
Abstract
Microbial ∆ 1 -dehydrogenation is one of the most important transformations in the synthesis of steroid hormones. In this study, a 3-ketosteroid-∆1-dehydrogenase (kstDF) involved in fusidane antibiotic biosynthesis from Aspergillus fumigatus CICC 40167 was characterized for use in steroid transformation. KstDF encodes a polypeptide consisting of 637 amino acid residues. It shows 51% amino acid identity with a kstD from Thermomicrobium roseum DSM 5159. Expression of kstDF in Escherichia coli and Pichia pastoris showed that all kstDF activity is located in the cytoplasm. This indicates that it is a soluble intracytoplasmic enzyme, unlike most kstDs from bacteria, which are membrane-bound. The expression of kstDF was performed in P. pastoris, both intracellularly and extracelluarly. The intracellularly expressed protein displayed good activity in steroid transformation, while the extracellularly expressed protein showed nothing. Interestingly, the engineered P. pastoris KM71 (KM71I) and GS115 (GS115I) showed different transformation activities for 4-androstene-3,17-dione (AD) when kstDF was expressed intracellularly. Under the same conditions, KM71I was found capable of transforming 1.0 g/l AD to 1,4-androstadiene-3,17-dione (ADD), while GS115I could transform 1.5 g/l AD to both ADD and boldenone (BD). The production of BD is attributed to a 17β-hydroxysteroid dehydrogenase in P. pastoris GS115I, which catalyzes the reversible reaction between C17-one and C17-alcohol of steroids. The conversion of AD by GS115I and KM71I may provide alternative means of preparing ADD or BD. In brief, we show here that kstDF is a promising enzyme in steroid ∆ 1 -dehydrogenation that is propitious to construct genetically engineered steroid-transforming recombinants by heterologous overexpression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
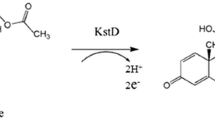
Microbial transformations play irreplaceable roles in the synthesis of steroid hormones. Among these, ∆ 1-dehydrogenation is one of the most crucial transformations. The introduction of C1,2 double bonds to some steroid hormones, such as adrenocortical hormones, may greatly increase their activities and decrease their side effects (Abul-hajj 1972). The ∆ 1-dehydrogenation of steroids by organisms is carried out by 3-ketosteroid-∆1-dehydrogenases (kstDs) [4-ene-3-oxosteroid:(acceptor)l-ene-oxidoreductase, EC 1.3.99.4]. These are flavoproteins functioning in the conversion of 4-ene-3-oxosteroid to 1,4-diene-3-oxosteroid by trans-axial elimination of the Cl and C2 hydrogens ( Itagaki et al. 1990). The transformation of 4-androstene-3,17-dione (AD) to 1,4-androstadiene-3,17-dione (ADD) is one example of this.
In general, isolated microorganisms with good kstD activity are bacteria, mainly actinobacteria. The physiological role of kstDs in these microorganisms involves the use of steroids as carbon sources. They initiate cleavage of the nucleus of steroids by the introduction of a double bond into ring A (Chiang et al. 2008; Florin et al. 1996). Therefore, these microorganisms are usually not good biocatalysts of steroids ∆ 1-dehydrogenation due to their powerful steroid-degradation capacities as well as the other side reactions (Chiang et al. 2008; Donova 2007; Szentirmai 1990). This is why molecular characterization and heterologous expression of kstDs have attracted such attention as means of creating recombinant kstD bacteria (Choi et al. 1995; Li et al. 2007; Morii et al. 1998; Plesiat et al. 1991). KstDs have been isolated and characterized from many actinobacteria, including Pseudomonas sp., Nocardia sp., Rhodococus sp., Mycobacterium sp., and Arthrobacter sp. So far, however, all of the kstDs found in bacteria have been membrane-bound and localized in the inner membrane fraction by two hydrophobic transmembrane domains. These include kstDs from Arthrobacter simplex, Pseudomonas testosterone, Rhodococcus erythropolis, and Mycobcterium sp. (Li et al. 2007; Molnar et al. 1995; Plesiat et al. 1991; Wagner et al. 1992). Many attempts have been made to express these kstDs heterologously in Escherichia coli, Streptomyces lividans, and Bacillus subtilis, and some experiments have achieved a 30 to 100-fold increases in kstD production over the original strains (Choi et al. 1995; Li et al. 2007; Morii et al. 1998; Plesiat et al. 1991). However, these expressions did not show themselves to be successful upon application. Expressed in E. coli, these bacterial kstDs were found to consist mainly of inclusion bodies, and only a trace activity was detected in the membrane (Li et al. 2007; Plesiat et al. 1991; Wagner et al. 1992). The secretory overexpression systems of S. lividans and B. subtilis were also employed to produce a kstD from A. simplex extracellularly. The results of this attempt indicated that the A. simplex kstD is produced intracelluarly instead of extracellularly (Li et al. 2007; Choi et al. 1995). The expression of the A. simplex kstD in B. subtilis showed itself to be the only version of the protein suitable to steroid conversion in practice. It could transform 1.0 g/l AD to ADD at a 45.3% conversion rate (Li et al. 2007). As well known, membrane-bound proteins are not easy to overexpress in their functional forms in heterologous hosts. Therefore, the membrane-bound attribute of these bacterial kstDs is likely to be what limits their heterogenous overexpression so seriously (Wagner et al. 1992; Freigassner et al. 2009; Linares et al. 2010).
In the course of our research on fusidane antibiotic biosynthesis in Aspergillus fumigatus, we discovered a soluble kstD (kstDF) responsible for ∆ 1-dehydrogenation in steroidal fusidane antibiotic biosynthesis. This may be a better kstD than bacterial kstDs for heterologous expression and application in steroid transformation. KstDF showed no obvious hydrophobic transmembrane domains, and it can be expressed as a soluble enzyme in E. coli and Pichia pastoris. The recombinant P. pastoris with overexpressed kstDF is promising in the conversion of AD to ADD or boldenone (BD).
Materials and methods
Strains, plasmids, media, and chemicals
A. fumigatus CICC 40167 was obtained from the China Center of Industrial Culture Collection (CICC, Beijing, China). E. coli BL21 (λDE3) and pET-28a(+) were from Merck Biosciences (Germany). P. pastoris GS115, P. pastoris KM71, and expression vectors pPIC3.5 k and pPIC9k were from Invitrogen (USA). Restriction endonucleases, T4 DNA ligase, and DNA polymerase were from Takara (Japan). Steroids including 4-androstene-3,17-dione, 1,4-androstadiene-3,17-dione, testosterone, and boldenone were from Sigma (USA). E. coli strains were cultured in Luria-Bertani (LB) medium (Difco, Detroit, MI, USA). Buffered gylcerol-complex medium (BMGY) and buffered methanol-complex medium (BMMY) were used for the growth and heterologous protein expression of P. pastoris strains. The BMGY was composed of 2% peptone, 1% yeast extract, 100 mM potassium phosphate pH 6.0, 1.34% yeast nitrogen base (w/o amino acid (AA)), 0.4 μg/ml biotin, and 1% glycerol. The BMMY was composed of 2% peptone, 1% yeast extract, 100 mM potassium phosphate pH 6.0, 1.34% yeast nitrogen base (w/o AA), 0.4 μg/ml biotin, and 0.5% methanol.
Heterologous expression of kstDF
According to the available genome sequence of A. fumigatus Af293 (GenBank AAHF00000000), primers kstD-UE (GAATTCCCTGATGCTGGAGATGGG) and kstD-DE (AAGCTTGACCTTGCCGTGGTTGTT) were designed to amplify the entire open reading frame (ORF) of kstD F from A. Fumigatus CICC 40167. The PCR product was ligated into the pMD19-T vector according to the manufacturer’s instructions (Takara). Then, the kstD F gene was digested with HindIII/EcoRI and ligated into the expression vector pET-28a(+), creating pET-28a(+)-kstD F . The resulting recombinant plasmid was then used to transform E. coli BL21 (λDE3) according to the manufacturer’s instructions, creating BL21-pET28a-kstD F .
The entire ORF of kstD F with the EcoRI/NotI sites was cloned with primers kstD-UP (GAATTCATGGCCGCAAGACAGCTC) and kstD-DP (GCGGCCGCCTATACATGCTCAGAAGCAATAT). The resulting fragment digested with EcoRI and NotI was ligated into the pPIC3.5 K and pPIC9K, resulting in the recombinant pPIC3.5 K-kstD F and pPIC9K-kstD F . Both pPIC3.5 K-kstD F and pPIC9K-kstD F were linearized with SalI and then electrotransformed (2.5 kV, 25 μF, 1000 Ω) into P. pastoris KM71 and GS115 cells. High-level expression transformants were screened from YPDS plates containing Geneticin at a final concentration of 0.5 g/l. These were named KM71I, GS115I, KM71E, and GS115E.
The expression of kstDF in E. coli BL21(λDE3) was performed in 30 ml LB per 250 ml Erlenmeyer flask at 37 °C with shaking at 200 rpm. When the optical density at 600 nm (OD600) reached a value of 0.5–0.6, IPTG was added to a final concentration of 0.5 mM. The cells were then grown for another 8 h with shaking at 200 rpm at different induction temperatures: 20 °C, 30 °C, and 37 °C. Thirty milliliters of culture was collected, centrifuged for cells, and resuspended in 50 mM Tris-HCl (pH 7.0) to adjust OD600 to 4.0. Resuspendant was prepared for the subsequent sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) using E. coli BL21(λDE3) transformed with pET-28a(+) as a control (BL21-pET28a).
The expression of kstDF in P. pastoris was conducted according to the protocol described by Handumrongkul et al. (1998). The intracellular expression cell lysate and the extracellular expression culture supernatant were analyzed separately by SDS-PAGE.
Whole-cell steroid biotransformation
The cultures of BL21-pET28a-kstD F grown in LB medium and induced at 20 °C, 30 °C, and 37 °C were incubated with AD at a final concentration of 0.1 g/l for 4 h at 30 °C with shaking at 200 rpm. The reaction mixture was extracted three times with the same volume of ethyl acetate. The organic phases were collected and detected by thin layer chromatography (TLC) and high-performance liquid chromatography (HPLC) using BL21-pET28a as a control.
KM71I and GS115I were cultured for 20–24 h at 30 °C in BMGY medium (30 ml per 250 ml flask) with shaking at 200 rpm. The cells were harvested and resuspended in BMMY medium (30 ml per 250 ml flask) for protein induction. After 6 days, AD dissolved in methanol was added into the BMMY medium to a final concentration of 0.25 g/l. The reaction mixtures were incubated at 30 °C for 2 h and 4 h, respectively, and then extracted for TLC and HPLC analysis. P. pastoris KM71 and GS115 were used as controls.
The culture process for KM71E and GS115E was identical to that used for KM71I and GS115I. After 6 days of induction in BMMY medium, the whole cell and the culture supernatants of KM71E and GS115E were collected, respectively, and used to transform AD. AD dissolved in methanol was added to a final concentration of 0.25 g/l, and phenazine methosulfate (PMS) (dissolved in 50 mM Tris-HCl buffer, pH 7.0) as an exogenous electronic acceptor for kstDF was added to final concentrations of 0.013 g/l and 0.039 g/l for KM71E and GS115E, respectively. The reaction mixtures were incubated at 30 °C for 4 h and then extracted for TLC and HPLC analysis. The culture supernatants of P. pastoris KM71 and GS115 were used as controls.
To test the AD conversion capabilities of KM71I and GS115I, AD dissolved in methanol was added to the BMMY cultures with substrate concentration gradients of 1 g/l, 1.5 g/l, 2 g/l, and 2.5 g/l. The reaction mixtures were incubated at 30 °C for 6 days and then extracted three times with the same volume of ethyl acetate. Samples were treated as described previously for TLC and HPLC analysis. P. pastoris KM71 and GS115 were set as controls.
Analytical methods
Steroid transformation was analyzed by TLC and HPLC. TLC was performed on a TLC plate (HSGF254, Qingdao Marine Chemical Factory, China), developed by petroleum ether/ethyl acetate (6:4 v/v), and visualized by spraying with 20% H2SO4 and heating at 100 °C for about 5 min until the colors developed.
HPLC was carried out with an Agilent 1100 instrument (Agilent Technologies, USA). The chromatographic conditions were as follows: an Agilent XDB-C18 column (4.6 × 250 mm) with a constant temperature of 40 °C, a mobile phase of methanol/water (70:30), and a flow rate of 1 ml/min. Analyses were performed simultaneously with UV detection at 254 nm. Liquid chromatography–mass spectrometry (LC–MS) was performed with an Agilent 1100 LC/MSD apparatus (Agilent Technologies, USA) with conditions as follows: electron spray impact; ion spray voltage, 30 v; ion source temperature, 100 °C; and desolvation temperature, 300 °C.
Sequence analysis and multiple sequence alignments were performed using Clustal X2.1 (Larkin et al. 2007). Analysis with basic local alignment search tool (BLAST) was conducted on a NCBI BLAST server (http://www.ncbi.nlm.nih.gov/BLAST/). The transmembrane regions and orientation were predicted by TMpred program (http://www.ch.embnet.org/software/TMPRED_form.html). The theoretical molecular mass and isoelectric point (pI) of the protein were calculated using the ProtParam tool (http://us.expasy.org/tools/protparam.html).
Isolation of subcellular fractions and localization of the soluble kstDF
The cellular distribution of kstDF in BL21-pET28a-kstDF, KM71I, and GS115I was determined by measuring the kstD activity in different subcellular fractions. Cells were collected by centrifugation at 4 °C (10 min, 5,000 × g). Precipitates were then dissolved in 50 mM Tris-HCl (pH 7.0). E. coli cells were disrupted by sonication for 5 min, while P. pastoris cells were broken by high-pressure cracking. The cell debris were removed by centrifugation at 4 °C (30 min, 10,000 × g). Membranes were sedimented by high-speed centrifugation (1 h, 100,000 × g) and subsequently resuspended in 50 mM Tris-HCl (pH 7.0). Proteins in the 100,000 × g supernatant were defined as soluble proteins. Each of these fractions was then assayed for the kstD activity, which was dissolved in Tris-HCl buffer and cultivated with substrate AD and electronic acceptor PMS at final concentrations of 0.013 g/ml and 0.039 mg/ml, respectively, at 30 °C for 4 h. Equal volumes of ethyl acetate were added three times to extract the steroids. The phase of ethyl acetate was collected and used for TLC and HPLC analyses.
Purification and characterization of kstDF
To determine the kinetic parameters of kstDF in E. coli, Ni2+-chelating sepharose column was used to purify the targeted protein. BL21-pET28a-kstDF cells were lysed by sonication. The cell lysate was centrifuged at 10,000 × g for 20 min, and the supernatant was filtered through 0.45-μm membranes. The flow-through was then applied to a 15-ml Ni2+-chelating Sepharose Fast Flow column (Amersham Biosciences, USA) which had been pre-equilibrated with buffer A (5 mM imidazole, 0.5 M NaCl, 20 mM phosphate buffer, pH = 8.0). The protein was eluted with buffer B (200 mM imidazole, 0.5 M NaCl, 20 mM phosphate buffer, pH = 8.0), followed by a 5-ml Hitrap desalting column (Amersham Biosciences) to remove salts. The desalting column was used according to the manufacturer’s instructions. The concentration of kstDF protein was estimated by UV absorption and the bandscan method.
To determine the kinetic parameters of kstDF expressed in P. pastoris, the kstDF protein was purified by hydrophobic interaction chromatography column with conditions as follows: HiPrep 16/10 Phenyl FF (high sub) column, AKTA explorer (Amersham Biosciences); mobile phase, 20 mM Tris-HCl (pH 7.0) containing 1 M (NH4)2SO4; and elution, 20 mM Tris-HCl (pH 7.0) containing 0.2 M (NH4)2SO4.
The kstDF enzyme activity assay was performed as described previously (van der Geize et al. 2002). One unit is defined as the amount of enzyme capable of transforming of 1 mM AD to ADD in 1 min.
Determination of the derivatives of AD and ADD
Two derivatives of AD and ADD were detected in the fermentation broth of GS115I, when GS115I was added with 2.5 g/l AD. Under the 254 nm UV light, we located the two steroids on silica gel TLC plates, scraped them from silica gel TLC plates, and then extracted them with ethyl acetate. The extract was evaporated and resuspended in methanol, which was analyzed by HPLC and LC–MS.
Results
KstDF from A. fumigatus CICC 40167
Antibiotic fusidanes are a category of steroidal secondary metabolites. They have been detected in about 20 fungal species of the Pezizomycotina. Helvolic acid is a typical fusidane. It has a C1,2-double bond and A. fumigatus is a known producer. In 2009, a gene cluster encoding helvolic acid biosynthesis in A. fumigatus Af293 was discovered by two separate labs. This cluster includes the gene Afu4g14850, which is speculated to encode a kstD responsible for the C1,2-double bond in helvolic acid ( Lodeiro et al. 2009; Mitsuguchi and Seshime 2009).
It has been demonstrated that the clusters of helvolic acid and the other fusidanes are distinguished for lack of introns (Lodeiro et al. 2009). Thus, the complete ORF of kstD (kstD F ) (gene accession number: XM_746255.1) was directly cloned from A. fumigatus CICC 40167 according to the sequence of Afu4g14850 from the genome sequence of A. fumigatus Af293 (Nierman et al. 2005). The kstD F is 99% identical to Afu4g14850, which consists of 1,791 nucleotides and encodes a deduced protein of 597 amino acids. The molecular weight of kstDF was estimated to be 64.37 kDa, and the pI value was calculated to be 7.81. A BLASTp search indicated that the deduced amino acid sequence of kstD F shared the highest identity, 51%, with the kstD from Thermomicrobium roseum (Genbank P12676). No other kstD from fungi had been deposited in GenBank. The BLASTp search also showed that the deduced amino acid sequence of kstD F shared lower similarities with well-defined bacterial kstDs from Rhodococcus rhodochrous (GenBank AB007847), A. Simplex (GenBank D37969.1), and M. neoaurum (GeneBank GQ228843) (34%, 36%, and 44%, respectively) (Molnar et al. 1995; Morii et al. 1998; Wei et al. 2010). Sequence comparison of kstDF with these bacterial kstDs (Fig. 1) revealed that residues 299 through 320 are an active center, and the putative N-terminal flavin adenine dinucleotide (FAD)-binding motif in kstDF is consistent with the sequence previously described: GSG(A/G)(A/G)(A/G)X17E (Wierenga et al. 1986). In the active site, the conserved residues Arg-320 in kstDF (Arg-242 in the A. simplex kstD) have been implicated to be involved in substrate binding, while the highly conserved residues Glu-318 (Glu-240 in the A. simplex kstD, Glu-239 in the R. rhodochrous kstD and Glu-282 in the M. neoaurum kstD) regarded as electrophilic zones are substituted by Gly-318 in kstDF (Molnar et al. 1995).

Sequence alignment analysis of kstDF against well-defined kstDs. The consensus amino acid residues for FAD binding and the active center are shown in the solid line box and underline, respectively. Hydrophobic stretches that may form transmembrane domains are shown in the dotted line box. A. fumigatus (A. f.), R. rhodochrous WO3338 (R. r.), A. simplex IFO 12069 (A. s.), and M. neoaurum NwIB-01 (M. n.)
Bacterial kstDs were reported to be membrane-bound proteins localized in the inner membrane fraction by two hydrophobic transmembrane domains (Plesiat et al. 1991; Molnar et al. 1995; Wagner et al. 1992). A kstD from A. simplex was found to have two long hydrophobic stretches that form transmembrane domains (amino acids 193–214 and 379–401) (Fig. 1) (Molnar et al. 1995). The kstD from M. neoaurum NwIB-01 was predicted to possess two transmembrane domains by TMpred program analysis (amino acids 11–31 and 544–561) (Wei et al. 2010). For a kstD from R. rhodochrous, the hydrophobic amino acids, residues 8–13, and residues 30–36 were found to form the β/α/β hydrophobic structure (Morii et al. 1998). However, the TMpred program predicted that kstDF would contain neither typical signal-anchor sequences nor obvious hydrophobic transmembrane stretches, implying that kstDF may be not a membrane-bound protein.
Expression and characterization of kstDF in E. coli BL21(λDE3)
To characterize kstDF, the expression of kstDF was first carried out in E. coli BL21(λDE3) by the expression vector pET-28a(+) to generate the expression strain BL21-pET28a-kstD F . SDS-PAGE results showed an obvious protein band close to the estimated molecular weight of kstDF in the protein profile of BL21-pET28a-kstD F (Fig. 2a). Compared to E. coli BL21(λDE3) with pET-28a(+), BL21-pET28a-kstD F displayed good kstD activity and was capable of transforming AD to ADD (Fig. 2b). The expression and activity of kstDF in BL21-pET28a-kstD F were affected by culture temperatures. As shown in Fig. 2, the expression level of kstDF increased progressively as the temperature increased from 20 °C to 30 °C to 37 °C (Fig. 2a). However, the activity of kstDF decreased progressively as temperature increased (Fig. 2b). The cell culture induced and cultured at 20 °C was capable of transforming 0.015 g/l AD to ADD at a 100% conversion rate in 4 h. In the same conditions, although the amount of kstDF induced and cultured in 37 °C was many times higher than that induced and cultured at 20 °C, the conversion rate of 0.015 g/l AD to ADD by the cell culture induced and cultured at 37 °C was no more than 50%. Therefore, low induction temperature was beneficial to the expression and maintenance of kstDF activity, and excessive expression of kstDF at higher culture temperatures could result in the inactivation of most of the kstDF. This implied that E. coli BL21(λDE3) was not an optimal host for kstDF expression.

Protein expression and kstDF activity for AD transformation by BL21-pET28a-ksdD F . a SDS-PAGE profile of protein. Lane 1: E. coli BL21(λDE3) with pET-28a(+) as a control incubated and cultured at 37 °C for 8 h. Lanes 2–3: BL21-pET28a-ksdDF induced and cultured at 20 °C for 8 h. Lanes 4–5: BL21-pET28a-ksdDF induced and cultured at 30 °C for 8 h. Lanes 6–7: BL21-pET28a-ksdDF induced and cultured at 37 °C for 8 h. The upload volume of samples 1–7 was 10 μl. b TLC chromatogram of AD transformation for 4 h. Lane 1: AD transformed by E. coli BL21(λDE3) with pET-28a(+) as a control. Lane 2: AD transformed by BL21-pET28a-ksdDF induced and cultured at 20 °C. Lane 3: AD transformed by BL21-pET28a-ksdDF induced and cultured at 30 °C. Lane 4: AD transformed by BL21-pET28a-ksdDF induced and cultured at 37 °C. Samples 1–4 were all 0.015 g/l AD transformed by equal volumes of cell culture
Given that the activity of kstDF expressed in E. coli BL21(λDE3) suffered great losses as culture temperature increased, the kstDF expressed at 20 °C was selected for characterization. The kstDF protein was purified by Ni2+-chelating chromatography. The Michaelis constant (K m) of kstDF for AD was determined to be 191 μM.
Almost of all the kstDF activity of BL21-pET28a-kstD F could be sedimented by centrifugation. This indicated that kstDF was located intracellularly in BL21-pET28a-kstD F . To determine whether kstDF might be a membrane-bound protein, the activity distribution of kstDF in BL21-pET28a-kstD F cell was investigated by separating subcellular fractions and determining the kstDF activity. The cell extract prepared from BL21-pET28a-kstD F was fractionated by fractional centrifugation, and the distribution of the kstDF in different fractions was examined. Of the total kstD activity, a major part (97%) resided in the cytosolic fraction, while only 3% was present in the membrane fraction (Table 1), indicating that kstDF is a soluble protein. Significantly, the activity distribution of kstDF in the E. coli expression host was entirely different from those of kstDs from P. testosterone and N. restrictus (Plesiat et al. 1991; Sih and Bennet 1962) (Table 1). This confirmed the TMpred prediction that kstDF would be soluble rather than membrane-bound.
Expression and characterization of kstDF in P. pastoris
The soluble expression of kstDF in E. coli implied that the kstDF may be more suitable for heterogenous expression than bacterial kstDs. For this reason, eukaryotic hosts P. pastoris strain KM71 and strain GS115 were then chosen to overexpress the fungi kstDF. pPIC3.5 K, a powerful intracellular expression plasmid, was firstly selected. The recombinant kstDF strains were designated as KM71I and GS115I. The expression profiles of KM71I and GS115I showed that the expressed kstDF was larger than 66 kDa (Fig. 3a). To test kstDF activity, KM71I and GS115I were cultured for 6 days and selected to transform AD. In both cases, 0.25 g/l AD was entirely transformed to products for no more than 4 h (Fig. 3b). This indicated a more powerful transformation capacity than BL21-pET28a-kstD F .

Protein expression and kstDF activity for AD transformation by KM71I and GS115I. a SDS-PAGE protein profile. Lane 1: wild-type P. pastoris KM71 cultured for 6 days after induction. Lane 2: KM71I cultured for 6 days after induction. Lane 3: GS115I cultured for 6 days after induction. b TLC chromatogram of AD transformation. Lane 1: AD transformed by wild-type GS115 for 2 h. Lane 2: AD transformed by GS115I for 2 h. Lane 3: AD transformed by KM71I for 2 h. Lane 4: AD transformed by GS115I for 4 h. Lane 5: AD transformed by KM71I for 4 h. c HPLC analysis of AD conversion by GS115I for 2 h. d Putative transformation relationship between AD, ADD, TS, and BD as assessed by GS115I
As shown in Fig. 3b, KM71I and GS115I transformed AD to different products. This may be attributed to the original AD transformation capacity of P. pastoris GS115 (Fig. 3b) and KM71 (data not shown). One AD product produced by P. pastoris GS115 and KM71 developed as celadon spots on TLC and was later identified as testosterone (TS) by LC–MS and an authentic sample. Another product, produced by GS115I, developed as brick-red spots on TLC in AD transformation and was identified as boldenone (BD). The AD transformed by GS115I was further analyzed by HPLC. The HPLC profiles of the samples transformed by GS115I for 2 h showed four chemical peaks: ADD (t = 7.5 min), boldenone (t = 10.1 min), AD (t = 11.2 min), and testosterone (t = 14.2 min) (Fig. 3c). The HPLC profiles of the samples transformed by GS115I for 4 h showed no remaining TS. The HPLC profiles also showed that the transformation of AD by KM71I produced mainly ADD without BD, and only trace TS was detected in the sample after 2 h, none after 4 h.
Structurally, biotransformation from AD to TS is usually carried out by 17β-hydroxysteroid-dehydrogenases (17Hsd), which are widespread in microorganisms such as yeasts. The 17Hsds have been characterized as reversible redox enzymes that can transform the reversible reduction/oxidation between C17-one and C17-alcohol of steroids (Donova et al. 2005). Therefore, the transformation relationship between AD, ADD, TS, and BD by GS115I can be described as shown in Fig. 3d. This was further verified by the transformation of ADD and TS to BD by GS115I.
Considering that kstDF was well expressed intracellularly in P. pastoris, the extracellular expression of kstDF in P. pastoris becomes more interesting because it might be more powerful for steroid transformation. pPIC9K, a derived plasmid of pPIC3.5 K that expresses proteins extracellularly, was then used to express kstDF in P. pastoris KM71 and GS115. The recombinant kstDF strains were designated as KM71E and GS115E, respectively. The expression profiles of KM71E and GS115E were consistent with those of kstDF expressed in KM71I and GS115I. Surprisingly, KM71E and GS115E showed no kstDF activity for AD transformation. To further confirm the solubility of kstDF in P. pastoris KM71I and GS115I, cells of KM71I and GS115I were fractionated, and each of these fractions was assayed for kstDF activity. The results of AD transformation demonstrated that both kstDFs from KM71I and GS115I were indeed active as soluble proteins: the distribution of kstDF activity in the cytosolic fraction accounted for 98% and 99% of the total kstDF activity for KM71I and GS115I, respectively. The distinct performances of intracellular and extracellular kstDF expressed in P. pastoris indicated that kstDF is not suitable for extracellular expression in P. pastoris.
To further characterize the activity of kstDF expressed in P. pastoris, kstDF was purified from GS115I by hydrophobic interaction chromatography. Its activity was determined to be 51.68 U/mg. Its K m value was determined to be 47 μM, lower than the K m value (82 μM) of kstD1 from R. erythropolis (van der Geize et al. 2002). The K m value of kstDF from P. pastoris is only a quarter of that from E. coli, indicating that P. pastoris is the more suitable host for kstDF expression.
Application of KM71I and GS115I in the transformation of AD
Steroids are minimally water-soluble. This seriously retards the efficiency of steroid biotransformation because of inadequate mass transfer. Therefore, strategies need to be developed to improve their dispersity and solubility in reaction media (Malaviya and Gomes 2008). P. pastoris is a methylotrophic yeast that can use menthol as a carbon source. In addition, menthol is the powerful inducer for kstDF overexpression under the control of methanol-inducible promoter in pPIC3.5 K. In this study, therefore, menthol can be just used as a good solvent to facilitate the addition of AD to the reaction medium. In combination with the classical cultivation process of P. pastoris, the transformation process of AD by KM71I or GS115I was designed as follows: KM71I and GS115I were precultivated at 30 °C, 200 rpm for 20–24 h in BMGY medium. Then AD dissolved in methanol was added (final methanol concentration, 0.5%). Over the following 6 days, the same amount of methanol (0.5% final concentration) was added every other day.
KM71I and GS115I showed different AD conversion capacities. In shake flask fermentation process (30 ml medium per 250 ml flask), KM71I could transform 1.0 g/l AD into ADD at a 100% conversion rate, while GS115I could completely transform 1.5 g/l AD to produce ADD and BD simultaneously (Fig. 4a). Although TS was produced from 1.0 g/l AD transformation by P. pastoris strain GS115 (Fig. 4b) and strain KM71 (data not shown), both the 1.0 g/l AD transformation by KM71I and the 1.5 g/l AD transformation by GS115I showed no TS production (Fig. 4b). Further analysis indicated that the transformation of AD by KM71I did not foster the accumulation of TS as an end product, though little TS was detected on the first day after the addition of AD. In contrast, TS was detected when above 1.5 g/l AD was transformed by GS115I (Fig. 4b).

Properties of AD conversion by KM71I and GS115I. a TLC chromatogram of AD transformation by KM71I and GS115I for 6 days. KM71I: lane 1, 1.0 g/l AD; lane 2, 1.5 g/l AD; lane 3, 2.0 g/l AD; and lane 4: 2.5 g/l AD. GS115: lane 1, 1.0 g/l AD; lane 2, 1.5 g/l AD; lane 3, 2.0 g/l AD; and lane 4, 2.5 g/l AD. b HPLC profile of AD transformation for 6 days. Graph 1: 1.0 g/l AD transformed by P. pastoris GS115. Graph 2: 1.0 g/l AD transformed by KM71I. Graph 3: 1.5 g/l AD transformed by GS115I. Graph 4: 2.5 g/l AD transformed by GS115I. Graph 5: 2.5 g/l AD transformed by KM71I
BD is an important steroid. To enhance the proportion of BD in the end products of AD transformation by GS115I, glucose as a provider of reducing power was added to the GS115I fermentation process. Moderate concentrations of glucose were added after 4 days of induction by methanol. Then the GS115I culture was continued for another 3 days. The results indicated that glucose significantly enhanced the transformation of AD to BD. It showed that 3% glucose was optimal for BD production, resulting in 75% BD in the end products (Fig. 5). In the glucose-free control, BD made up of only 41%.

Effects of glucose on the transformation of AD by GS115I. a TLC profile. Lane 1: without glucose. Lane 2: 2% glucose. Lane 3: 3% glucose. Lane 4: 4% glucose. b HPLC profile. Graph 1: without glucose. Graph 2: 3% glucose
Discussion
Generally, bacterial kstDs are known as key enzymes functioning in the initial stage of steroid catabolism. These are widespread in the microorganisms that can utilize steroids as carbon and energy sources. The bacterial kstDs, especially those from actinobacteria such as Mycobacterium sp., Rhodococcus sp., Arthrobacter sp., and Nocardia sp., have been studied in terms of molecular, catalytic, and physiological properties, as well as their applications in steroid transformation (Donova 2007). In this study, we revealed a fungal kstD from A. fumigatus (kstDF) and investigated its value to steroid transformation. KstDF and its orthologous counterparts in fungi are important tailoring enzymes involved in the biosynthesis of steroidal fusidane antibiotics, such as helvolic acid (Lodeiro et al. 2009; Mitsuguchi and Seshime 2009). Therefore, they are distinctly different from bacterial kstDs in physiological function. In addition, the stereochemistry of steroidal fusidanes is not typical of common steroids—ring B adopts a boat conformation instead of a chair conformation. However, kstDF showed good activity for the common steroid AD. Its K m value was determined to be 47 μM, which is lower than that of kstD1 from R. erythropolis (82 μM). KstDF also showed good activity for other steroids, such as progesterone and testosterone. In this study, we used AD as a type substrate to investigate the properties of kstDF.
Based on molecular property analysis of kstDF by TMpred program prediction and determination of the subcellular activity distribution in heterologous expression hosts, we supposed kstDF to be a soluble protein because it had no detectable hydrophobic transmembrane domains, and most of its activity was found in the cytosol. In this way, kstDF is distinct from the well-defined membrane-bound kstDs found in bacteria. The difference in molecular properties between kstDF and bacterial kstDs may be associated with their physiological roles. As well known, steroids are minimally water-soluble compounds. Concentrations usually remain below 0.1 mM. The uptake and degradation of steroids by microorganisms are closely associated with the direct contact of the cell membrane and steroid particles (Atrat et al. 1991; Fernandes et al. 2003; Rajkhowa et al. 2000). Bacterial kstDs are the initial enzymes in steroid degradation. It may therefore be required for them to bind to the cellular membrane tightly by hydrophobic transmembrane domains to make it easier to contact with steroids in cellular membrane. Obviously, the biosynthesis of steroidal antibiotic fusidanes in fungi occurs intracelluarly, which may not require the kstDF to bind tightly with the cellular membrane. Nevertheless, the extracellular expression of kstDF in P. pastoris showed no activity, which may be because of misfolding or of some required intracelluar components to support its active state. The physiological properties of kstDF merit further investigation.
For many steroid hormones, the ∆ 1 double bonds are a pivotal moiety for pharmacological activity. Therefore, the development of engineered microorganisms with powerful kstD activity is important to the pharmaceutical industry. Nevertheless, well-characterized kstDs from actinobacteria were all found to be membrane-bound enzymes (Donova 2007). The construction of genetically engineered recombinant strains for bacterial kstDs has not yet been very successful (Choi et al. 1995; Li et al. 2007; Morii et al. 1998; Plesiat et al. 1991; Molnar et al. 1995; Wagner et al. 1992). The expression of kstD of A. simplex in B. subtilis was found to be the only case that was in any way effective in practice with regard to steroid conversion. It could transform 1.0 g/l AD to ADD at a 45.3% conversion rate (Li et al. 2007). In this study, kstDF was identified as a soluble enzyme. It was successfully expressed intracellularly in E. coli and P. pastoris. The engineered recombinant P. pastoris strains, KM71I and GS115I, were found to be more powerful under actual use than recombinant B. subtilis reported by Li et al. (2007). In the shake flask fermentation (Fig. 4a), KM71I was found capable of transforming more than 1.0 g/l AD into ADD at a 100% conversion rate, and GS115I was found capable of transforming 1.5 g/l AD to ADD and BD at a 100% conversion rate. This indicates that these two strains may be more promising than other engineered recombinant kstD strains in steroid ∆ 1 -dehydrogenation. Especially, strain KM71I showed good properties in preparing ADD without by-products. Fermentation optimization may further enhance the AD transformation efficiency of KM71I and GS115I.
P. pastoris GS115I was found to be capable of transforming AD to both ADD and BD, and in some case, TS. The transformation of AD to ADD, TS, and BD is certainly the result of synergism between exogenous kstDF and endogenous 17Hsd (Fig. 3d). Both P. pastoris strains, KM71 and GS115, could transform AD to TS, indicating that both KM71 and GS115 contain the gene for 17Hsd. 17Hsd in yeast and other microorganisms are well defined as reversible redox enzymes (Donova et al. 2005). The different performances of AD transformation seen in KM71I and GS115I should be attributed to the differences in physiological redox status between strain KM71I and strain GS115I. As shown in Fig. 3d, conversion of steroidal C17-one to C17-alcohol is a reduction reaction, which may be promoted by reducing power. Glucose is often used as a provider of reducing power in the microbial transformation process. Here, to enhance the proportion of BD in the end products, glucose was added to the GS115I fermentation process to regulate the redox status of GS115I. The addition of glucose in the late stages of AD transformation by GS115I suggested that the relative amount of end product BD can be greatly enhanced. Thus, it can be expected that the transformation of AD to BD or ADD by GS115I can be regulated by optimizing the physiological redox status of GS115I. In optimal redox status, the conversion of AD by GS115I may provide an alternative means of preparing BD.
References
Abul-hajj YJ (1972) Stereochemistry of C-l,2 dehydrogenation of 5β-pregnane-3,11,20-trione by Septomyxa affinis. J Biol Chem 247:689–691
Atrat P, Hösel P, Richter W, Meyer H, Hörhold C (1991) Interactions of Mycobacterium fortuitum with solid sterol substrate particles. J Basic Microb 31:413–422
Chiang YR, Ismail W, Gallien S, Heintz D, Dorsselaer AV, Fuchs G (2008) Cholest-4-en-3-one-Δ1-dehydrogenase, a flavoprotein catalyzing the second step in anoxic cholesterol metabolism. Appl Environ Microbiol 74:107–113
Choi KP, Molnar IJ, Yamashita M (1995) Purification and characterization of the 3-ketosteroid-Δ1-dehydrogenase of Arthrobacter simplex produced in Streptomyces lividans. J Biochem 117(5):1043–1049
Donova MV (2007) Transformation of steroids by actinobacteria: a review. Appl Biochem Microbiol 43:1–14
Donova MV, Egorova OV, Nikolayeva VM (2005) Steroid 17β-reduction by microorganisms: a review. Process Biochem 40:2253–2262
Fernandes P, Cruz A, Angelova B, Pinheiro HM, Cabral JMS (2003) Microbial conversion of steroid compounds: recent developments. Enzyme Microb Tech 32:688–705
Florin C, Köhler T, Grandguillot M, Plesiat P (1996) Comamonas testosteroni 3-ketosteroid-Δ4(5α)-dehydrogenase: gene and protein characterization. J Bacteriol 178:3322–3330
Freigassner M, Pichler H, Glieder A (2009) Tuning microbial hosts for membrane protein production. Microb Cell Fact 8:69
Handumrongkul C, Ma DP, Silva JL (1998) Cloning and expression of Candida guilliermindii xylose reductase gene (xyll) in Pichia pastoris. Appl Microbiol Biotechnol 49:399–404
Itagaki E, Matushita H, Hata T (1990) The enzymatic transformation of water-insoluble reactants in nonaqueous solvents. Conversion of cholesterol to cholest-4-ene-3-one by a Nocardia sp. J Biochem 108:122–127
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Li Y, Lu F, Sun T (2007) Expression of ksdD gene encoding 3-ketosteroid-Δ1-dehydrogenase from Arthrobacter simplex in Bacillus subtilis. Lett Appl Microbiol 44:563–568
Linares DM, Geertsma ER, Poolman B (2010) Evolved Lactococcus lactis strains for enhanced expression of recombinant membrane proteins. J Mol Biol 40:45–55
Lodeiro S, Xiong Q, Wilson WK, Ivanova Y, Smith ML, May GS, Matsuda SPT (2009) Protostadienol biosynthesis and metabolism in the pathogenic fungus Aspergillus fumigatus. Org Lett 11:1241–1244
Malaviya A, Gomes J (2008) Androstenedione production by biotransformation of phytosterols. Biores Technol 99:6725–6737
Mitsuguchi H, Seshime Y (2009) Biosynthesis of steroidal antibiotic fusidanes: functional analysis of oxidosqualene cyclase and subsequent tailoring enzymes from Aspergillus fumigatus. J Am Chem Soc 131:6402–6411
Molnar I, Choi KP, Yamashita M, Murooka Y (1995) Molecular cloning, expression in Streptomyces lividans, and analysis of a gene cluster from Arthrobacter simplex encoding 3-ketosteroid-Δ1-dehydrogenase, 3-ketosteroid-Δ5-isomerase and a hypothetical regulatory protein. Mol Microbiol 15:895–905
Morii S, Fujii C, Miyoshi T (1998) 3-Ketosteroid-Δ1-dehydrogenase of Rhodococcus rhodochrous sequencing of the genomic DNA and hyperexpression purification, and characterization of the recombinant enzyme. J Biochem 124:1026–1032
Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, Bennett J, Bowyer P, Chen D, Collins M, Coulsen R, Davies R, Dyer PS, Farman M, Fedorova N, Fedorova N, Feldblyum TV, Fischer R, Fosker N, Fraser A, García JL, García MJ, Goble A, Goldman GH, Gomi K, Griffith-Jones S, Gwilliam R, Haas B, Haas H, Harris D, Horiuchi H, Huang J, Humphray S, Jiménez J, Keller N, Khouri H, Kitamoto K, Kobayashi T, Konzack S, Kulkarni R, Kumagai T, Lafton A, Latgé JP, Li W, Lord A, Lu C, Majoros WH, May GS, Miller BL, Mohamoud Y, Molina M, Monod M, Mouyna I, Mulligan S, Murphy L, O’Neil S, Paulsen I, Peñalva MA, Pertea M, Price C, Pritchard BL, Quail MA, Rabbinowitsch E, Rawlins N, Rajandream MA, Reichard U, Renauld H, Robson GD, Córdoba SR, Rodríguez-Peña JM, Ronning CM, Rutter S, Salzberg SL, Sanchez M, Sánchez-Ferrero JC, Saunders D, Seeger K, Squares R, Squares S, Takeuchi M, Tekaia F, Turner G, Aldana CRV, Weidman J, White O, Woodward J, Yu JH, Fraser C, Galagan JE, Asai K, Machida M, Hall N, Barrell B, Denning DW (2005) Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151–1156
Plesiat P, Grandguillot M, Harayama S (1991) Cloning, sequencing, and expession of Pseudomonas testosteroni gene encoding 3-oxosteroid-Δ1-dehydrogenase. J Bacteriol 9:7210–7219
Rajkhowa RC, Goswami P, Singh HD (2000) Mode of uptake of sterol by Arthrobacter simplex. World J Microb Biotech 16:63–68
Sih CJ, Bennet RE (1962) Steroid 1-dehydrogenase of Nocardia restrictus. Biochim Biophys Acta 56:584–592
Szentirmai A (1990) Microbial physiology of sidechain degradation of sterols. J Ind Microbiol 6:101–116
van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L (2002) Molecular and functional characterization of the kstD2 gene of Rhodococcus erythropolis SQ1 encoding a second 3-ketosteroid-Δ1-dehydrogenase isoenzyme. Microbiol 148:3285–3292
Wagner M, Atrat PG, Wagner B, Hanemann V, Clark-Curtiss JE (1992) Overexpression of a Rhodococcus erythropolis protein in Escherichia coli with immunological identity to the Rhodococcus steroid-1-dehydrogenase. Immunoelectron microscopic localization and electrophoretic studies. J Basic Microbiol 32:269–277
Wei W, Wang F, Fan S, Wei D (2010) Inactivation and augmentation of the primary 3-ketosteroid-Δ1-dehydrogenase in Mycobacterium neoaurum NwIB-01: biotransformation of soybean phytosterols to 4-androstene-3,17-dione or 1,4-androstadiene-3,17-dione. Appl Environ Microbiol 76:4578–4582
Wierenga RK, Terpstra P, Hoi WGJ (1986) Prediction of the occurrence of the ADP-binding flap-fold in proteins, using amino acid sequence fingerprint. J Mol Biol 187:101–107
Acknowledgments
The work was supported by the National Basic Research Program of China (no. 2009CB724703), the National High Technology Research and Development Program of China (no. 2008AA02Z209), and the National Special Fund for State Key Laboratory of Bioreactor Engineering, grant no. 2060204.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Miao-Miao Chen and Feng-Qing Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Chen, MM., Wang, FQ., Lin, LC. et al. Characterization and application of fusidane antibiotic biosynethsis enzyme 3-ketosteroid-∆1-dehydrogenase in steroid transformation. Appl Microbiol Biotechnol 96, 133–142 (2012). https://doi.org/10.1007/s00253-011-3855-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3855-5