
Abstract
Poly(ethylene terephthalate) (PET) is one of the most consumed plastics in the world. The development of efficient technologies for its depolymerization for monomers reuse is highly encouraged, since current recycling rates are still very low. In this study, 16 commercial lipases and cutinases were evaluated for their abilities to catalyze the hydrolysis of two PET samples. Humicola insolens cutinase showed the best performance and was then used in reactions on other PET sources, solely or in combination with the efficient mono(hydroxyethyl terephthalate)-converting lipase from Candida antarctica. Synergy degrees of the final titers of up to 2.2 (i.e., more than double of the concentration when both enzymes were used, as compared to their use alone) were found, with increased terephthalic acid formation rates, reaching a maximum of 59,989 µmol/L (9.36 g/L). These findings open up new possibilities for the conversion of post-consumer PET packages into their minimal monomers, which can be used as drop in at existing industrial facilities.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
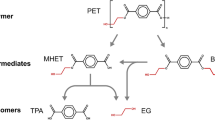
Poly(ethylene terephthalate) (PET) is a synthetic aromatic polyester composed of ethylene glycol (EG) and terephthalic acid (TPA) units [15], which is extremely versatile and used in a variety of applications, such as clothing and technical textiles [23] and packages (e.g., water and soft drink bottles, salad domes and biscuit trays) [30], with an annual worldwide production over 50 million tons [5]. Efficient PET recycling is an urgent global need from both environmental and economic aspects, since plastics production is expected to grow faster than petroleum production in the next decades and their share on oil use is forecasted to rise significantly, from 6 (in 2014) to 20% (in 2050). This is even more crucial for short first-use materials, as bottles, which accumulates in the environment, damaging ocean’s fauna and slowing down the transition to a circular economy [30]. Mechanical PET recycling technologies, although in a higher readiness level, offer limited applicability, such as a rapid and drastic drop in the polyester ductility, which restricts the use of the recycled polymer to low-value uses, as carpets [13]. After decades being considered non-biodegradable, a paradigm change has occurred in the past few years with the publication of studies reporting in vivo or in vitro PET depolymerization to its monomers [4, 22, 26, 32] with faster reaction rates being observed in enzyme-catalyzed processes [32].
The enzymes reported for PET hydrolysis belong to the sub–sub class carboxylic acid hydrolases (EC 3.1.1.-) [7], such as cutinases (EC 3.1.1.74) [3, 15, 28], lipases (EC 3.1.1.3) [15, 17], serine esterases (EC 3.1.1.56, 3.1.1.12) [14] and carboxylesterases (EC 3.1.1.1) [23]. However, previous works have demonstrated that enzymes may show different specificities during the depolymerization process, being more capable to attack certain substrates. For example, Yoshida et al. [32] identified two different catalytic properties from Ideonella sakaiensis enzymes, defining them as PETase and MHETase. PETase was shown to efficiently breakdown PET to mono(hydroxyethyl) terephthalate (MHET), whereas MHETase catalyzed the conversion of MHET to TPA and EG. Similar properties were observed by Carniel et al. [8], who demonstrated that the Humicola insolens cutinase and the Candida antarctica lipase B act synergistically to convert PET into its final monomers. Also, Barth et al. [2] reported a dual enzyme reaction system consisted with a polyester hydrolase, LC-cutinase, hydrolyzing PET film chains and an immobilized carboxylesterase, TfCa KW3 from Thermobida fusca, as the second enzyme applied to eliminate inhibitory intermediates produced from PET hydrolysis. These studies were still preliminary, but opened up a range of possibilities for the development of efficient technologies for PET depolymerization.
Therefore, the objective of the present study was to compare the catalytic performance of several enzymes from microbial (bacterial, fungal) and plant sources for PET depolymerization, and to investigate the synergy behavior of selected biocatalysts during hydrolysis of different PET samples from industrial facilities.
Materials and methods
Substrates and enzymes
PET bottle film used was from post-consume non-carbonated mineral water bottles (brand Crystal©). It was cut into squares of aprox. 0.5 cm and it presented 0.1 mm thickness. Amorphous, Mineral Water (MW), Carbonated Soft Drink (CSD), CSD-Plus and CSD-Low Intrinsic Viscosity (CSD-LIV) PET resins were generously provided as chips by the industrial PET production plant PetroquímicaSuape (Suape, Brazil). All these PET chips grades were cold-milled in a blade mill (Ika). PET flakes from a local plastic industrial recycling plant (PIRP) were milled in a knife mill and provided by Professor Marcos Lopes (Federal University of Rio de Janeiro).
Lipases from Aspergillus oryzae (AoL), Burkholderia cepacia (BcL), Candida antarctica (CaL), porcine pancreas (PPL), Candida rugosa (CrL), Mucor miehei (MmL), Pseudomonas fluorescens (PfL), Rhizomucor miehei (RmL), Rhizopus niveus (RnL), Rhizopus oryzae (RoL), Thermomyces lanuginosus (TlL) and wheat (WL—Triticum aestivum) as well as high-purity standards of BHET and TPA were purchased from Sigma–Aldrich. RmL and TlL were purchased as liquid preparations. All the other enzymes above were obtained as powder and were solubilized in sodium phosphate buffer 200 mM pH 7.0 prior to their applications. C. antarctica lipase B (CALB, product Lipozyme© CALB L), Humicola insolens cutinase (HiC, product Novozym© 51,032), Lecitase Ultra© (LU) and Lipozyme TL© 100L (LTL) were liquid preparations kindly provided by Novozymes (Araucária, Brazil).
Screening of enzymes for PET hydrolysis
All enzymes were screened on PET bottle films and milled amorphous PET. 10 mL phosphate buffer 200 mM pH 7.0 with 200 mg initial PET (PET bottle films and amorphous PET granules) and enzyme amount of 0.01 gprotein/gPET were incubated in a hybridization incubator (Combi-D24, FINEPCR) at 37 °C under an agitation speed of 25 rpm. Control reactions (with no enzyme addition) were carried out at the same conditions used in each experiment. Protein amount was determined according to Bradford method [6].
Application of the selected enzymes for the hydrolysis of different PET samples
The two best enzymes from screening reactions were applied to catalyze hydrolysis of different milled PET samples, either in separate or simultaneously. Reactions were carried out in a hybridization incubator (Combi-D24, FINEPCR) at 60 °C, 25 rpm in 10 mL phosphate buffer 200 mM pH 7.0 with 200 mg initial PET (bottle, amorphous, MW, CSD, CSD-Plus, CSD-LIV and PIRP). The enzyme loading was 0.01 gprotein/gPET in the reactions carried out with each biocatalyst in separate and 0.02 gprotein/gPET when both enzymes were used (being 0.01 g/g of each enzyme).
Analyses
The quantification of TPA, MHET and BHET (bis(hydroxyethyl terephthalate)) was carried out in a Thermo Scientific Dionex UltiMate 3000 HPLC using an Agilent Zorbax SB-C18 pre-column and an Eclipse Plus C18 column (at 30 °C). A gradient mixture of acetonitrile (TEMED) and 0.05% formic acid (Sigma–Aldrich) was used as mobile phase at a flowrate of 0.5 mL/min. An UV detector was used for the detection of the hydrolysis product, at a wavelength of 254 nm. TPA and BHET peak areas were correlated to the ones of high-purity analites, whereas for MHET a high-purity (>99%) powder obtained from a quantitative BHET hydrolysis with HiC was used.
For the observation of the samples using scanning electron microscopy (SEM), PET substrates were metallized with Au/Pd in a Quorum Q150TES coater. The images of the prepared samples were then captured in a Zeiss EVO-LS15 microscope, using 1 kV voltage.
Crystallinity of PET substrates was determined in a Perkim-Elmer DSC8000 calorimeter, according to the standard ASTM D 3418-2012, employing heating (to 290 °C) and cooling (to 0 °C) rates of 10 °C/min. The heat of fusion (DHm) of each sample was correlated to the one of a 100% crystalline polymer (estimated as 140 J/g) [14, 16], for the determination of their degree of crystallinity.
Intrinsic viscosity (iv) was determined using a mixture of phenol/1,1,2,2-tetrachoroethane (PTCE) as solvent and it was correlated to the polymer molar mass using Mark-Houwink-Sakurada equation [17].
Analysis of surface area and particles diameter of selected PET samples was performed in a Mastersizer 2000 analyzer (Malvern Instruments).
Calculations
Mole fraction of each reaction component (χ i , where i = BHET, MHET or TPA) was expressed as a ratio between the mole concentration (n) of the component and the sum of the mole concentrations of the three measured products, as shown in Eq. 1. The calculation of PET conversion into released TPA (%) was according to Eq. 2 where TPA and PET concentrations used were in mass basis (mg/L) and the theoretical yield is 0.8643 mgTPA/mgPET. The synergy degree (SD) between the two selected enzymes in the PET depolymerization reaction was defined as the ratio between TPA mole concentration (n TPA) when the two selected enzymes (E1, E2) were used simultaneously and the sum of TPA mole concentration when the enzymes were used in separate, at the same reaction conditions, as shown in Eq. 3.
Results and discussion
Screening of enzymes for PET hydrolysis
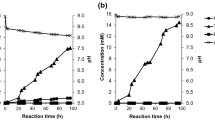
Enzymes were first screened for their ability to catalyze the hydrolysis of either bottle or amorphous PET. For this study, it was prioritized a condition in which all enzymes could be active and stable, more than an ideal condition of PET availability, such as near to its glass transition temperature. Although not at the highest hydrolysis rates, such mild condition would be effective to show the potential of possibly enzymes with low thermal stability. As shown in Figs. 1 and 1S, HiC lead to significantly higher amounts of TPA and MHET (up to 98 and 199 µmol/L in PET bottle and 402 and 950 µmol/L in amorphous PET, respectively) than the other 15 biocatalysts, which use resulted in no more than 20 µmol/L of each of these compounds. On the other hand, BHET concentrations in the reaction media were very similar for all biocatalysts, including HiC. In most cases, it was accumulated during the first 7 days of reaction and afterward converted into the final monomers. The exception was when HiC was employed for amorphous PET hydrolysis, yielding a continuously increasing BHET profile. MHET was the predominant depolymerization product during all the reaction time investigated, on both substrates, when HiC was used.

Profiles of TPA released from screening of PET bottle and amorphous PET hydrolysis reactions at 37 °C catalyzed by HiC (time course a) and maximum TPA concentrations in the reactions catalyzed by all the other 15 enzymes over PET bottle (b) and amorphous PET (c)
Interestingly, cutinases usually show lower molecular weight (20–30 kDa) than lipases [19], in accordance to the results we found in this present study, with up to 70 kDa bands (from RnL) (Table 1S). This suggests that the mobility of smaller proteins along PET chains may play an important role for hydrolysis rate, being necessary more specific experiments to prove this hypothesis, since other enzyme structure characteristics, such as the catalytic triad, the flexibility of the active site and key residues in the oxyanion hole are related to the specific ability of each enzyme for the biocatalysis [9, 14]. As reviewed by Ferrario et al. [10] from a large list of microbial sources, cutinases present molar mass of up to 40 kDa. Yang et al. [31] reported a low mass cutinase (~25.3 kDa) from Thielavia terrestris which demonstrated to be more efficient to hydrolyze several polyesters than higher enzymes, such as the wild-type Thermobifida fusca cutinase (~33.8 kDa). Moreover, glycosylation patterns have been reported in the literature to influence hydrolysis rates due to steric hindering that difficult the access of the catalytic site to the PET chains [27].
Even when compared at an enzyme molar basis (µmolTPA/µmolenzyme), HiC presented higher performance than the other biocatalysts, being by 29 times higher than the second best enzyme (Table 2S).
At the end of the tests (14 days) on PET bottle and amorphous PET, χ MHET was 0.659 and 0.692, whereas χ TPA at the same conditions was 0.327 and 0.293, respectively. Oppositely, although resulting in very low concentrations, CALB led to the final TPA mole fractions of 1.000 and 0.754, when in contact with PET bottle and amorphous PET, respectively. This is in agreement with the behavior of these enzymes, previously observed by our group [8], which suggested a limiting rate for the hydrolysis of MHET to TPA during catalysis by HiC, especially at lower temperatures, and higher hydrolysis rates of MHET to TPA during catalysis by the enzyme from C. antarctica.
TPA release rates were maintained constant throughout hydrolysis of both substrates (PET bottle and amorphous PET) catalyzed by HiC, as follows: 6.9 µmol/L/d (n = 7, R 2 = 0.998) and 29.6 µmol/L/d (n = 7, R2 = 0.986), respectively. These results indicate that the enzyme keeps active during the considered reaction time. Therefore, the experiments with this biocatalyst were continued for over 2 months and continuously increasing TPA and MHET concentrations were still observed, in both substrates (Fig. 3S). It was observed, however, a trend in the proportions of TPA and MHET mole fractions. On bottle PET, χ TPA (0.55 ± 0.01) surpassed χ MHET (0.44 ± 0.01) after 52 days of reaction, and the final monomer continued to be the predominant compound until the end of the test. On amorphous PET, MHET mole fraction continuously decreased from 0.74 ± 0.02 at 3 days of reaction to 0.52 ± 0.01 at 73 days of reaction, whereas TPA mole fraction increased from 0.22 ± 0.01 to 0.47 ± 0.01 during the same period. χ BHET was no more than 0.06 during reactions on both substrates.
In a screening of seven microbial enzymes for PET film hydrolysis, Korpecka et al. [20] observed the highest TPA release in the reaction catalyzed by a Fusarium solani cutinase, followed by H. insolens and T. fusca enzymes. In the reaction carried out with T. lanuginosus enzyme, the highest MHET concentration was found, but TPA was very low, suggesting a similar behavior as noticed in the present study for HiC. The proportions of MHET and TPA found in the study of Korpecka et al. [20] for T. lanuginosus (more MHET than TPA) and P. fluorescens (more TPA than MHET) are in accordance with the final mole fractions observed in the present study, where we found the final χ TPA = 0.20 and χ MHET = 0.80 using TlL for amorphous PET depolymerization and the final χ TPA = 0.58 and χ MHET = 0.41 using PfL for amorphous PET hydrolysis (Fig. 2S), although corresponding to very low titers.
In the present screening study, MHET was the predominant product at the end of eight reactions in PET bottle and nine reactions in amorphous PET, attesting that the final hydrolysis step to TPA is commonly limiting. Similarly, a proportion of MHET/TPA of about 2.7 was found after hydrolysis of PET film by a PETase from a newly isolated and promising Ideonela sakaiensis strain [32].
Synergy studies on different PET samples
HiC was then selected for further investigations, along with CALB, due to the ability of this latter enzyme to more quickly catalyze the conversion of MHET to TPA [8]. Also, reaction temperature was increased to 60 °C, based on preliminary studies that indicated the thermal tolerance of HiC during PET hydrolysis [8, 24]. As the enzymes present complementary activity profiles at the final stages of PET depolymerization, the synergy between HiC and CALB was investigated on seven different PET samples, being five from an industrial PET production plant, one from an industrial PET recycling plant and one from commercial PET bottle. The enzymes were then used in separate or simultaneously for the hydrolysis of each substrate. Time course of TPA and MHET concentrations is shown in Fig. 2, whereas BHET data are shown in Fig. 4S.

Time course of TPA and MHET release during hydrolysis reaction at 60 °C of amorphous (a), bottle (b), MW (c), CSD (d), CSD-Plus (e), CSD-LIV (f) and PIRP (g) PET samples catalyzed by HiC, CALB or both enzymes simultaneously
In all substrates, MHET was confirmed as a reaction intermediate, showing decreasing concentrations from the first half of the test. At 14 days of reaction, TPA was the main product of the reactions in the seven substrates (χ TPA = 0.552–0.938), with the only exception being when HiC was used as the only biocatalyst for amorphous PET hydrolysis (χ TPA and χ MHET were 0.494 and 0.495, respectively). Interestingly, the highest amounts of MHET (31,877 µmol/L in 10d) were found in this condition. Thus, the stabilization of MHET concentration from 10 to 14 days of reaction may suggest that this is a limiting condition for MHET accumulation; however, systematic enzyme kinetic studies are needed to support the hypothesis of inhibition by MHET, as done by Barth et al. [2]. Even by other hydrolysis product, such as EG, may change the active site dynamics, as recently detected by Groß et al. [13]. The enzyme continued acting after this point, since TPA concentration increased from 23,050 to 31,536 µmol/L (i.e., up to 7.45 g/L), but as MHET was stable, the MHET formation and consumption rates must had been very similar. On this substrate, when CALB was used concomitantly, MHET consumption and TPA formation were more intense, reinforcing the role of this enzyme to complement PET hydrolyzing activity of HiC. The impact of this enzyme, however, was not as fast as expected based on previous results obtained at 37 °C [8], and did not depleted MHET from reactional medium, and this may be related to the nominal optimum temperature range of this enzyme (30–60 °C), which is lower than that of HiC (35–70 °C) [21].
Oppositely, on MW and the three CSD PET samples, a lower, but continuous MHET consumption rate was observed from the third day of reaction (18.5–33.6 µmol/L/d). As in these substrates the reaction occurred at lower extent, MHET concentration was far from limiting conditions.
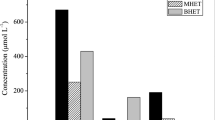
It can be observed that the combined use of the two enzymes increased substantially TPA and MHET concentrations only when PET bottle was used (684 and 432 µmol/L after 14 days of reaction), and at the end of the test with amorphous PET (44,842 and 12,256 µmol/L, respectively). On CSD PET samples (regular, LIV and Plus), only a marginal positive synergy effect was observed (SD up to 1.10), and not during all reaction time. On MW PET and PIRP, the highest final TPA concentrations were achieved when HiC was used solely (1003 and 8693 µmol/L, respectively), representing a SD below 1 (Fig. 3). It can be noted from almost all profiles that SD tends to decrease during the reaction, being higher in the first half of incubation period. Similar profiles were observed by Arias et al. [1] during synergy studies between cellulolytic cocktails from the fungi Penicillium funiculosum, Aspergillus niger and Trichoderma harzianum, when higher SD (around 1.8, expressed as glucose released) occurred at 12 h of hydrolysis and thereafter decreased to 1.4 by 48 h of reaction.

Time course of the synergy degree (SD) between HiC and CALB during reactions in seven PET samples
The simultaneous use of two enzymes for PET film hydrolysis was investigated by Barth et al. [2]. Authors used a T. fusca carboxylesterase (TfCa) to complement the good PET depolymerizing activity of either a T. fusca cutinase or a metagenome-derived LC-cutinase, and observed that the higher the amount of TfCa, the higher the proportion of TPA over MHET.
On PET bottle, TPA release rates were 32.8 and 52.0 µmol/L/d when HiC was used solely and when it was used in combination with CALB, respectively. This represents 4.7- and 7.5-fold increases in reaction rate, when compared to the results observed in the screening, at 37 °C, corroborating the positive effect of temperature within this range.
On amorphous PET, SD was up to 2.0 (at 3-day reaction time) and TPA release rate was increased from 2352 to 3226 µmol/L/d when CALB was present in the reaction along with HiC. When both enzymes were employed, the TPA release rate was similar to the highest rate observed in the study of the effect of different buffers on the hydrolysis of PET film by a Thermobifida fusca KW3 cutinase (around 3480 µmol/L/d, based on the sum of the concentrations of TPA, MHET and BHET) [25]. In this same study, the effect of phosphate buffer concentration on the initial hydrolysis rate of PET films was investigated, being the maximal rates observed at a concentration of 0.9 M (around 700 µM cm2/h/µg). This suggests that the results obtained in the present study may be further increased through the adjustment of buffer concentration.
A variable that may significantly influence PET depolymerization degree is particle size (or surface area). Aiming to understand this impact, a new set of experiments was conducted. Amorphous PET was sieved, resulting in two particle size ranges, one below and another above 1 mm. For these experiments, reactions were maintained at 60 °C during 14 days and thereafter temperature was dropped to 37 °C and CALB was added. Reactions progress was monitored during the subsequent 24 h. For comparison purposes, the same strategy was employed for PIRP PET depolymerization, which is the realest post-consumer PET sample used in this study. The results shown in Fig. 4 indicate that this was an efficient approach. Within the 24 h evaluated, CALB was able to completely convert MHET to TPA, increasing TPA concentration by 39% (PIRP PET) and 88% (amorphous >1 mm PET). At the end of the 24 h of the second reaction stage, TPA concentrations in the reactions with amorphous <1 mm, amorphous >1 mm and PIRP PET were 59,989 ± 1408 µmol/L, 27,061 ± 191 µmol/L and 13,640 ± 108 µmol/L, respectively. The sequential addition of CALB was also effective in terms of enrichment of the final products composition in TPA. At the end of the reactions with amorphous <1 mm, amorphous >1 mm and PIRP PET, χ tpa 0.993, 0.994 and 0.993, respectively. Compared to the χ tpa at the end of the reactions in which both enzymes were acting simultaneously during all the time on amorphous PET and PIRP samples (0.782 and 0.606), the media were enriched by 50% in TPA.

Time course of products released during hydrolysis of amorphous <1 mm (a), amorphous >1 mm (b) and PIRP (c) PET samples catalyzed by sequential incubation with HiC (at 60 °C) and CALB (at 37 °C)
The very distinct results obtained when small and large amorphous PET granules were tested (Fig. 4) support that particle size significantly influenced reaction progress, since the molar mass and crystallinity properties of these samples did not statistically differ (Table 1). The paper by Gamerith et al. [11] reported a study on the effect of particle size of PET samples during depolymerization catalyzed by a T. cellulosilytica cutinase. At 50 °C, the concentrations of released products in the reaction over 0.05–0.1 mm powder were 2.8 times higher than the concentration achieved using 0.25–0.5 mm powders (around 5 mM).
It is worthy to mention the broad range of products concentrations in the reactions on different PET samples (Figs. 2, 4) may also be related to the polymer properties. As can be seen in Table 1, amorphous PET showed the lowest molar mass and crystallinity (as expected), thus being more accessible for the enzymatic catalysis. On the other hand, MW and the three CSD PET samples were shown to present higher molar mass and also high crystallinity. Bottle and PIRP PET had intermediate molar mass and medium to high crystallinity. Melting and crystallization temperature also varied significantly between PET samples. T m ranged from 230.66 ± 0.07 °C (CSD-LIV PET) to 250.98 ± 0.47 °C (amorphous PET) and Tc ranged from 200.49 ± 0.32 °C (PIRP PET) to 210.17 ± 0.56 °C (CSD-LIV PET). The influence of PET crystallinity during hydrolysis by three commercial cutinases (from H. insolens, Fusarium solani and Pseudomonas mendocina) was investigated by Ronkvist et al. [24]. NaOH consumption rates (as a titrimetric measure of carboxylic acid release) was up to 23-times increased when a low-crystallinity PET (7.0% crystallinity) was used, as compared to a biaxially oriented PET (35.0% crystallinity), thus supporting the crucial role of PET crystallinity on the extension of its depolymerization.
In Fig. 4, a comparison between the results obtained employing amorphous PET < 1 mm or PIRP PET as substrate corroborates the importance of the degree of crystallinity for depolymerization efficiency. This is because they presented relatively similar surface properties, i.e., surface area (0.0132 m2/g for amorphous <1 mm and 0.0110 m2/g for PIRP PET) and surface weighted mean diameter (454.6 µm for amorphous <1 mm and 545.4 µm for PIRP PET). These samples, however, differed significantly regarding the degree of crystallinity (4.9 ± 0.9% for amorphous <1 mm and 41.1 ± 0.3% for PIRP PET), as shown in Table 1. Similarly, amorphous PET powder (12% crystallinity) and semi-crystalline PET powder (24% crystallinity) were employed in depolymerization reactions, under catalysis by a T. cellulosilytica cutinase [11]. Interestingly, higher TPA concentrations were observed during the 72 h of reaction using the semi-crystalline sample, which also presented larger particle size (0.5–1.0 µm) than the amorphous PET (<0.5 µm). Authors supported their findings based on the differences of purity of each sample, since additives may change the glass transition temperature of the polymer and increase its chain mobility.
The addition of CALB was an effective strategy to boost TPA formation, even at lower reaction temperature. Data shown in Table 2 reveal that TPA formation rate was up to 141-fold increased after CALB addition (from 0.61 µmol/(L min) to 85.8 µmol/(L min), for amorphous PET > 1 mm), which sustain the findings previously reported by Carniel et al. [8]. It is worthy to notice also that MHET consumption rate was lower than TPA formation rate for all three reaction conditions. Once the stoichiometry is 1 mol of MHET forming 1 mol of TPA, these results suggest that possibly CALB was further acting on substrates with higher molar mass present in the reaction medium, more rapidly catalyzing the release of MHET, which is still a reaction intermediate. The MHET consumption rates observed are, therefore, a result of its formation rates (from higher substrates) and its conversion rates to TPA.
Amorphous PET (as a film) was also the substrate used for depolymerization studies with a T. fusca cutinase. The best variant of the engineered enzyme yielded a conversion of 42.6%, considerably higher than that found with the wild-type cutinase (15.9%), and the increase was attributed to relieved product inhibition [29].
In the two-step depolymerization reactions, the highest TPA-specific production (achieved on amorphous <1.0 mm) was 3565 µ/µenzyme, which is 98.6 times higher than the initial specific production using solely HiC, observed during the screening studies. This indicates that the overall process strategy here described was very effective to a better utilization of the commercial enzyme.
At the end of incubation with CALB at 37 °C, remaining particles of the substrates were used for SEM analysis. Images shown in Fig. 5 reveal vast morphological changes in the two amorphous PET samples (Fig. 5b, d), as compared to their corresponding controls (Fig. 5a, c). PIRP PET was also visualized presenting degraded surface, but less extensively than in the other samples. The deep erosion pattern, however, was similar to that previously observed in PET bottle [8]. The extent of degradation was in agreement with the conversion of PET to TPA found at the end of the reactions in amorphous <1 mm, amorphous >1 mm and PIRP PET: 57.6 ± 1.4%, 27.0 ± 1.8% and 13.6 ± 0.8%, respectively.

SEM images of PET samples after 14-day reaction with HiC at 60 °C followed by 24-h reaction with CALB at 37 °C. Control reactions (a, c, e) and depolymerization reactions (b, d, f) of amorphous <1 mm, amorphous >1 mm and PIRP PET, respectively
Conclusions
In this study, 16 commercial enzymes were screened for their ability to catalyze the depolymerization of PET bottle and amorphous PET samples. In many cases, the hydrolysis intermediate mono(hydroxyethyl) terephthalate (MHET) was the predominant product, confirming a common behavior reported in the literature regarding its inhibition effect to cutinases. In this step of the study, the cutinase from Humicola insolens (HiC) stood out as the best biocatalyst, and terephthalic acid (TPA) titers of up to 3377 µmol/L were found at the end of an extended test performed during over 2 months, in which the enzyme was shown to be active all along. Although not effective for PET depolymerization, based on previous knowledge that the Candida antarctica lipase (CALB) can efficiently catalyze the hydrolysis of MHET to TPA, synergy studies between these enzymes were conducted, over five industrial PET resin and two post-consumer PET samples. When the enzymes were used simultaneously at 60 °C, the highest effective synergy degrees (SD) were observed in amorphous and in the two post-consumer PET (SD up to 2.2) and they decreased during the reaction, as similarly observed in synergy studies with cellulases. When the enzymes were employed sequentially, time course of products release with predominant MHET concentrations was observed during incubation solely with HiC, but after CALB addition and temperature reduction to 37 °C, it was rapidly converted to TPA, resulting in up to 141-times higher reaction rates and yielding a 1.7-fold higher titer than that found when the two enzymes were used at the same temperature in the PET sample from and industrial recycling plant. The findings reported in this study reinforce the different and complementary catalytic behavior of HiC and CALB, supporting the meaningful use of these enzymes in combination for increased conversion of real PET substrates to TPA.
References
Arias JM, Modesto LFA, Polikarpov I, Pereira N (2016) Design of an enzyme cocktail consisting of different fungal platforms for efficient hydrolysis of sugarcane bagasse: optimization and synergism studies. Biotechnol Progr 32(5):1222–1229. doi:10.1002/btpr.2306
Barth M, Honak A, Oeser T, Wei R, Belisário-Ferrari MR, Then J, Schmidt J, Zimmermann W (2016) A dual enzyme system composed of a polyester hydrolase and a carboxylesterase enhances the biocatalytic degradation of polyethylene terephthalate films. Biotechnol J 11:1082–1087. doi:10.1002/biot.201600008
Barth M, Oeser T, Wei R, Then J, Schmidt J, Zimmermann W (2015) Effect of hydrolysis products on the enzymatic degradation of polyethylene terephthalate nanoparticles by a polyester hydrolase from Thermobifida fusca. Biochem Eng J 93:222–228. doi:10.1016/j.bej.2014.10.012
Barth W, Wei R, Oeser T, Then J, Schmidt J, Wohlgemuth F et al (2015) Enzymatic hydrolysis of polyethylene terephthalate films in an ultrafiltration membrane reactor. J Memb Sci 494:182–187. doi:10.1016/j.memsci.2015.07.030
Bornscheuer UT (2016) Feeding on plastic. Science 351:1154–1155. doi:10.1126/science.aaf2853
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Brenda (2017) http://www.brenda-enzymes.org/. Accessed 27 March 2017
Carniel A, Valoni E, Nicomedes N Jr, Gomes AC, Castro AM (2016) Lipase from Candida antarctica (CALB) and cutinase from Humicola insolens act synergistically for PET hydrolysis to terephthalic acid. Proc Biochem. doi:10.1016/j.procbio.2016.07.023
Chen S, Su L, Chen J, Wu J (2013) Cutinase: chacacteristics, preparation and application. Biotechnol Adv 31:1754–1767. doi:10.1016/j.biotechadv.2013.09.005
Ferrario V, Pellis A, Cespugli M, Guebitz GM, Gardossi L (2016) Nature inspired solutions for polymers: will cutinase enzymes make polyesters and polyamides greener? Catalysts 6:205. doi:10.3390/catal6120205
Gamerith C, Zartl B, Pellis A, Guillamot F, Marty A, Acero EH, Guebitz GM (2017) Enzymatic recovery of polyester building blocks from polymer blends. Proc Biochem. doi:10.1016/j.procbio.2017.01.004
García JM (2016) Catalyst: design challenges for the future of plastics recycling. Chem 1:813–819. doi:10.1016/j.chempr.2016.11.003
Groß C, Hamacher K, Schmitz K, Jager S (2017) Cleavage product accumulation decreases the activity of cutinase during PET hydrolysis. J Chem Inf Model 57:243–255
Guebitz GM, Cavaco-Paulo A (2008) Enzymes go big: surface hydrolysis and functionalisation of synthetic polymers. Trends Biotechnol 26:32–38. doi:10.1016/j.tibtech.2007.10.003
Hansen F, Atwood KB (2005) Polyester Fibers. In: Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, New York. doi:10.1002/0471238961.1615122508011419.a01.pub2
Kaisersberger E, Möhler H. (1991) DSC on polymeric materials. In: NETZSCH annual for science and industry, vol. 1. Selb: ©NETZSCH-Gerätebau GmbH, D 8672
Kamide K, Miyazaki Y, Kobayashi H (1977) Unperturbed chain dimensions of polyethylene terephthalate and polyethylene 1,2-diphenoxyethane p, p’-carboxylate. Polym J 9:317–327. doi:10.1295/polymj.9.317
Kim HR, Song WS (2006) Lipase treatment of polyester fabrics. Fibers Polym 7:39–343. doi:10.1108/09556221011008785
Kold D, Dauter Z, Laustsen AK, Brzozowski AM, Turkenburg JP, Nielsen AD et al (2014) Thermodynamic and structural investigation of the specific SDS binding of Humicola insolens cutinase. Protein Sci 23:1023–1035. doi:10.1002/pro.2489
Korpecka J, Heumann S, Billig S, Zimmermann W, Zinn M, Ihssen J et al (2010) Hydrolysis of cutin by PET-hydrolases. Macromol Symp 296:342–346. doi:10.1002/masy.201051047
Novozymes® Lipases enzymes. http://www.novozymes.com/en/solutions/pharmaceuticals/biocatalysis/Pages/lipase-enzymes.aspx. Accessed 05 April 2016
Nowak B, Pajak J, Labuzek S, Rymarz G, Talik E (2011) Biodegradation of poly(ethylene terephthalate) modified with polyester “Bionolle” by Penicillium funiculosum. Polimery/Polymers. 56:35–44
Ribitsch D, Heumann S, Trotscha E, Herrero Acero E, Greimel K, Leber R et al (2011) Hydrolysis of polyethylene terephthalate by p-nitrobenzylesterase from Bacillus subtilis. Biotechnol Prog 27:951–960. doi:10.1002/btpr.610
Ronkvist AM, Xie W, Lu W, Gross RA (2009) Cutinase-catalyzed hydrolysis of poly(ethylene terephthalate). Macromolecules 42:5128–5138. doi:10.1021/ma9005318
Schmidt J, Wei R, Oeser T, Belisário-Ferrari MR, Barth M, Then J, Zimmermann W (2016) Effect of Tris, MOPS, and phosphate buffers on the hydrolysis of polyethylene terephthalate films by polyester hydrolases. FEBS OpenBio 6:919–927
Sharon C, Sharon M (2012) Studies on biodegradation of polyethylene terephthalate: a synthetic polymer. J Microbiol Biotechnol Res 2:248–257
Shirke AN, Basore D, Holton S, Su A, Baugh E, Butterfoss GL, Makhatadze G, Bystroff C, Gross RA (2016) Influence of surface charge, binding site residues and glycosylation on Thielavia terrestris cutinase biochemical characteristics. Appl Microbiol Biotechnol 100:4435–4446
Sulaiman S, Yamato S, Kanaya E, Kim JJ, Koga Y, Takano K et al (2012) Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach. Appl Environ Microbiol 78:1556–1562. doi:10.1128/AEM.06725-11
Wei R, Oeser T, Schmidt J, Meier R, Barth M, Then J, Zimmermann W (2016) Engineered bacterial polyester hydrolases efficiently degrade polyethylene terephthalate due to relieved product inhibition. Biotechnol Bioeng 113:1658–1665
World Economic Forum© (2016) The New Plastics Economy—Rethinking the future of plastics, http://www3.weforum.org/docs/WEF_The_New_Plastics_Economy.pdf. Acessed 05 April 2016
Yang S, Xu H, Yan Q, Liu Y, Zhou P, Jiang Z (2013) A low molecular mass cutinase of Thielavia terrestris efficiently hydrolyze polu(esters). J Ind Microbiol Biotechnol 40:217–226
Yoshida S, Hiraga K, Takehana T, Taniguchi I, Yamaji H, Maeda Y et al (2016) A bacterium that degrades and assimilates poly(ethylene terephthalate). Science 351:1196–1199. doi:10.1126/science.aad6359
Acknowledgements
Authors wish to acknowledge PETROBRAS for the financial support. To Rogério Martins for his support in SEM analyses. To Thalmo Azevedo for his assistance in particle size analyses. To Aline Lima for her collaboration in intrinsic viscosity and DSC analyses. To Rafael Dias and Wellison Muniz for their help in the milling of PET samples. To Novozymes for providing some of the enzymes used in this study. To PetroquímicaSuape and Prof. Marcos Lopes for providing the PET samples.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
de Castro, A.M., Carniel, A., Nicomedes Junior, J. et al. Screening of commercial enzymes for poly(ethylene terephthalate) (PET) hydrolysis and synergy studies on different substrate sources. J Ind Microbiol Biotechnol 44, 835–844 (2017). https://doi.org/10.1007/s10295-017-1942-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-017-1942-z