo3 were all disrupted in Mut(S1&S2&S3), cell growth was severely affected, indicating that the catabolism of sterols was greatly blocked. However, the complemented strains of Smo-deletion mutants showed better growth state on 4-cholesten-3-one agar plates and their activities toward 4-cholesten-3-one were restored to some extent. In addition, Smo2-complemented mutant exhibited better cell-growth properties than Smo1 and Smo3-complemented mutants. These results showed that a combination of SMO1, SMO2 and SMO3 was required for M. neoaurum cell growth and SMO2 was more important than SMO1 and SMO3 for cell growth and enzyme activity.
To further assess the effects of Smo1, Smo2 and Smo3 on the catabolism of sterols, a fermentation medium containing sufficient nutriment supplemented with 4-cholesten-3-one and HP-β-CD was used for the degradation of 4-cholesten-3-one. There was no obvious difference in the cell growth between the wild-type strain M. neoaurum ATCC 25795 and its mutants MutS1, MutS2, MutS3, Mut(S1&S2), Mut(S1&S3), Mut(S2&S3) and Mut(S1&S2&S3) in the nutriment medium (data not shown). As shown in Fig. 2, the wild type could completely degrade 1 g/L 4-cholesten-3-one within 100 h, while the Smo1-deleted mutant MutS1, Smo2-deleted mutant MutS2 and Smo3-deleted mutant MutS3 degraded 79%, 66% and 87% 4-cholesten-3-one within 100 h. The mutants MutS1, MutS2 and MutS3 required 156 h, 180 h, and 120 h to completely degrade 1 g/L 4-cholesten-3-one. Among the mutants with the two Smo gene deletion (Mut(S1&S2) Mut(S2&S3) and Mut(S1&S3)), Mut(S1&S2) retarded 4-cholesten-3-one degradation most seriously, while Mut(S1&S3) retarded the degradation slightly. Besides, the results of Fig. 2 also provided an additional clue that there should be other enzymes in M. neoaurum which exhibit a similar function to SMOs, as Mut(S1&S2&S3) could still retain a partial capacity to degrade 4-cholesten-3-one. Recent studies have indicated that some other steroid C27 monooxygenases, such as CYP142A1 in M. tuberculosis and CYP142A2 in M. smegmatis, play a similar role to SMOs for the initial degradation of sterol side chain [5, 21]. All these results suggested that SMO was the key enzyme in the sterol catabolism pathway, and SMO2 showed stronger functions in sterol metabolism. Therefore, it would be helpful to enhance the sterol conversion efficiency to valuable steroids by improving the enzymatic activity through metabolic engineering method. Since there was no obvious difference in cell growth between ATCC25795 and Smo-deleted mutants (data not shown), the difference in the behavior in 4-cholesten-3-one degradation was mainly caused by Smo deletion. In this sense, these results were in accordance with that identified on the 4-cholesten-3-one agar plates.

The utilization of 4-cholesten-3-one by the Smo-deleted mutants and their corresponding complements. The strains were cultured in the fermentation medium added with 1 g/L 4-cholesten-3-one. The residue of 4-cholesten-3-one was detected by HPLC. All assays were performed in triplicate with three independent measurements. Error bars represented standard deviations of the biological replicates
Improved ADD production by SMO2 expression in M. neoaurum
Since SMO1, SMO2 and SMO3 play important roles in the sterol catabolism pathway, it is intriguing to determine whether the ADD yield could be significantly enhanced by improving the activity of SMO in M. neoaurum JC-12. Therefore, in this study, to detect the effects of SMO isoenzymes on ADD production, recombinant strains JC-12S1 overexpressing SMO1, JC-12S2 overexpressing SMO2 and JC-12S3 overexpressing SMO3 were constructed, respectively.
As shown in Fig. 3, SMOs overexpression has no obvious effects on cell growth and glucose consumption, while the SMO-augmented strains could greatly increase the phytosterol conversion rate and ADD production. Compared with the parent strain M. neoaurum JC-12, the sterol conversion rate of the recombinant strains JC-12S1, JC-12S2 and JC-12S3 was increased from 47.6% to 59.4%, 65.1% and 54.7%, respectively. Accordingly, the ADD yield of the recombinant strains JC-12S1, JC-12S2 and JC-12S3 was enhanced from 5.2 g/L to 6.5 g/L, 7.3 g/L and 6.1 g/L with an increase of 25.0%, 40.4% and 17.3%. These results suggested that ADD production could be enhanced by SMO over expression, and SMO2-augmentation exhibited the highest level of ADD yield improvement, which further verified that SMO2 was more important than SMO1 and SMO3 in sterol catabolism. In a previous study, although ChoM2 is more important than ChoM1 in the sterol catabolism in M. neoaurum, the ChoM1-augmented strains and the ChoM2-augmented strains exhibited no obvious difference in their transformation capacity to accumulate ADD or AD [45]. Unlikely, in this study, the more important role of SMO2 than SMO1 and SMO3 in sterol catabolism lead to higher ADD yield of SMO2-augmented strain than that of SMO1 or SMO3-augmented strains. All these results confirmed that SMOs play an important role in the sterol catabolic pathway, and SMOs augmentation is beneficial to ADD accumulation in M. neoaurum. This is the first report about the application of SMOs in improving ADD production, and these results strongly suggested that the SMOs over expression in the sterols transforming mycobacteria might be a viable way to enhance sterol transformation to valuable steroid intermediates. However, the ADD concentration gradually decreased after 144 h, and this was mainly due to the ADD degradation catalyzed by the KSH enzyme [4]. Thus, it is imperative to block the ADD degradation pathway by ksh gene disruption to realize ADD accumulation.

Effects of the augmentation of SMOs on strain growth (a), glucose consumption (b), phytosterol conversion (c) and ADD production (d) in M. neoaurum. 20 g/L phytosterol (added with 60 g/L HP-β-CD) was supplemented as the substrate into the fermentation medium to conduct the transformation. N the number of CFU (colony forming units) per 1 mL of culture fluid. Glucose was fed when needed. All assays were performed in triplicate with three independent measurements. Error bars represented standard deviations of the biological replicates
Blocking the pathway of ADD degradation by kshA gene disruption
As is well known, KSH activity is represented by two components: terminal oxygenase (KshA) and ferredoxin reductase (KshB) in steroid 9α-hydroxylation [46]. A diversity of KshA activity can be usually observed in some strong sterol-using strains, indicating its significant role in the 9α-hydroxylation process of catalyzing the conversion of AD to 9α-OH-AD and degradation of ADD to 9α-OH-ADD in M. neoaurum [23, 38]. Therefore, we attempted to inactivate the KSH activity by disrupting the KshA component in M. neoaurum JC-12.
In this work, the kshA gene was knocked out to construct the mutant strain JC-12ΔkshA. Compared with M. neoaurum JC-12, KSH enzyme activity of JC-12ΔkshA was hardly detected, which verified the successful disruption of KSH (Fig. S4). As shown in Fig. 4, fermentation curves showed that there were no difference in biomass and residual glucose between JC-12ΔkshA and M. neoaurum JC-12, indicating that kshA gene disruption has no obvious effect on M. neoaurum cell growth. During sterol transformation by M. neoaurum JC-12, ADD reached the highest production of 5.2 g/L and decreased gradually after 144 h. This was because sterols could not be transformed into ADD, while ADD incessantly degraded into 9α-OH-ADD by catalysis of KSH. On the contrary, during the JC-12ΔkshA sterol transformation process, ADD production was higher than that of M. neoaurum JC-12 and the highest yield reached 6.5 g/L at 144 h with no reduction later. This was because the KSH enzyme inactivation lead to the obstruction of the ADD 9α-hydroxylation pathway, which resulted in the higher ADD accumulation. Therefore, it is beneficial for ADD production through KSH disruption.

Fermentation curves of M. neoaurum JC-12 (hollow symbols) and JC-12ΔkshA (solid symbols). The fermentation was carried out with 20 g/L phytosterol (added with 60 g/L HP-β-CD). Glucose was fed when necessary. The parameters, such as glucose (inverted triangle), the biomass (square) and ADD concentration (triangle) were concurrently recorded during the fermentation. All assays were performed in triplicate with three independent measurements. Error bars represented standard deviations of the biological replicates
In the sterol catabolism process, ADD is converted to 9α-OH-ADD by KSH catalysis, while 9α-OH-ADD is an unstable compound followed by non-enzymatic ring B destruction with further full degradation [41, 42]. To protect the steroidal nucleus, previous efforts mainly focused on the chemical inhibition of KSH and the screening and improvement of microorganisms for higher AD/ADD yield [19]. In this work, we disrupted the kshA gene to block the KSH-catalyzed pathway from AD/ADD to 9α-OH-AD/9α-OH-ADD. The results showed that kshA deletion had no effect on cell growth, while ADD yield of JC-12ΔkshA increased to some extent and ADD did not degrade. Therefore, inactivation of KSH activity was supposed to be a fundamental premise to develop a promising ADD biocatalyst.
Significantly enhanced ADD production by coexpression of ChoM, SMO and KSDD in KSH-disrupted mutant JC-12ΔkshA
It has been reported that ChoM and KSDD were key enzymes in sterol transformation [26, 48], while SMO2 was also identified as a key enzyme in sterol catabolism and played a more important role than SMO1 and SMO3. Thus, we attempted to strengthen the sterol metabolic flux by coexpressing ChoM, SMO2 and KSDD in mutant JC-12ΔkshA to realize a higher ADD production.
The successful construction of the recombinant strain JC-12S2-choM-ksdd/ΔkshA was confirmed by DNA sequencing and SDS-PAGE analysis (data not shown), and the enzyme activity assay further certified the functional expression of these three enzymes (Table S4). As shown in Fig. 5a, the biomass of JC-12S2-choM-ksdd/ΔkshA was slightly lower than that of JC-12ΔkshA. This was mainly because the over expression of three enzymes possibly increased the burden of cell growth, but this effect was not obvious. As shown in Fig. 5b, compared with JC-12ΔkshA, sterol conversion amount of JC-12S2-choM-ksdd/ΔkshA enhanced from 49.6% to about 90%. Accordingly, the ADD yield increased from 6.5 to 11.6 g/L, almost a 78% increase (Fig. 5c). These results showed that the coexpression of ChoM, SMO2 and KSDD significantly improved the ADD production of M. neoaurum JC-12. In summary, it is a feasible metabolic engineering means to increase the transformation efficiency of sterols to the valuable steroid intermediate ADD by augmentation of ChoM, SMO2 and KSDD and disruption of KSH in M. neoaurum.

Effects of the coexpression of ChoM, SMO and KSDD on ADD production in mutant JC-12ΔkshA. The fermentation was carried out with 20 g/L phytosterol (added with 60 g/L HP-β-CD). Glucose was fed when necessary. The parameters, such as the biomass, phytosterol conversion rate and ADD yield were concurrently recorded during the fermentation. All assays were performed in triplicate with three independent measurements. Error bars represented standard deviations of the biological replicates
Some efforts have been made to identify the key enzymes involved in sterol transformation and improve the yield of the valuable steroid intermediates. Wei et al. reported the inactivation and augmentation of the primary KSDD in M. neoaurum NwIB-01 to increase the AD or ADD production from soybean phytosterol biotransformation [39]. Yao et al. identified two cholesterol oxidases involved in the initial step of sterol catabolism in M. neoaurum, and the augmentation of the ChoM2 activity achieved the increased AD and ADD production in M. neoaurum NwIB-R10 and in M. neoaurum NwIB-01MS, respectively [45]. However, this work only used genetic engineering by expressing or disrupting the gene in host strains, and no metabolic engineering strategy was used for enhancing ADD production. In the present work, we firstly identified the function of SMOs in sterols catabolism in M. neoaurum JC-12. Then, we coexpressed ChoM, SMO2 and KSDD in the KSH-disrupted strain to strengthen the metabolic flux. The final ADD production improved significantly. This is the first report for improving ADD yield through metabolic engineering strategy.
To evaluate the applicability of the strain JC-12S2-choM-ksdd/ΔkshA in industry scale, the performance of this recombinant strain was carried out in a 5-L fermentor using 30 g/L phytosterol as substrate. As shown in Fig. 6, compared with the ADD production (7.9 g/L) by M. neoaurum JC-12 at 144 h, the recombinant strain JC-12S2-choM-ksdd/ΔkshA produced the maximum ADD yield of 20.1 g/L at 120 h, with the conversion rate of 0.168 g/L/h, molar yield of 91.6% and ADD/AD molar ratio of 20:1. To our knowledge, this is the highest ADD production ever reported (Table 3). These results indicated that metabolic engineering of sterol catabolic pathway could be an effective strategy to increase the production of valuable steroidal intermediates from low-cost sterols in pharmaceutical industry scale.

Time profiles of ADD fermentation of parent strain M. neoaurum JC-12 (a) and the recombinant strain JC-12S2-choM-ksdd/ΔkshA (b) in a 5-L fermentor. The fermentation was carried out with 30 g/L phytosterol (added with 90 g/L HP-β-CD). Glucose was fed when necessary. All assays were performed in triplicate with three independent measurements. Error bars represented standard deviations of the biological replicates
Conclusion
Here, we identified three C27 monooxygenase isoenzymes as the key enzymes involved in sterol catabolism and determined SMO2 as the strongest one in M. neoaurum C27-hydroxylation. By blocking KSH to prevent the ADD degradation pathway and coexpressing three key enzymes, ChoM, SMO2 and KSDD, the final ADD yield reached 20.1 g/L, which is the highest ever reported. This work provides new insight into the redesigned microreactor, which efficiently produces ADD through metabolic engineering strategy. This strategy also paves the way for developing M. neoaurum as a microbial factory for the efficient production of other valuable steroid metabolites from sterols in the pharmaceutical industry.
Abbreviations
- AD:
-
4-Androstene-3,17-dione
- ADD:
-
Androst-1,4-diene-3,17-dione
- 9α-OH-AD:
-
9α-Hydroxy-4-androstene-3,17-dione
- 9α-OH-ADD:
-
9α-Hydroxy-androst-1,4-diene-3,17-dione
- 3β-HSD:
-
3β-Hydroxysteroid dehydrogenase
- CFU:
-
Colony forming units
- ChoM:
-
Cholesterol oxidase
- SMO:
-
Steroid C27 monooxygenase
- HP-β-CD:
-
Hydroxymethyl-β-cyclodextrin
- KSDD:
-
3-Ketosteroid-Δ1-dehydrogenase
- KSH:
-
3-Ketosteroid 9α-hydroxylase
References
Bao T, Zhang X, Rao ZM, Zhao XJ, Zhang RZ, Yang TW, Xu ZH, Yang ST (2014) Efficient whole-cell biocatalyst for acetoin production with NAD+ regeneration system through homologous co-expression of 2,3-butanediol dehydrogenase and NADH oxidase in engineered Bacillus subtilis. PLoS One 9:e102951. https://doi.org/10.1371/journal.pone.0102951
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Brzostek A, Śliwiński T, Rumijowska-Galewicz A, Korycka-Machała M, Dziadek J (2005) Identification and targeted disruption of the gene encoding the main 3-ketosteroid dehydrogenase in Mycobacterium smegmatis. Microbiology 151:2393–2402
Capyk JK, Casabon I, Gruninger R, Strynadka NC, Eltis LD (2011) Activity of 3-ketosteroid 9α-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J Biol Chem 286:40717–40724
Capyk JK, Kalscheuer R, Stewart GR, Liu J, Kwon H, Zhao R, Okamoto S, Jacobs WR, Eltis LD, Mohn WW (2009) Mycobacterial cytochrome P450 125 (Cyp125) catalyzes the terminal hydroxylation of C27-steroids. J Biol Chem 284:35534–35542. https://doi.org/10.1074/jbc.M109.072132
Chaudhari P, Chaudhari B, Chincholkar S (2010) Cholesterol biotransformation to androsta-1,4-diene-3,17-dione by growing cells of Chryseobacterium gleum. Biotechnol Lett 32:695–699. https://doi.org/10.1007/s10529-010-0206-z
Chen MM, Wang FQ, Lin LC, Yao K, Wei DZ (2012) Characterization and application of fusidane antibiotic biosynthesis enzyme 3-ketosteroid-∆ 1-dehydrogenase in steroid transformation. Appl Microbiol Biotechnol 96:133–142
Chen YR, Huang HH, Cheng YF, Tang TY, Liu WH (2006) Expression of a cholesterol oxidase gene from Arthrobacter simplex in Escherichia coli and Pichia pastoris. Enzyme Microb Technol 39:854–860. https://doi.org/10.1016/j.enzmictec.2006.01.018
Chiang YR, Ismail W, Heintz D, Schaeffer C, Van Dorsselaer A, Fuchs G (2008) Study of anoxic and oxic cholesterol metabolism by Sterolibacterium denitrificans. J Bacteriol 190:905–914. https://doi.org/10.1128/Jb.01525-07
Donova MV (2007) Transformation of steroids by actinobacteria: a review. Appl Biochem Microbiol 43:1–14. https://doi.org/10.1134/s0003683807010012
Donova MV, Egorova OV (2012) Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol 94:1423–1447. https://doi.org/10.1007/s00253-012-4078-0
Gordhan BG, Parish T (2001) Gene replacement using pretreated DNA. In: Parish T, Stoker NG (eds) Mycobacterium tuberculosis protocols, vol 54. Methods in molecular medicine. Humana Press Inc, Totowa, pp 77–92. https://doi.org/10.1385/1-59259-147-7:077
Gulla V, Banerjee T, Patil S (2010) Bioconversion of soysterols to androstenedione by Mycobacterium fortuitum subsp. fortuitum NCIM 5239, a mutant derived from total sterol degrader strain. J Chem Technol Biotechnol 85:1135–1141
Hanson JR (2005) Steroids: reactions and partial synthesis. Nat Prod Rep 22:104–110
Huang CL, Chen YR, Liu WH (2006) Production of androstenones from phytosterol by mutants of Mycobacterium sp. Enzyme Microb Technol 39:296–300
Li Y, Lu F, Sun T, Du L (2007) Expression of ksdD gene encoding 3-ketosteroid-Δ1-dehydrogenase from Arthrobacter simplex in Bacillus subtilis. Lett Appl Microbiol 44:563–568
Liu YC, Chen GY, Ge FL, Li W, Zeng L, Cao W (2011) Efficient biotransformation of cholesterol to androsta-1,4-diene-3,17-dione by a newly isolated actinomycete Gordonia neofelifaecis. World J Microbiol Biotechnol 27:759–765. https://doi.org/10.1007/s11274-010-0513-5
Mahato SB, Garai S (1997) Advances in microbial steroid biotransformation. Steroids 62:332–345
Malaviya A, Gomes J (2008) Androstenedione production by biotransformation of phytosterols. Bioresour Technol 99:6725–6737. https://doi.org/10.1016/j.biortech.2008.01.039
Molchanova MA, Andryushina VA, Savinova TS, Stytsenko TS, Rodina NV, Voishvillo NE (2007) Preparation of androsta-1,4-diene-3,17-dione from sterols using Mycobacterium neoaurum VKPM Ac-1656 strain. Russ J Bioorganic Chem 33:354–358. https://doi.org/10.1134/s1068162007030132
Ouellet H, Guan S, Johnston JB, Chow ED, Kells PM, Burlingame AL, Cox JS, Podust LM, De Montellano PRO (2010) Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol Microbiol 77:730–742. https://doi.org/10.1111/j.1365-2958.2010.07243.x
Ouellet H, Johnston JB, Ortiz de Montellano PR (2011) Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol 19:530–539. https://doi.org/10.1016/j.tim.2011.07.009
Petrusma M, Hessels G, Dijkhuizen L, van der Geize R (2011) Multiplicity of 3-ketosteroid-9α-hydroxylase enzymes in Rhodococcus rhodochrous DSM43269 for specific degradation of different classes of steroids. J Bacteriol 193:3931–3940
Rosłoniec K (2010) Steroid transformation by Rhodococcus strains and bacterial cytochrome P450 enzymes. Dissertation, University of Groningen
Shao M, Chen Y, Zhang X, Rao Z, Xu M, Yang T, Li H, Xu Z, Yang S (2017) Enhanced intracellular soluble production of 3-ketosteroid-Δ1-dehydrogenase from Mycobacterium neoaurum in Escherichia coli and its application in the androst-1,4-diene-3,17-dione production. J Chem Technol Biotechnol 92:350–357. https://doi.org/10.1002/jctb.5012
Shao ML, Rao ZM, Zhang X, Xu MJ, Yang TW, Li H, Xu ZH, Yang ST (2014) Bioconversion of cholesterol to 4-cholesten-3-one by recombinant Bacillus subtilis expressing choM gene encoding cholesterol oxidase from Mycobacterium neoaurum JC-12. J Chem Technol Biotechnol. https://doi.org/10.1002/jctb.4491
Shao ML, Rao ZM, Zhang X, Xu MJ, Yang TW, Li H, Xu ZH, Yang ST (2015) Bioconversion of cholesterol to 4-cholesten-3-one by recombinant Bacillus subtilis expressing choM gene encoding cholesterol oxidase from Mycobacterium neoaurum JC-12. J Chem Technol Biotechnol 90:1811–1820. https://doi.org/10.1002/jctb.4491
Shao ML, Zhang X, Rao ZM, Xu M, Yang T, Li H, Xu Z, Yang S (2016) A mutant form of 3-ketosteroid-Δ1-dehydrogenase gives altered androst-1,4-diene-3, 17-dione/androst-4-ene-3,17-dione molar ratios in steroid biotransformations by Mycobacterium neoaurum ST-095. J Ind Microbiol Biotechnol 43:691–701. https://doi.org/10.1007/s10295-016-1743-9
Shao ML, Zhang X, Rao ZM, Xu MJ, Yang TW, Li H, Xu ZH (2015) Enhanced production of androst-1,4-diene-3, 17-dione by Mycobacterium neoaurum JC-12 using three-stage fermentation strategy. PLoS One 10:e0137658
Shao ML, Zhang X, Rao ZM, Xu MJ, Yang TW, Li H, Xu ZH, Yang ST (2016) Efficient testosterone production by engineered Pichia pastoris co-expressing human 17β-hydroxysteroid dehydrogenase type 3 and Saccharomyces cerevisiae glucose 6-phosphate dehydrogenase with NADPH regeneration. Green Chem 18:1774–1784. https://doi.org/10.1039/c5gc02353j
Sharma P, Slathia P, Somal P, Mehta P (2012) Biotransformation of cholesterol to 1,4-androstadiene-3,17-dione (ADD) by Nocardia species. Ann Microbiol 62:1651–1659. https://doi.org/10.1007/s13213-012-0422-y
Shen YB, Wang M, Zhang LT, Ma YH, Ma B, Zheng Y, Liu H, Luo JM (2011) Effects of hydroxypropyl-β-cyclodextrin on cell growth, activity, and integrity of steroid-transforming Arthrobacter simplex and Mycobacterium sp. Appl Microbiol Biotechnol 90:1995–2003. https://doi.org/10.1007/s00253-011-3214-6
Sripalakit P, Wichai U, Saraphanchotiwitthaya A (2006) Biotransformation of various natural sterols to androstenones by Mycobacterium sp. and some steroid-converting microbial strains. J Mol Catal B Enzym 41:49–54. https://doi.org/10.1016/j.molcatb.2006.04.007
Szentirmai A (1990) Microbial physiology of sidechain degradation of sterols. J Ind Microbiol 6:101–115
Tong WY, Dong X (2009) Microbial biotransformation: recent developments on steroid drugs. Recent Pat Biotechnol 3:141–153
Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci USA 104:1947–1952. https://doi.org/10.1073/pnas.0605728104
Wang ZL, Zhao FS, Chen DJ, Li D (2006) Biotransformation of phytosterol to produce androsta-diene-dione by resting cells of Mycobacterium in cloud point system. Process Biochem 41:557–561. https://doi.org/10.1016/j.procbio.2005.09.014
Wei W, Fan SY, Wang FQ, Wei D (2010) A new steroid-transforming strain of Mycobacterium neoaurum and cloning of 3-ketosteroid 9α-hydroxylase in NwIB-01. Appl Biochem Biotechnol 162:1446–1456. https://doi.org/10.1007/s12010-010-8919-y
Wei W, Wang FQ, Fan SY, Wei DZ (2010) Inactivation and augmentation of the primary 3-ketosteroid-Δ1-dehydrogenase in Mycobacterium neoaurum NwIB-01: biotransformation of soybean phytosterols to 4-androstene-3,17-dione or 1,4-androstadiene-3,17-dione. Appl Environ Microbiol 76:4578–4582. https://doi.org/10.1128/aem.00448-10
Wilbrink MH (2011) Microbial sterol side chain degradation in Actinobacteria. University of Groningen
Wovcha MG, Antosz FJ, Knight JC, Kominek LA, Pyke TR (1978) Bioconversion of sitosterol to useful steroidal intermediates by mutants of Mycobacterium fortuitum. Biochim Biophys Acta 531:308–321. https://doi.org/10.1016/0005-2760(78)90213-8
Wovcha MG, Brooks KE, Kominek LA (1979) Evidence for two steroid 1,2-dehydrogenase activities in Mycobacterium fortuitum. Biochim Biophys Acta 574:471–479. https://doi.org/10.1016/0005-2760(79)90243-1
Xu XX, Jin FL, Yu XQ, Ji SX, Wang J, Cheng HX, Wang C, Zhang WQ (2007) Expression and purification of a recombinant antibacterial peptide, cecropin, from Escherichia coli. Protein Expr Purif 53:293–301. https://doi.org/10.1016/j.pep.2006.12.020
Yang X, Dubnau E, Smith I, Sampson NS (2007) Rv1106c from Mycobacterium tuberculosis is a 3beta-hydroxysteroid dehydrogenase. Biochemistry 46:9058–9067. https://doi.org/10.1021/bi700688x
Yao K, Wang FQ, Zhang HC, Wei DZ (2013) Identification and engineering of cholesterol oxidases involved in the initial step of sterols catabolism in Mycobacterium neoaurum. Metab Eng 15:75–87. https://doi.org/10.1016/j.ymben.2012.10.005
Yao K, Xu LQ, Wang FQ, Wei DZ (2014) Characterization and engineering of 3-ketosteroid-∆1-dehydrogenase and 3-ketosteroid-9α-hydroxylase in Mycobacterium neoaurum ATCC 25795 to produce 9α-hydroxy-4-androstene-3,17-dione through the catabolism of sterols. Metab Eng 24:181–191. https://doi.org/10.1016/j.ymben.2014.05.005
Zhang LT, Wang M, Shen YB, Ma YH, Luo JM (2009) Improvement of steroid biotransformation with hydroxypropyl-β-cyclodextrin induced complexation. Appl Biochem Biotechnol 159:642–654. https://doi.org/10.1007/s12010-008-8499-2
Zhang WQ, Shao ML, Rao ZM, Xu MJ, Zhang X, Yang TW, Li H, Xu ZH (2013) Bioconversion of 4-androstene-3,17-dione to androst-1,4-diene-3,17-dione by recombinant Bacillus subtilis expressing ksdd gene encoding 3-ketosteroid-Δ1-dehydrogenase from Mycobacterium neoaurum JC-12. J Steroid Biochem Mol Biol 135:36–42. https://doi.org/10.1016/j.jsbmb.2012.12.016
Zhang X, Zhang RZ, Bao T, Rao ZM, Yang TW, Xu MJ, Xu ZH, Li HZ, Yang ST (2014) The rebalanced pathway significantly enhances acetoin production by disruption of acetoin reductase gene and moderate-expression of a new water-forming NADH oxidase in Bacillus subtilis. Metab Eng 23:34–41. https://doi.org/10.1016/j.ymben.2014.02.002
Acknowledgements
We sincerely appreciate Professor W. R. Jacobs, Jr. (Howard Hughes Medical Institute, USA) for providing plasmids pMV261 and pMV306, and Professor T. Parish (Department of Infectious & Tropical Diseases, United Kingdom) for providing plasmids of p2NIL and pGOAL19. This work was supported by the National Natural Science Foundation of China (31700041 and 31570085), the China Post-doctoral Science Foundation Funded Project (2017M610297), the Jiangsu Province Post-doctoral Science Foundation (1701100B), The Jiangsu Province Science Fund for Distinguished Young Scholars (BK20150002), the 111 Project (111-2-06) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shao, M., Zhang, X., Rao, Z. et al. Identification of steroid C27 monooxygenase isoenzymes involved in sterol catabolism and stepwise pathway engineering of Mycobacterium neoaurum for improved androst-1,4-diene-3,17-dione production. J Ind Microbiol Biotechnol 46, 635–647 (2019). https://doi.org/10.1007/s10295-018-02135-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-02135-5