
Abstract
A gene (agrP) encoding a β-agarase from Pseudoalteromonas sp. AG4 was cloned and expressed in Escherichia coli. The agrP primary structure consists of an 870-bp open reading frame (ORF) encoding 290 amino acids (aa). The predicted molecular mass and isoelectric point were determined at 33 kDa and 5.9, respectively. The signal peptide was predicted to be 21 aa. The deduced aa sequence showed 98.6% identity to β-agarase from Pseudoalteromonas atlantica. The recombinant protein was purified as a fusion protein and biochemically characterized. The purified β-agarase (AgaP) had specific activity of 204.4 and 207.5 units/mg towards agar and agarose, respectively. The enzyme showed maximum activity at 55°C and pH 5.5. It was stable at pH 4.5 to 8.0 and below 55°C for 1 h. The enzyme produced neoagarohexaose and neoagarotetraose from agar and in addition to that neoagarobiose from the agarose. The neoagarooligosaccharides were biologically active. Hence, AgaP is a useful enzyme source for use by cosmetic and pharmaceutical industries.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alternating 3-linked β-D-galactopyranosyl and 4-linked α-D- or L-galactopyranosyl residues comprising sulfated, methylated, pyruvated galactans are the major structural cell wall components of marine red algae (Rhodophycea). Polymers with the 4-linked residues in the L configuration are known as agars, and polymers having the D-configuration are known as carrageenans. The neutral component of agar, agarose, is an alternating 3-linked β-D-galactopyranosyl and 4-linked 3,6-anhydro-α- L-galactopyranosyl residue and has the thermoreversible aqueous gelation property. However, in agar, due to the presence of naturally occurring sulfate substituent, the strength of the gel can be weakened [1, 2].
Agarase is the first enzyme in the agar catabolic pathway and is mainly found in marine organisms [3, 4]. Depending on the type of bonds hydrolyzed, agarases are grouped into α-agarases and β-agarases [5, 6]. The α-agarases hydrolyze the α-(1 → 3) linkages of agarose, producing agaro-oligosaccharides such as agarobiose, while β-agarases, the best studied and most numerous, hydrolyze β-(1 → 4) linkages, resulting in neoagarooligosaccharides (NAOs) such as neoagarohexaose (NA6), neoagarotetraose (NA4) or neoagarobiose (NA2) as their main products. These oligomers exhibit a variety of physiological and biological activities that show potential for use in a commercial setting including the food, cosmetics, medical, biochemistry and aquaculture industries [7].
Microorganisms are the main sources for agarase production, and with the exception of a few cases, attention has focused on marine bacteria because of the presence of a wide multiplicity of enzymes with a broad range of biotechnological applications. To date, many genera of bacteria producing agarase activities have been found, including the genera Vibrio [8–11], Pseudomonas [12–14], Pseudoalteromonas [15, 16], Alteromonas [4, 6, 17–19], Agarivorans [20, 21], Cytophage [22, 23], Saccharophagus [24] and Bacillus [25]. Moreover, cloning and expression of genes encoding to β-agarase have been reported from Agarivorans [26, 27], Microbulbifer [28–30]), Pseudoalteromonas [31]; Pseudomonas [14, 32, 33], Vibrio [9, 34]), Streptomyces [35] and Zobellia [36].
To date, despite the discovery of an increasing number of agarases, only a few of these enzymes have been successfully applied in scientific research or in industry. Indeed, the only commercially available agarase is the β-agarase II from P. atlantica T6c (P13734; Sigma-Aldrich Ref. A6306) belonging to family 86 of glycoside hydrolases. Hence, the identification of other enzymes hydrolyzing agar and elucidation of their mechanism of hydrolysis could enable researchers to find enzymes that allow an efficient production of specific agarooligosaccharides.
Therefore, the β-agarase gene (agrP) from Pseudoalteromonas sp. AG4 isolated from the marine environment of Jeju Island was studied. The gene was cloned and characterized, and the overexpressed recombinant AgrP (rAgrP) was purified. The biochemical properties of the enzyme and activity of NAOs, obtained after hydrolysis of agar by purified agarase, were studied. As a significant contribution, this paper describes the unique characteristics of the rAgrP, which possesses a high optimum temperature and broad range of pH stability, with the potential for industrial application described.
Methodology
Isolation of agarolytic bacteria strains
Agarase-producing bacteria were isolated from the red algae, Chondrus crispus, at the south coast of Jeju Island, Republic of Korea. The crushed algae were spread on SWT (0.3% Tryptone and 1.5% agar in seawater), SWY (0.3% yeast extract and 1.5% agar in seawater) and marine agar plates (Difco, Detroit, MI). Positive colonies showing pits or clear zones were picked out from the selection plates and re-streaked. The pure colonies were selected by repeat streaking under the same conditions and inoculated in their respective broths including 0.2% agar, then incubated at 30°C. The stock samples were prepared using 20% glycerol and kept at −70°C. The genomic DNA was extracted from the isolated bacteria, and polymerase chain reaction (PCR) was performed for 16S rDNA sequence amplification. As shown in Table 1, the universal primers (16S-27F as forward and 16S-1492R as reverse) were used for the amplification. The sequences were analyzed using the National Center for Biotechnology Information (NCBI) Blast N program and DNAssist program. Furthermore, the partial 16S rDNA sequence from Pseudoalteromonas sp. AG4 was aligned with the corresponding 16S rDNA partial sequences of Pseudoalteromonas spp., available in GenBank data bank, in order to perform a phylogenetic analysis.
PCR amplification of the agrP from Pseudoalteromonas sp. AG4
All of the cloning experiments were carried out according to Sambrook et al. (1989) [37] with slight modifications. For the amplification of the agarase gene, agrP-F1 forward and agrP-R2 reverse primers were designed using other known agarase gene from Pseudoalteromonas spp. present in the GenBank database (Table 1). The PCR amplification was carried out using genomic DNA as template and Ex Taq DNA polymerase (TaKaRa, Japan). After amplification, the resultant PCR product was purified using the Accuprep™ PCR purification kit (Bioneer Co., Korea).
Sequence characterization
The resultant PCR product was sequenced, and the corresponding gene was analyzed by nucleotide BLAST and Protein BLAST available on the NCBI webpages. The signal peptide sequence of agrP was predicted using the SignalP program (http://www.cbs.dtu.dk/services/SignalP/). Identity and similarity of the full-length amino acid sequences and 16S rDNA partial nucleotide sequences were calculated using the FASTA program and expressed as percent [38]. Multiple sequence alignment of the agarase gene was performed using the ClustalW version 1.8 [39]. A phylogenetic tree was constructed for 16S rDNA sequences and agarase sequences using the neighbor-joining (NJ) method from MEGA 3.1 [40]. The bootstrap values were replicated 1,000 times to obtain the confidence value for analysis.
Cloning of the agrP coding sequence into the expression vector
To clone the agrP agarase gene without its signal sequence into the pMal-c2x vector (New England Biolabs, USA), agrP-F3 and agrP-R4 primers were designed with their corresponding restriction enzyme sites at the N-terminus and C-terminus of agrP, respectively (Table 1). The vector and PCR products were restriction digested, ligated and transformed into Escherichia coli DH5α cells, and the correct recombinants (confirmed by restriction enzyme digestion and sequencing) were transformed into E. coli BL21(DE3). E. coli BL21(DE3) harboring pMal-c2x-agrP (AgrP fused with the maltose binding protein) was induced for overexpression with 0.5 mM isopropyl-ß-thiogalactopyranoside (IPTG) at the mid-exponential growth phase and incubated at 20°C for 8 h. Cells were harvested by centrifugation (4,000×g for 20 min at 4°C), resuspended in column buffer (Tris-HCl, pH 7.4, 200 mM NaCl) and frozen at −20°C overnight. After thawing on ice, cells were disrupted by sonication. After centrifugation (13,000 rpm for 30 min at 4°C), the supernatant was taken as crude enzyme. The AgrP was purified from the crude extract using pMALTM protein fusion and purification system. Proteins were collected in 500-μl fractions and run on 12% SDS-PAGE. The concentrations of the purified proteins were determined by the method of Bradford using bovine serum albumin (BSA) as the standard.
Agarase enzyme assay
The reaction mixture, containing appropriately diluted enzyme with 1% agar in pH 5.5 acetate buffer, was incubated at 45°C for 30 min. The amount of reducing sugar in the supernatant was determined using the dinitrosalicylic acid (DNS) method [41] with minor modifications. The D-galactose was used as the standard. One unit of the enzyme activity was defined as the amount of enzyme required to produce 1 μmol of reducing sugar per min.
Biochemical characterization of purified rAgrP
Agarase activity was measured at different temperatures ranging from 40 to 65°C in 5°C intervals at pH 5.5 in acetate buffer. To determine its thermostability, AgrP was incubated at temperatures ranging from 40 to 55°C for 30, 60 and 120 min. The residual agarase activity was assayed by the DNS method at 45°C and pH 5.5. AgrP activity was assayed at different pHs (pH 4.5–9) using two different buffers: acetate buffer (pH 4.5–6) and sodium phosphate buffer (pH 6.5–9.0). The agar solution (1%) and purified rAgrP were mixed in buffers with different pHs, incubated at 45°C for 30 min, and the activity was measured under standard conditions. The effects on the enzyme activity of various metal ions (Na, Mg, K, Ca, Cu, Fe and Mn; 2 mM of each) and EDTA (2 mM) were studied by incubating the purified enzyme in phosphate buffer, pH 7.0, at 45°C for 30 min, and the residual activity was subsequently determined under standard assay conditions. The substrate specificity and specific activity of purified rAgrP were determined by incubating the enzyme with different substrates such as 1% agar, agarose and carrageenan in acetate buffer (pH 5.5) under standard assay conditions.
Identification of hydrolysis products of purified rAgrP
The purified rAgrP (50 μl) was added to 200 μl of 1.5% food-grade agar solution, and the mixture was incubated at 45°C for 0.5, 1, 2, 12 and 24 h. Similar experiments were conducted with 1.5% agarose solution. NA4 and NA6 used as substrates were tested separately with the AgrP (50 μl) at 45°C for at least 24 h. After reaction, NAOs were identified by thin layer chromatography (TLC) using silica gel 60 plates (Merck, Germany) that were developed by a solvent system composed of n-butanol:acetic acid:water (2:1:1, v/v). The resultant oligosaccharide spots were visualized by spraying 10% H2SO4 onto the plate and heating on a hot plate. NA6 and D-(+)-galactose were purchased from Sigma (USA). The NA6 was digested using commercial β-agarase (New England Biolabs, USA) to obtain NA4 with a smaller amount of NA2. All mentioned oligosaccharides were used as standards.
Preparation of NAOs
The purified rAgrP was mixed with a 1.5% agar solution, and the reaction was performed at 45°C for 24 h. The agar hydrolysis was confirmed by TLC analysis. Finally, the resultant product mixture was freeze dried and used for the analysis of respective biological activities.
Whitening effect
The B16F10 murine melanoma cells were seeded in a 24-well plate at a density of 2 × 105 cells/ml. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (BioWhittaker, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% CO2 for 24 h. The cells were washed two times with phosphate buffered saline (PBS) and fed 2 ml of fresh media. Lastly, the cells were treated with 200 nM α-melanocyte stimulating hormone (MSH). NAOs and a positive control (arbutin) were added to the cells at a final concentration of 0.1, 1, 10 and 100 μg/ml, and incubated for 72 h under the same conditions. After centrifugation (1,700×g, 4°C for 5 min), the cells were treated with 200 μl of 1 N NaOH for 1 h at 56°C into a 96-well plate. The melanin content was determined at 405 nm using a microplate reader ELISA.
Antioxidant activity
The antioxidant activity of the NAOs was determined by utilizing the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay. A 100-μl volume of 0.01, 0.1 and 1 mg/ml neoagarooligosaccharide mixture was added to 100 μl of DPPH and incubated at 25°C for 60 min in darkness. Then by using 0.15 mM of DPPH in ethanol as the colorimetric reagent, the absorbance was measured at 492 nm using a microplate reader ELISA.
Cytotoxicity assay
A cytotoxicity assay was conducted using B16F10 murine melanoma and normal human fibroblast cells. Cells were cultured in a 96-well plate and maintained in DMEM containing 10% FBS for 24 h. Cells were washed two times with 1 ml PBS, fed fresh media and treated with α-melanocyte stimulating hormone (α-MSH) at 200 nM. NAOs and the positive control (arbutin) were added to the cells at the final concentration of 0.1, 1, 10 and 100 μg/ml and incubated for 72 h as above. Cells were washed twice with PBS and fed 200 μl of fresh media. Then, 20 μl of 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) solution (5 mg/ml in PBS) was added to each well, and the cells were incubated for 12 h at 37°C. After removing the media, cells were dissolved in 100 μl of DMSO. The amount of formazan converted from MTT was measured by absorbance at 570 nm using a microplate reader ELISA.
Nucleotide sequence accession number
The 16S rDNA and agrP sequences from Pseudoalteromonas sp. AG4 were deposited in the GenBank database with the accession numbers GU143092 and GQ243743, respectively.
Results
Isolation, cloning and nucleotide sequence analysis of the agrP
On the basis of an apparition of a clear zone around colonies on SW and MA selection plates, an agarolytic bacterium was isolated. After 16S rDNA sequence amplification, the nearest neighbors were identified by BLASTN against the Genbank database, and the bacterial strain was identified by phylogenetic analysis. Figure 1 shows an unrooted phylogenetic tree of the 16S rDNA sequences of different Pseudoalteromonas species and the selected 16S rDNA sequence in this study. The phylogenetic tree results demonstrated that the 16S rDNA sequence from AG4 strain clustered with Pseudoalteromonas marina, P. gracilis, P. translucida and P. undina, with a 72% bootstrap confidence value. BLASTN analysis results showed that the identified strain had 99.8% identity to Pseudoalteromonas sp. BSw20092. In addition, when the corresponding partial sequence was aligned with the identified strains, it showed 99.8, 99.3, 99.0 and 97% identity to P. gracilis, P. marina, P. translucida and P. undina, respectively. Hence it was grouped into the genera Pseudoalteromonas spp. and named as Pseudoalteromonas sp. AG4.

Phylogenetic tree of the relatedness of Pseudoalteromonas sp. AG4 with other Pseudoalteromonas spp. based on the partial 16S rDNA sequences. The unrooted tree was constructed by NJ analysis. The numbers indicate the bootstrap confidence values of 1,000 replicates. The GenBank accession numbers are shown next to each species
After PCR amplification using specific primers (Table 1), the β-agarase gene was identified from the Pseudoalteromonas sp. AG4 and named agrP. The nucleotide and amino acid sequence of the agrP is shown in Fig. 2. The open reading frame (ORF) has 870 bp and encodes 290 amino acids. The AgrP has a theoretical molecular mass and an isoelectric point (pI) of 33 kDa and 5.9, respectively. Analysis of the deduced amino acid sequence of AgrP revealed that the protein possesses an N-terminal signal peptide of 21 aa. The mature protein displays the catalytic residues classically found in the enzymes belonging to family 16 of glycoside hydrolases (Glu148-Asp150-Glu153) [42]. The protein shows 99.3 and 92.8% similarity, and 98.6 and 85.9% identity to P. atlantica β-agarase I and Aeromonas sp. β-agarase, respectively, both enzymes belonging to family 16 of glycoside hydrolases. The comparison of AgrP with other GH16 agarases by multiple alignment is shown in Fig. 3. The overall comparisons of the deduced amino acid sequence of AgrP with other agarases in the Genbank database are shown in Table 2. The unrooted phylogenetic tree was constructed based on the amino acid sequences of β-agarases in NCBI database using the NJ method (Fig. 4). Analysis of the results revealed that AgrP was closer to the clade of Zobellia galactanivorans AgaA than that of Z. galactanivorans AgaB. All data considered, these results show that the AgrP belongs to family 16 of glycoside hydrolases, in the subfamily of galactanases.

The nucleotide and deduced amino acid sequences of the agrP of Pseudoalteromonas sp. AG4. The predicted signal peptide sequence is highlighted in gray. The start (ATG) and stop (TAA) codons are in bold, and the stop codon is marked with an asterisk. The GHF-16 β-agarase domain is in italics. Active sites and calcium binding residues are in boxes and bold letters, respectively. The catalytic residues in signature of the GH16 family (E × D × E) is dotted line boxed

Multiple sequence alignment of the AgrP amino acid sequence of Pseudoalteromonas sp. AG4 with known agarases. The conserved catalytic residues are highlighted in dark gray with black circles (filled circle) and conserved residues involved in calcium ion binding are represented by filled squares. Identical residues in all sequences are shaded in light gray and indicated by an asterisk under the column; conserved substitutions are indicated by a colon, and semi-conserved substitutions are indicated by a dot. Deletions are indicated by dashes. Sequence sources: Pseudoalteromonas atlantica β-agarase I (AAA91888), Aeromonas sp. β-agarase (AAF03246), Saccharophagus degradans β-agarase I (AAT67062), Pseudomonas sp. ND137 agarase (BAB79291), Zobellia galactanivorans β-agarase B (AAF21821)

Phylogenetic analysis of AgrP compared with known agarases based on amino acid sequences. Phylogenetic analysis was done by NJ method using MEGA3.1 based on sequence alignment using ClustalW (1.81). The numbers indicate the bootstrap confidence values of 1,000 replicates. The accession numbers of the selected agarase sequences are as follows: AAA91888, β-agarase I (Pseudoalteromonas atlantica); AAP49346, AguB; AAP70390, AguH; AAP70365, AguK; AAP49316, AguD from uncultured bacterium; AAF03246, β-agarase (Aeromonas sp.); AB124837, agarase (Microbulbifer thermotolerans); BAC99022, agarase (Microbulbifer elongatus); BAB79291, agarase, (Pseudomonas sp. ND137; AAF21821, β-agarase B precursor (Zobellia galactanivorans); AAF21820, β-agarase A precursor (Zobellia galactanivorans); AAN39119, extracellular agarase precursor, (Pseudoalteromonas sp. CY24); CAB61795, extracellular agarase precursor (Streptomyces coelicolor A3); BAH16616, agarase (Cellvibrio sp. OA-2007); AAT67062, β-agarase I (Saccharophagus degradans)
Purification of rAgrP
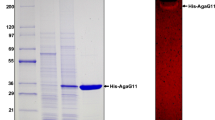
The mature AgrP sequence was cloned into the pMal-c2x expression vector by fusion with a MBP at the N-terminal. The rAgrP was overexpressed in E. coli BL21 (DE3), and the soluble fraction of induced cells was purified as a fusion protein. The different steps of the rAgrP purification are shown by 12% SDS-PAGE analysis in Fig. 5. The purified rAgrP was identified as an approximately 72.5-kDa strong specific band (lane 4). Given that the molecular mass of MBP is 42.5 kDa, the observed molecular mass of the recombinant protein is in agreement with the theoretical molecular mass of the mature protein.

SDS-PAGE of the AgrP. Protein samples were separated on 12% SDS-PAGE and stained with Coomassie brilliant blue. M: molecular mass marker (BioRad, USA). Lane 1: total cellular extract from E. coli BL21 (DE3) before induction; lane 2: total cellular soluble extract after induction; lane 3: total cellular insoluble extract after induction; lane 4: purified AgrP
Biochemical characterization of purified rAgrP
The enzyme showed specific activities towards agar and agarose with 204.4 and 207.5 unit/mg, respectively. However, no activity was observed when carrageenan was used as substrate. The rAgrP was maximally active at 55°C and was stable below 55°C for 1 h (Fig. 6a). The purified enzyme showed its maximum activity at pH 5.5 and exhibited broad pH stability, which retained its activity up to 60% at pH 4.5–7.5 (Fig. 6b). Temperature stability analysis of rAgrP showed that the enzyme was stable after 2 h at 45°C with retention of its maximum activity of 90% (Fig. 5c). Furthermore, the enzyme remained stable up to 75% after 1 h at 55°C. At 45°C, the addition of 2 mM CaCl2 (final concentration) into the reaction mixture did not affect the thermostability compared to the samples where no calcium was added (Fig. 5d). On the contrary, at 55°C, the calcium caused a decrease of the thermostability from 70 to 30%. Furthermore, Na+, K+, Mg2+ and Ca2+ stimulated agarase activity by 5%, whereas Fe2+ stimulated the activity by 20% (Fig. 7). The Cu2+, Zn2+ and Mn2+ inhibited the activity by 100, 80 and 40%, respectively, and EDTA at 2 mM concentration inhibited the agarase activity by 90%.

Biochemical characterization of purified rAgrP. a The effect of temperature on the AgrP. The effect of temperature on enzyme activity was determined under standard assay conditions at temperatures ranging between 40 and 65°C. b The effect of pH on the activity of AgrP. Optimum pH for AgrP activity was examined from pH 4.5–9.0 at pH 0.5 intervals at 45°C under standard assay conditions using acetate (pH 4.5–6.0) and phosphate buffer (pH 6.5–9.0). c The effect of thermostability on AgrP at different temperatures for different time points. Thermostability was determined by measurement of residual activity under standard assay conditions at temperatures between 40 and 55°C for 30, 60 and 120 min. d The effect of 2 mM CaCl2 on thermostability of AgrP

Effect of various ions and EDTA on the activity of purified AgrP. Various ions (MgSO4, CaCl2, NaCl, KCl, CuSO4, FeSO4, MnCl2) and EDTA at a final concentration of 2 mM were added into the reaction buffer to test the activity of AgrP at 45°C for 30 min. The data presented are the average of three replicates. Means with the different letters are significantly different at P < 0.05, based on ANOVA. Error bars represent ± SD
Identification of hydrolysis products of the rAgrP on TLC
Hydrolysis patterns of the food grade agar, agarose, NA6 and NA4, obtained from purified rAgrP, are shown in Fig. 8. When food grade agar and agarose were used as a substrate, reaction products were clearly observed on the TLC plate. The results showed that the final product obtained from agar hydrolysis by rAgrP was mainly the NA4 with a small amount of agarobiose after 24 h of reaction time. The agarose hydrolysis also showed the same result with amounts of neoagarobiose higher than in the agar hydrolysis case at 24 h. Finally, the rAgrP could not hydrolyze NA4, but could hydrolyze NA6 to generate NA4 with a slight amount of neoagarobiose.

TLC of hydrolysis products of the purified AgrP on food grade agar, agarose and NAOs. Assays were performed in 250-μl reactions containing 50 μl of purified agarase and 200 μl of 1.5% agar at 45°C for 0.5, 1, 2, 12 and 24 h. NA6 and NA4 were separately tested with purified AgrP agarase (50 μl) at 45°C for 120 min. NA6, NA4, NA2 and D-(+)-galactose (G) were used as standards (STD)
Functional activity of NAOs
The whitening effects of NAOs and arbutin were evaluated by measuring the melanin content in melanocytes (Fig. 9a). In this study, B16F10 murine melanoma cells were whitened by 10 and 100 μg/ml of NA4 (20%), and a similar effect was observed with the commercial whitening compound arbutin at the same concentrations.

Functional properties of NAOs produced by purified AgrP. a Whitening effect of NAOs and arbutin at different concentrations in B16F10 cells. NAOs and positive control (arbutin) were treated to the 200 nM MSH treated cells in the final concentration of 0.1, 1, 10 and 100 μg/ml and incubated for 72 h at 37°C. After incubation of the cells with 1 N NaOH at 56°C for 1 h, the melanin content was determined at 405 nm. b The effect of NAOs on the DPPH scavenging activity. The 100 μl of 0.01, 0.1 and 1 mg/ml NAO was added into 100 μl of DPPH, followed by incubation at 25°C for 60 min in darkness and the OD measured at 492 nm. The data presented are an average of three independent experiments. Means with the different letters are significantly different at P < 0.05, based on ANOVA. Error bars represent +SD
The level of antioxidant activity was determined by measuring the DPPH radical scavenging activity assay (Fig. 9b). The results showed that NAOs had lower activity than commercial antioxidant vitamin C and E. However, the antioxidant activity was increased in a dose-dependent manner. The NAOs showed about 50% DPPH scavenging activity, whereas commercial antioxidants showed about 90% activity.
A cell cytotoxicity assay showed that NAOs were not cytotoxic to B16F10 melanoma and normal human fibroblast cells at concentrations ranging from 0.1 to 100 μg/ml (data not shown).
Discussion
The agarolytic Pseudoalteromonas sp. AG4 was isolated from Chondrus crispus at the south coast of Jeju Island in the Republic of Korea. agrP ORF consists of 870 bp, encoding a protein of 290 amino acids with a predicted molecular mass of 33 kDa. The 21 amino acids located at the N-terminus of the deduced amino acid show the typical attributes of a signal peptide: a positively charged region, a hydrophobic region and a signal sequence cleavage site. The sequence analysis showed that the AgrP is restricted to a single catalytic domain, revealing that it is not a modular enzyme like P. atlantica β-agarase I and Aeromonas sp. β-agarase. The AgrP contains the strictly conserved catalytic residues, Glu148 and Glu153, which act as nucleophile and acid residues, respectively, in catalysis, as reported previously for sequences of the GH16 family [31, 43, 44]. The BLAST P and motif scan analysis together with phylogenetic analysis results confirmed that AgrP is a β-agarase that belongs to family 16 of glycoside hydrolases.
Most of the GHF-16 β-agarase degrades agarose and oligoagarosaccrides comprising six sugars to yield four sugars as their main products [16, 43]. Similarly, the main products of agar hydrolysis by rAgrP were NA6 and NA4 with a small amount of agarobiose. Moreover, TLC results indicate that AgrP hydrolyzes agar, agarose and neo-agarooligosaccharides larger than NA6, and NA6 to form NA4 as the main product. In contrast to this, AgaB from Pseudoalterommonas sp. CY24 degrades agarose to neoagarooctaose and neoagarodecaose, suggesting that it contains a large catalytic cleft consisting of 12 glycosyl binding subsites [31]. However, for the precise structure function relationships of the enzyme, 3D and crystal structural analyses need to be conducted in the future.
The rAgrP fusion protein, when fused with MBP, was purified by the pMALTM protein fusion and purification system. The rAgrP showed a strong single band on 12% SDS-PAGE, indicating the complete purification of the enzyme. The molecular mass of the protein was found to about 72.5 kDa, which was in agreement with our predicted molecular mass of 30 kDa (mature protein), since MBP has a 42.5-kDa molecular mass. The size is closer to that reported for the β-agarases of Pseudoalteromonas sp. N-1 (33 kDa) [15] and Pseudomonas atlantica (32 kDa) [13]. To date, the previously reported β-agarases belonging to the GH16 family have been characterized to have an optimum temperature of 40°C and neutral to mild alkaline pH [34]. For example, the β-agarase from Pseudoalteromonas sp. strain N-1 was reported to have a pH and temperature optima of 7 and 30°C, respectively [15]. In another example, the pH and temperature optima for recombinant β-agarase AgaB from Pseudoalteromonas sp. CY24 were pH 6.0 and 40°C, respectively [31]. In this paper, we describe the rAgrP that is active over a broad pH range (4.5–7.5 with an optimum pH of 5.5) and that has an optimum temperature of 55°C, higher than the agarose gelling temperature (40°C). Therefore, rAgrP would be very useful for the industrial production of oligoagarosaccharides from agar. In contrast to the optimum temperature of AgrP, the protein thermal stability profiles, which show stability up to 80% below 55°C after 1 h incubation, indicate that the enzyme could be used under moderate heating conditions. The existence of a calcium-binding site in the family of GH16 β-agarases has been experimentally studied by the structure of the AgaA from Z. galactanivorans complexed with a Ca2+ [45], and it has been reported that this calcium-binding site is conserved in family GH16 enzymes [44]. Moreover, this Ca2+ is not involved in the catalytic machinery, but has been shown to have a stabilizing effect in family GH16 [46]. The addition of CaCl2 did not show much effect on AgrP activity (increases the activity of only 5%). This confirms the previously published results [29, 34]. In contrary to previously reported data, addition of EDTA decreases the AgrP activity of 90% [18, 25]. An exact explanation of the activity in the presence of iron needs to be elucidated by further experiments. Finally, due to the affinity of the heavy metals for the SH, CO and NH moieties present in the amino acids, structural alterations could occur and explain the inhibitory effects of metal ions like Zn2+. Mixtures of NAOs were produced from rAgrP and examined for the degree of cytotoxicity, whitening and their antioxidant effect, and compared with the known commercial tyrosinase inhibitor, arbutin, and antioxidants, vitamin C and E. NAOs were not cytotoxic to B16F10 murine melanoma and normal human fibroblast cells at 0.1–100 μg/ml and showed a whitening effect at 10 and 100 μg/ml, with no significant difference observed with arbutin. These findings were consistent with the functional activities of oligoagrosaccharides produced by β-agarase from Agarivorans sp. JA-1 by Lee and his group [47]. Inhibition of the production of lipid peroxidation and inhibition of the production of NO were some of the antioxidant effects of oligoagarosaccharides [48]. However, herein we report a lower (50% at 1.0 mg/ml) antioxidant activity than that of the commercial antioxidants showing a free radicals scavenging ability of NAOs prepared by AgrP.
In conclusion, marine Pseudoalteromonas sp. AG4 was isolated from red algae found within Jeju’s coastal environment. AgrP features a homologous catalytic domain belonging to family 16 of glycoside hydrolases with characteristic biochemical properties used for NAO production. Since the simple NAOs generated from complex polysaccharides have whitening and antioxidant activities with no cytotoxicity, the hydrolysis products identified here have potential use in cosmetics as well as medical industries.
References
Craigie JS (1990) Cell walls. In: Cole KM, Sheath RG (eds) Biology of the red algae. Cambridge University Press, Cambridge, pp 221–257
Usov AI (1998) Structural analysis of red seaweed galactans of agar and carrageenan groups. Food Hydrocol 12:301–308
Araki C, Arai K (1967) Studies on the chemical constitution of agar-agar XXIV. Isolation of a new disaccharide as a reversion product from acidic hydrolysate. Bull Chem Societ Japan 40:1452–14564
Potin P, Richard C, Rochas C, Kloareg B (1993) Purification and characterization of the alpha-agarase from Alteromonas agarlyticus (Cataldi) comb. nov., strain GJ1B. Eur J Biochem 214:599–607
Araki C (1959) Seaweed polysaccharides. In: Wolfrom ML (ed) Carbohydrate chemistry of substances of biological interests. Pergamon Press, London, pp 15–306
Hassairi I, Ben AR, Nonus M, Gupta BB (2001) Production and separation of alpha-agarase from Alteromonas agarlyticus strain GJ1B. Biores Technol 79:47–517
Kobayashi R, Takisada M, Suzuki T, Kirimura K, Usami S (1997) Neoagarobiose as a novel moisturizer with whitening effect. Biosci Biotechnol Biochem 61:162–1638
Aoki T, Araki T, Kitamikado M (1990) Purification and characterization of a novel beta-agarase from Vibrio sp. AP-2. Eur J Biochem 187:461–465
Sugano Y, Terada I, Arita M, Noma M, Matsumoto T (1993) Purification and characterization of a new agarase from a marine bacterium, Vibrio sp. strain JT0107. Appl Environ Microbiol 59:1549–155410
Araki T, Lu Z, Morishita T (1998) Optimization of parameters for isolation of protoplasts from Gracilaria verrucosa (Rhodophyta). J Mar Biotechnol 6:193–197
Fu W, Han B, Duan D, Liu W, Wang C (2008) Purification and characterization of agarases from a marine bacterium Vibrio sp. F-6. J Ind Microbiol Biotechnol 35:915–922
Groleau D, Yaphe W (1977) Enzymatic hydrolysis of agar: purification and characterization of beta-neoagarotetraose hydrolase from Pseudomonas atlantica. Can J Microbiol 23:672–679
Morrice LM, McLean MW, Williamson FB, Long WF (1983) Beta-agarases I and II from Pseudomonas atlantica. Purifications and some properties. Eur J Biochem 135:553–558
Ha JC, Kim JT, Kim SK, Oh TK, Yu JH, Kong IS (1997) Beta agarase from Pseudomonas sp. W7: purification of the recombinant enzyme from Escherichia coli and the effects of salt on its activity. Biotechnol Appl Biochem 26:1–6
Vera J, Alvarez R, Murano E, Slebe JC, Leon O (1998) Identification of a marine agarolytic Pseudoalteromonas isolate and characterization of its extra cellular agarase. Appl Environ Microbiol 64:4378–4383
Schroeder DC, Jaffer MA, Coyne VE (2003) Investigation of the role of a beta (1–4) agarase produced by Pseudoalteromonas gracilis B9 in eliciting disease symptoms in the red alga Gracilaria gracilis. Microbiol 149:2919–2929
Leon O, Quintana L, Peruzzo G, Slebe JC (1992) Purification and properties of an extracellular agarase from Alteromonas sp. strain C-1. Appl Environ Microbiol 58:4060–4063
Kirimura K, Masuda N, Iwasaki Y, Nakagawa H, Kobayashi R, Usami S (1999) Purification and characterization of a novel beta agarase from an alkalophilic bacterium, Alteromonas sp. E-1. J Biosci Bioeng 87:436–441
Wang J, Mou H, Jiang X, Guan H (2006) Characterization of a novel beta-agarase from marine Alteromonas sp. SY37–12 and its degrading products. Appl Microbiol Biotechnol 71:833–839
Fu XT, Lin H, Kim SM (2008) Purification and characterization of a novel beta-agarase, AgaA34, from Agarivorans albus YKW-34. Appl Microbiol Biotechnol 78:265–273
Hu Z, Lin BK, Xu Y, Zhong MQ, Liu GM (2009) Production and purification of agarase from a marine agarolytic bacterium Agarivorans sp. HZ105. J Appl Microbiol 106:181–190
Duckworth M, Turvey JR (1969) The action of a bacterial agarase on agarose, porphyran and alkali treated porphyran. Biochem J 113:687–692
Van der Meulen HJ, Harder W (1975) Production and characterization of the agarase of Cytoplaga flevensis. Antonie van Leeuwenhoek 41:431–447
Ekborg NA, Taylor LE, Longmire AG, Henrissat B, Weiner RM, Hutcheson SW (2006) Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2–40. Appl Environ Microbiol 72:3396–3405
Suzuki H, Sawai Y, Suzuki T, Kawai K (2003) Purification and characterization of an extracellular beta agarase from Bacillus sp. MK03. J Biosci Bioeng 93:456–463
Lee DG, Park GT, Kim NY, Lee EJ, Jang MK, Shin YG, Park GS, Kim TM, Lee JW, Lee JH, Kim SJ, Lee SH (2006) Cloning, expression, and characterization of a glycoside hydrolase family 50 beta-agarase from a marine Agarivorans isolate. Biotechnol Lett 28:1925–1932
Ohta Y, Hatada Y, Ito S, Horikoshi K (2005) High-level expression of a neoagarobiose-producing beta-agarase gene from Agarivorans sp. JAMB-A11 in Bacillus subtilis and enzymic properties of the recombinant enzyme. Biotechnol Appl Biochem 41:183–191
Ohta Y, Hatada Y, Nogi Y, Li Z, Ito S, Horikoshi K (2004) Cloning, expression, and characterization of a glycoside hydrolase family 86 beta agarase from a deep-sea Microbulbifer-like isolate. Appl Microbiol Biotechnol 66:266–275
Ohta Y, Hatada Y, Nogi Y, Miyazaki M, Li Z, Akita M, Hidaka Y, Goda S, Ito S, Horikoshi K (2004) Enzymatic properties and nucleotide and amino acid sequences of a thermostable beta-agarase from a novel species of deep-sea Microbulbifer. Appl Microbiol Biotechnol 64:505–514
Ohta Y, Hatada Y, Nogi Y, Miyazaki M, Li Z, Hatada Y, Ito S, Horikoshi K (2004) Enzymatic properties and nucleotide and amino acid sequences of a thermostable beta agarase from the novel marine isolate, JAMB-A94. Biosci Biotechnol Biochem 68(5):1073–1081
Ma C, Lu X, Shi C, Shi C, Li J, Gu Y, Ma Y, Chu Y, Han F, Gong Q (2007) Molecular cloning and characterization of a novel beta-agarase, AgaB, from marine Pseudoalteromonas sp. CY24. J Biol Chem 282:3747–3754
Lee S, Park J, Yoon S, Kim J, Kong I (2000) Sequence analysis of a beta-agarase gene (pjaA) from Pseudomonas sp. isolated from marine environment. J Biosci Bioeng 89:485–488
Ryu SK, Cho SJ, Park SR, Lim WJ, Kim MK, Hong SY, Baed W, Park YW, Kim BK, Kim H, Yun HD (2001) Cloning of the cel9A gene and characterization of its gene product from marine bacterium Pseudomonas sp. SK38. Appl Microbiol Biotechnol 57:138–145
Zhang WW, Sun L (2007) Cloning, characterization, and molecular application of a beta-agarase gene from Vibrio sp. strain V134. Appl Environ Microbiol 73:2825–2831
Kendall K, Cullum J (1984) Cloning and expression of an extracellular-agarase from Streptomyces coelicolor A3(2) in Streptomyces lividans 66. Gene 29:315–321
Jam M, Flament D, Allouch J, Potin P, Thion L, Kloareg B, Czjzek M, Helbert W, Michel G, Barbeyron T (2005) The endo-beta-agarases AgaA and AgaB from the marine bacterium Zobellia galactanivorans: two paralogue enzymes with different molecular organizations and catalytic behaviours. Biochem J 385:703–713
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Pearson WR (1990) Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol 183:63–98
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic alignment search tool. J Mol Biol 215:403–410
Allouch J, Jam M, Helbert W, Barbeyron T, Kloareg B, Henrissat B, Czjzek M (2003) The three-dimensional structures of two beta agarases. J. Biol Chem 278:47171–47180
Michel G, Chantalat L, Duee E, Barbeyron T, Henrissat B, Kloareg B, Dideberg O (2001) The kappa-carrageenase of Pseudoalteromonas carrageenovora features a tunnel-shaped active site: a novel insight in the evolution of clan-B glycoside hydrolases. Structure 9:513–525
Allouch J, Helbert W, Henrissat B, Czjzek M (2004) Parallel substrate binding sites in a β-agarase suggest a novel mode of action on double-helical agarose. Structure 12:623–632
Keitel T, Meldgaard M, Heineman U (1994) Cation binding to a Bacillus (1, 3–1, 4)-β-glucanase Geometry, affinity and effect on protein stability. Eur J Biochem 222:203–214
Lee DG, Jang MK, Lee OH, Kim NY, Ju S, Lee SH (2008) Over-production of a glycoside hydrolase family 50 β-agarase from Agarivorans sp. JA-1 in Bacillus subtilis and the whitening effect of its product. Biotechnol Lett 30:911–918
Chen HM, Zheng L, Yan XJ (2005) The preparation and bioactivity research of agaro-oligosaccharides. Food Technol Biotechnol 43(1):29–36
Acknowledgment
This research was financially supported by the Ministry of Education, Science Technology (MEST) and Korea Institute for Advancement of Technology (KIAT) through the Human Resource Training Project for Regional Innovation, and by a research grant (PP00740) from Korea Ocean Research & Development Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oh, C., Nikapitiya, C., Lee, Y. et al. Cloning, purification and biochemical characterization of beta agarase from the marine bacterium Pseudoalteromonas sp. AG4. J Ind Microbiol Biotechnol 37, 483–494 (2010). https://doi.org/10.1007/s10295-010-0694-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-010-0694-9