
Abstract
An agar-degrading bacterium, strain JAMB-A7, was isolated from the sediment in Sagami Bay, Japan, at a depth of 1,174 m and identified as a novel species of the genus Microbulbifer. The gene for a novel β-agarase from the isolate was cloned and sequenced. It encodes a protein of 441 amino acids with a calculated molecular mass of 48,989 Da. The deduced amino acid sequence showed similarity to those of known β-agarases in glycoside hydrolase family 16, with only 34–55% identity. A sequence similar to a carbohydrate-binding module was found in the C-terminal region of the enzyme. The recombinant agarase was hyper-produced extracellularly using Bacillus subtilis as the host, and the enzyme purified to homogeneity had a specific activity of 398 U (mg protein)–1 at pH 7.0 and 50°C. It was thermostable, with a half-life of 502 min at 50°C. The optimal pH and temperature for activity were around 7 and 50°C, respectively. The pattern of agarose hydrolysis showed that the enzyme was an endo-type β-agarase, and the final main product was neoagarotetraose. The activity was not inhibited by NaCl, EDTA, and various surfactants at high concentrations. In particular, sodium dodecyl sulfate had no inhibitory effect up to 2%.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
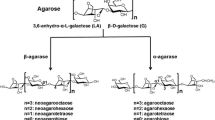
Agar, a well-characterized polysaccharide present in the cell walls of some red algae, is composed of agarose and agaropectins. Agarose consists of a linear chain of alternating residues of 3-O-linked β-d-galactopyranose and 4-O-linked 3,6-anhydro-α-l-galactose (Duckworth and Yaphe 1971). Agaropectins have the same basic disaccharide-repeating units as agarose, although some hydroxyl groups of 3,6-anhydro-α-l-galactose residues are substituted by sulfoxy or methoxy and pyruvate residues (Araki 1966).
Agar-degrading bacteria isolated from marine and other environments to date have been assigned to the genera Pseudomonas (Ha et al. 1997), Pseudoalteromonas (Vera et al. 1998), Streptomyces (Kendall and Cullum 1984), Alteromonas (Potin et al. 1993), Microscilla (Naganuma et al. 1993; Zhong et al. 2001), Vibrio (Sugano et al. 1993a, 1993b, 1994), and Cytophaga (Van der Meulen and Harder 1975). Agarases produced by these bacteria are classified into two groups based on their mode of action: α-agarase and β-agarase, which hydrolyze α-1,3 linkages and β-1,4 linkages in agarose, respectively. Most agarases that have been purified and characterized belong to the β-agarase group except for an α-agarase from Alteromonas agarlyticus GJ1B (Potin et al. 1993). Biochemical and genetic studies of β-agarases show high degrees of heterogeneity in terms of their amino acid sequences, molecular masses, substrate specificities, and catalytic properties even though the enzymes are functionally similar in the hydrolysis of β-1,4 linkages in the agar backbone.
Agarases have applications in the food, cosmetic, and medical industries for the production of oligosaccharides from agar. Neoagaro-oligosaccharides produced by β-agarase inhibit the growth of bacteria, slow the rate of degradation of starch, and are used as low-calorie additives to improve food qualities. Moreover, neoagarobiose is a rare reagent with both moisturizing and whitening effects on melanoma cells (Kobayashi et al. 1997). The polysaccharide fractions prepared from marine algae by β-agarase also have macrophage-stimulating activity and are suitable as a source of physiologically functional foods with protective and immunopotentiating activity (Yoshizawa et al. 1995). Moreover, agarase can be used to degrade the cell walls of marine algae for extraction of labile substances with biological activities and for the preparation of protoplasts (Araki et al. 1998). However, agarases reported to date have not been used widely for industrial applications due to their low activity, stability, and productivity.
To understand the molecular basis for the catalytic mechanism and diversity of agarases, further biochemical and genetic studies on the enzyme are required. We have isolated a number of deep-sea, agar-degrading microorganisms, and sequenced the genes encoding agarases and characterized the enzymes. In this report, we describe the identification of an agar-degrading bacterium—designated strain JAMB-A7—isolated from Sagami Bay, Japan, at a depth of 1,174 m, and the sequencing of the gene for the agarase (agaA7) that it produced. Moreover, we show that the recombinant enzyme expressed in Bacillus subtilis cells (RagaA7) is thermostable and resistant to various chemical reagents.
Materials and methods
Bacterial strains, plasmids, and culture conditions
The sediment samples in Sagami Bay at a depth of 1,174 m were collected using the submersible “Shinkai 2000”. The samples were suspended in marine broth 2216 (Difco, Detroit, Mich.). The suspensions were suitably diluted with the broth and spread on marine agar (Difco), followed by incubation at 25°C for several days. Colonies that formed pits or shallow craters around them were picked up. One of the bacteria exhibiting agarolytic activities was chosen and named strain JAMB-A7. Microbulbifer elongataus DSM 6810 and Microbulbifer hydrolyticus ATCC700072 were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ; Braunschweig, Germany). Microbulbifer salipaludis JCM11542 was obtained from the Japan Collection of Microorganisms (JCM; Saitama, Japan). All strains were propagated at 25°C in marine agar 2216. Escherichia coli HB101 (F′ supE44 hsdS20 recA13 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 leuB6 thi-1) was used as the host for cloning and routinely grown at 37°C in Luria-Bertani (LB) broth (Difco) supplemented with ampicillin (100 µg ml–1) or tetracycline (15 µg ml–1) when required. Plasmid pUC18 was used for cloning and sequencing. B. subtilis ISW1214 (leuA8 metB5 hsrM1) harboring plasmids was cultured at 30°C, with shaking, in an optimized medium composed of (w/v) 12% corn steep liquor (Nihon Syokuhin Kako, Shizuoka, Japan), 0.2% Lab-Lemco powder (Oxoid, Hampshire, UK), 0.1% yeast extract (Difco), 0.1% KH2PO4, 0.02% MgSO4·7H2O, 0.05% CaCl2, 6% maltose, and tetracycline (15 µg ml–1) (CLT medium).
Isolation of DNA, transformation, and sequencing
Genomic DNA and plasmids were prepared as described by Saito and Miura (1963) and Birnboim and Doly (1979), respectively. Restriction digestion and ligation were carried out using standard methods (Sambrook et al. 1989). Transformation of E. coli and B. subtilis with plasmids followed the methods of Hanahan (1983) and Chang and Cohen (1979), respectively. Double-stranded DNA sequencing was performed with custom oligonucleotide primers using an ABI Prism Big Dye Terminator Cycle sequencing kit and an ABI 377 Sequencer (Applied Biosystems, Foster City, Calif.). Computer sequence analysis was carried out using the GENETYX-MAC program ver. 10.1 (SDC Software Development, Tokyo, Japan).
16S rDNA analysis and DNA-DNA hybridization
16S rDNA analysis was performed by amplifying the 16S rRNA genes by polymerase chain reaction (PCR) using the eubacterial primers 27f and 1492r. The PCR mixtures (50 µl) contained 2.5 mM MgCl2, 250 µM each of deoxynucleoside triphosphates, 50 pmol of each primer, 2.5 U LA Taq DNA polymerase (TaKaRa, Kyoto, Japan), and ~10 ng DNA template in the manufacturer’s buffer. PCR was performed using a T Gradient 96 thermocycler (Whatman BiometraR, Goettingen, Germany) programmed as follows: 1 min of denaturation at 96°C, followed by 30 cycles at 96°C for 1 min, 63°C for 1 min, and 72°C for 1.5 min, with a final extension at 72°C for 2 min. The amplified 1.5-kb PCR product was purified using the High Pure PCR product purification kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. The DNA sequences were determined directly from the purified PCR product as described above using the primers described by Weisburg et al. (1991). The 16S rDNA sequence of the isolate was analyzed using FASTA programs (http://www.ddbj.nig.ac.jp) to obtain closely matched species. DNA-DNA hybridizations were performed fluorometrically following the method of Ezaki at al. (1989) using photobiotin-labeled DNA probes and microdilution wells.
Phenotypic analysis of the strains
Respiratory lipoquinones were analyzed as described by Komagata and Suzuki (1987) using a reversed-phase column (Bondasphere C18, 3.9×150mm, Waters, Milford, Mass.) and an LC-10Avp with CLASS-VP HPLC system (Shimadzu, Kyoto, Japan). The G+C content of the isolates was determined according to the method of Tamaoka and Komagata (1984) using a reversed-phase column (Cosmosil 5C18, 4.6×150 mm, Nacalai Tesque, Kyoto, Japan) and the HPLC system. For quantitative analysis of cellular fatty acid composition, a loop of cell masses were harvested and fatty acid methyl esters were prepared and identified according to the method of Waino et al. (1999) using a mass spectrometer (GCMA-QP5050A; Shimadzu) and a gas chromatograph (GC-17A; Shimadzu). Catalase activity was determined by bubble production in 3% (v/v) hydrogen oxide solution. Oxidase activity was determined by oxidation of 1% (w/v) p-aminodimethylaniline oxalate. Hydrolysis of gelatin and starch were determined as described by Cowan and Steel (1965). The production of indole and H2S were tested on SIM agar (Nissui, Tokyo, Japan). Acid production from carbohydrates was determined as described by Leifson (1963). Growth at various NaCl concentrations was examined in 1:10-diluted marine broth 2216. Growth at various temperatures was measured in marine broth 2216.
Cloning of the agaA7 gene
The genomic DNA of strain JAMB-A7 was digested with HindIII and EcoRI. The digests were purified using a High Pure PCR product purification kit (Roche Diagnostics) and ligated into pUC18 (pUA7), which had been digested with HindIII and EcoRI and then treated with shrimp alkaline phosphatase (Roche Diagnostics), using a DNA ligation kit ver. 2 (TaKaRa). The ligation mixture was transformed into competent E. coli HB101 cells, and transformants were selected on LB agar supplemented with ampicillin (100 µg ml–1). Clones expressing agarolytic activity were detected as colonies forming shallow craters around them on the agar. To confirm the agarolytic activity expressed in E. coli HB101 cells harboring plasmid pUA7, 2% (w/v) iodine solution (Wako Pure Chemical, Kyoto, Japan) was then poured onto the agar. Positive clones were visualized as having a clear zone around the colony with a dark brown background.
Expression and purification of RagaA7
Recombinant agarase, the product of the agaA7 gene, was designated as RagaA7. To produce RagaA7, the agaA7 gene was amplified from genomic DNA of strain JAMB-A7 by PCR using LA Taq DNA polymerase. Two primers, 5′-TAGTCGACCCCTGATGGCCGCCGACTGGG-3′ and 5′-GGGATCCTCAGTTGCTCAACGTAAATTTGTCG-3′, were constructed incorporating SalI and BamHI restriction sites (underlined) into the 5′ and 3′ ends of the agaA7 gene, respectively. The amplification product was digested with SalI and BamHI, and the digest was ligated into the SalI-BamHI site of the expression shuttle vector pHSP64 (Sumitomo et al. 1995). The recombinant plasmid, designated pA7AG, was then introduced into both E. coli HB101 and B. subtilis ISW1214 cells. E. coli HB101 cells harboring pA7AG were grown for 16 h on LB agar supplemented with ampicillin (100 µg ml–1). The agarolytic activities of E. coli HB101 cells harboring pA7AG were confirmed by pit formation in the agar. B. subtilis ISW1214 cells harboring the plasmid were grown for 72 h in CLT medium.
Cells and supernatants of the cultures were separated by centrifugation at 12,000 g for 10 min. The supernatant obtained was used for enzyme purification. All procedures for enzyme purification were carried out at a temperature below 4°C. The centrifugal supernatant (83 ml) was brought to 60% saturation with solid ammonium sulfate. The precipitates formed were collected by centrifugation (8,000 g, 25 min) and redissolved in a small volume of 20 mM Tris-HCl buffer (pH 7.5). The solution was dialyzed against the same buffer overnight. After removal of insoluble materials by centrifugation at 8,000 g for 15 min, the retentate was applied to a DEAE-Toyopearl 650 M column (2.5×15 cm; Tosoh, Tokyo, Japan) equilibrated with 20 mM Tris-HCl (pH 7.5). After washing the column with 200 ml 20 mM Tris-HCl (pH 7.5) containing 50 mM NaCl, the enzyme was eluted over 500 ml with a linear 50–500 mM NaCl gradient in the same buffer. The active fractions were combined and concentrated to 5 ml on a PM-10 membrane (10,000-Mr cutoff; Millipore, Bedford, Mass.). The buffer of this enzyme preparation was exchanged for 2.5 mM sodium phosphate buffer (pH 7.0) by 10-fold dilutions with 2.5 mM sodium phosphate buffer and concentration to 5 ml twice on the membrane. The retentate was loaded onto a hydroxyapatite column (2.5×15 cm; Seikagaku Kogyo, Tokyo, Japan) equilibrated with 2.5 mM sodium phosphate buffer (pH 7.0). Agarase activity passed through the column, and the non-adsorbed active fractions were pooled and concentrated by ultrafiltration on a PM-10 membrane. The concentrate was dialyzed overnight against 50 mM 3-morpholinopropanesulfonic acid-NaOH buffer (MOPS; pH 7.0). The dialyzate (6.3 ml) was used as the final preparation of purified enzyme in all experiments reported here.
Enzyme assay
A suitably diluted solution of 100 µl enzyme preparation was incubated in 1 ml 50 mM MOPS (pH 7.0) containing 0.2% (w/v) agar (Nacalai Tesque) at 50°C. Activity was expressed as the initial rate of agar hydrolysis by measuring the release of reducing ends using the 3,5-dinitrosalicylic acid procedure (Miller 1959) with d-galactose as the standard. One unit (U) of enzymatic activity was defined as the amount of protein that produced 1 µmol reducing sugar as d-galactose per minute under the conditions of the assay. The kinetic parameters of K m and k cat for agar and neoagarohexaose (Dextra Laboratories, Reading, UK) were determined at 50°C in 50 mM MOPS (pH 7.0). The initial rates of hydrolysis of agar were determined at seven different substrate concentrations, ranging from 0.15- to 2.5-times the estimated K m value. Hydrolysis of neoagarohexaose was monitored quantitatively by gel filtration chromatography on an Asahipak GS220 G7 column (6.7×500 mm; Asahi Kasei, Tokyo, Japan) using an LC-10Avp with CLASS-VP HPLC system (Shimadzu) equipped with a refractive index detector (RID-10A, Shimadzu). Reaction conditions were chosen so that <10% of the substrate was hydrolyzed, and the initial rates were determined based on the rates of substrate disappearance. Protein was determined using a protein assay kit (Bio-Rad, Hercules, Calif.) with bovine serum albumin as the standard protein. For comparison, we also used a commercially available β-agarase from Pseudomonas atlantica (PSA; BioWhittaker Molecular Applications, Rockland, Me.).
Electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed essentially as described by Laemmli (1970) on slab gels [12.5% (w/v) acrylamide, 70×50 mm, 2.0-mm thick], and the gels were stained for protein with Coomassie Brilliant Blue R-250 (CBB, Bio-Rad). The molecular mass was estimated by SDS-PAGE 12% (w/v) acrylamide gels with low-range molecular mass standards (Bio-Rad), which included phosphorylase b (97.4 kDa), serum albumin (66.2 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), trypsin inhibitor (21.5 kDa), and lysozyme (14.4 kDa).
Liquid chromatography-tandem mass spectrometry
The amino acid sequence of the C-terminal region, and molecular mass of the purified enzyme were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) after SDS-PAGE. The CBB-stained protein was manually excised from the gel and then digested in situ with 12.5 ng μl–1 trypsin (Promega, Madison, Wis.) using established protocols (Shevchenko et al. 1996). The tryptic digests were extracted from the gel slices with four 15-min washes with 50% acetonitrile/1% formic acid (v/v). The peptides were dried and suspended in a solution of 2% acetonitrile/98% H2O/0.1% trifluoroacetic acid (v/v). For on-line microcapillary LC-MS/MS analysis, the peptide mixtures were then chromatographed in a MAGIC 2002 HPLC system (Michrom Bioresources, Auburn, Calif.). All tandem mass spectra were recorded on a Thermo Finnigan LCQ Deca-plus ion-trap mass spectrometer (Thermo Finnigan, San Jose, Calif.) equipped with a nanospray ion source, using borosilicate PicoTips (Econo 10, New Objective, Woburn, Mass.) MS/MS spectra were acquired over the range of 450–2,000 m/z. The tandem MS/MS raw data were subsequently analyzed using TurboSEQUEST software (Thermo Finnigan) (Ducret et al. 1998; Washburn et al. 2001) that had been “indexed for speed” with carbamidomethylation as a static modification of cysteine (+57.0 Da), with and without oxidation of methionine (+16.0 Da). To identify the N- and C-terminal amino acids of RagaA7, a database was created based on the deduced amino acid sequence of the agaA7 gene and used for TurboSEQUEST searching.
Sequencing of amino-terminal regions of protein
The enzyme sample was blotted on a polyvinylidene difluoride membrane (Applied Biosystems) that had been wetted with methanol. The N-terminal amino acid sequence of the protein was determined directly using a protein sequencer (model 476A, Applied Biosystems).
Activity staining
SDS-PAGE was performed as described above. After electrophoresis, SDS in the gels was removed by soaking the gels in 50 mM MOPS (pH 7.0) three times for a total of 30 min. The gels were then overlaid onto agar sheets containing 1.5% (w/v) agar and 50 mM MOPS (pH 7.0) and incubated for 30 min at 37°C. Following incubation, the agar sheets were stained by flooding with 2% (w/v) iodine solution. Agarase activity was visualized as clear zones on a brown background. Proteins were visualized by staining with CBB.
Chromatographic analysis of the products of agar and neoagarooligosaccharides hydrolysis
Thin-layer chromatography (TLC) was used to identify products. Enzymatic hydrolysis of agarose (TaKaRa), neoagarohexaose and neoagarotetraose (Dextra Laboratories) were carried out at 40°C in 50 mM MOPS containing 1.0% (w/v) of each substrate. The reaction mixtures were applied to silica gel 60 TLC plates (Merck, Darmstadt, Germany) according to the method of Aoki et al. (1990). The plates were developed using solvent A [1-butanol-acetic acid-H2O (2:1:1, v/v)]. Spots of oligosaccharides resulting from the hydrolysis of agarose were visualizd by spraying with 10% (v/v) H2SO4 and heating. d-Galactose (Sigma, St. Louis, Mo.), neoagarotetraose, and neoagarohexaose were used as standards and as substrates. Reaction products was quantified by gel filtration chromatography using the HPLC system described above.
Chemicals
Unless otherwise stated, all chemicals used were from Wako Pure Chemical. Nonidet P-40 (nonylphenylpolyethylene glycol), Triton X-100 (polyethylene glycol mono-p-isooctylphenyl ether), Tween 20 (polyoxyethylene sorbitan monolaurate), p-chloromercuribenzoate, and 2-mercaptoethanol were from Nacalai Tesque. SDS was from Bio-Rad. Iodoacetoamide was from Kanto Chemical (Tokyo, Japan). Dithiothreitol was from Amersham Biosciences (Piscataway, N.J.).
Nucleotide sequence accession numbers
The 16S rDNA of strain JAMB-A7 and agaA7 nucleotide sequence data have been submitted to the DDBJ, EMBL, and GenBank databases under accession nos. AB107975 and AB107974, respectively.
Results
Taxonomic analysis of the isolate
Strain JAMB-A7, which was isolated as an agarolytic bacterium from deep-sea sediment, occurs as non-motile Gram-negative rods 1.5–4.5 μm in length and 0.5–0.8 μm in width. The temperature range for growth is 10–43°C. It is positive for catalase, oxidase, gelatinase, and amylase, and negative for the production of indole and H2S, and nitrate reduction. It produces acid from l-arabinose, cellobiose, d-fructose, d-galactose, and d-glucose. The G+C content was 56 mol%. The predominant respiratory lipoquinone was unsaturated ubiquinone with eight isoprene units (Q-8), and the major cellular fatty acid was iso-C15:0 at a ratio of 23% of total fatty acids.
To determine the phylogenetic position of the agar-degrading JAMB-A7, its 16S rDNA sequence was determined and analyzed using comparative sequence analysis against known 16S rDNA sequences. The 16S rDNA sequence of JAMB-A7 had the closest match (99.5% similarity) with that of M. elongatus DSM6810 (recently transferred from Pseudomonas elongata; Yoon et al. 2003). Our isolate exhibited 16S rDNA similarity levels of 98.3% and 97.2% to M. salipaludis JCM11542 and M. hydrolyticus ATCC700072, respectively. Therefore, JAMB-A7 should be classified as belonging to the genus Microbulbifer.
However, there are some differences between JAMB-A7 and the closely-matched M. elongatus DSM6810 in physiological characteristics, such as the range of NaCl concentration [0.3–11% for JAMB-A7, 3–9% for M. elongatus DSM6810 (w/v)] and temperature (39°C for JAMB-A7, 25–30°C for M. elongatus DSM6810) optimal for growth.
DNA-DNA hybridization
Stackebrandt and Goebel (1994) emphasized the importance of DNA-DNA relatedness in species definition, even if almost identical 16S rDNA sequences are obtained. Therefore, the genomic relatedness among strain JAMB-A7 and related strains of the genus Microbulbifer was determined. Strain JAMB-A7 showed DNA-DNA relatedness levels of 52, 25, and 34% with M. elongatus DSM6810, M. salipaludis JCM11542, and M. hydrolyticus ATCC700072, respectively. These values are lower than 70%, the value recognized as delineating a species (Wayne et al. 1987). Therefore, on the basis of phenotypic properties and genomic data, strain JAMB-A7 should be placed in the genus Microbulbifer as a member of a novel species. This strain has been deposited as a patent strain (FERM BP-8320) in the National Institute of Advanced Industrial Science and Technology (AIST) of Japan.
Cloning of the agarase gene
The agarase gene from strain JAMB-A7 was cloned into E. coli HB101 by the shotgun method as described in Materials and methods. The clone expressed agarolytic activity, which was confirmed by the formation of shallow crater around it on LB agar supplemented with 100 µg ml–1 ampicillin. The recombinant plasmid in the positive clone was designated pUA7. It had an insert of approximately 2.7 kb, and the insert was sequenced in both directions. The insert comprised 2,747 bp with a G+C content of 55 mol%. A single open reading frame (ORF) was found in the sequence, which begins with an ATG at nucleotide 874 and ends with TGA at nucleotide 2,197 (Fig. 1). This gene, agaA7, encodes a protein of 441 amino acids with a calculated molecular mass of 48,989 Da. A putative ribosome binding site of 5′-AAGGAG-3′ is present 8 bp upstream from the initiation codon ATG. A putative sigma 70-type promoter sequence, 5′-TTCAAA-3′ for the −35 region and 5′-TAACCT-3′ for the −10 region, is located 64 bp upstream from the initiation codon with 17 bp spacing. An inverted-repeat sequence is found 36 bp downstream of the TGA stop codon. The free energy value of this sequence for a stem-loop structure was calculated to be −122.6 kJ mol–1, which would be sufficient for termination of transcription. The deduced amino acid sequence was analyzed using the program PSORT (http://psort.nibb.ac.jp). A possible signal sequence of 19 amino acids is present, with a potential cleavage site between amino acids Ala19 and Ala20, suggesting that AgaA7 is localized on the outer membrane or in the periplasmic space, as in the case of Gram-negative bacteria. The calculated molecular mass and isoelectric point of the mature enzyme deduced from its amino acid sequence were 47,009 Da (422 amino acid residues) and pH 4.40, respectively.

Complete nucleotide sequence of the agaA7 gene and its flanking regions. Sequences similar to the −35 and −10 consensus promoters of Escherichia coli are underlined. A putative ribosome-binding site is boxed. The deduced amino acid sequence of AgaA7 is in single letter code under the nucleotide sequence. The putative signal peptide is shown by a dotted line. Amino acid residues from Met18 to Val27 (closed circles) refer to the N-terminal end of the extracellular recombinant enzyme secreted by Bacillus subtilis. Amino acid residues from Phe289 to Ser307 (the C-terminus) (closed triangles) were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Convergent arrows Inverted repeats downstream of the stop codon TGA (*) of the ORF
Comparison of the deduced amino acid sequence of the agaA7 gene product with those of other agarases
Database searches using FASTA (http://ddbj.nig.ac.jp) with the full-length deduced amino acid sequence of AgaA7 showed homology to other known agarases that are members of the glycoside hydrolases family 16 (http://afmb.cnrs-mrs.fr/CAZY/index.html). The overall sequence identity between AgaA7 and agarases from Pseudomonas sp. strain ND137 (AB063259-1), Aeromonas sp. strain B9 (U61972-1), Pseudoalteromonas atlantica ATCC19262 (M73783-1), Zobellia galactaninovorans Dsij (AF098954-1, AF098955-1), Microscilla sp. strain PRE1 (AF339846-17), and Streptomyces coelicolor A3(2) (AL133236-6) were only 55.3%, 54.3%, 52.6%, 47.5%, 41.2%, 37.5%, and 34.5%, respectively. Multiple alignments showed that the homologous regions in all the agarases were located in the regions corresponding to Ala20-Val293 in AgaA7.
A separate BLAST search of the C-terminal half—Pro294-Asn441—in AgaA7 showed low but substantial amino acid homology of 41% to some regions of an α-agarase from A. agarlyticus GJ1B (AF121273), of 37% to each of the two putative β-agarases MS115 and MS116 from Microscilla sp. strain PRE1 (AF339846-16 and AF339846-17), of 27% to cellulase B from Cellvibrio mixtus (AF003697), and of 24% to an acetyl xylan esterase from Fibrobacter succinogens S85 (AF180369). When regions in agarases homologous to the C-terminal part of AgaA7 are suitably aligned using the GENETYX-MAC 10.1 program, seven aromatic amino acids (Phe315, Tyr352, Tyr362, Tyr366, Trp400, Trp430, Trp432), two hydrophobic amino acids (Leu422, Leu439), three negatively charged residues (Glu313, Asp324, Asp348), and six neutral amino acids (Ala312, Gly319, Asn342, Gly416, Gly320, Asn433) are found to be highly conserved (Fig. 2). Some of these conserved amino acids might be involved in substrate binding.

A multiple alignment of amino acid sequences of the C-terminal Pro294–Asn441 in AgaA7 with those of related carbohydrate-binding modules (CBMs). Open circles Amino acid residues identical in all sequences, bold letters residues conserved in at least three of the enzymes. Sequence sources: AgaA7 Microbulbifer sp. strain JAMB-A7 (AB107974) (this study); AgaA an α-agarase from Alteromonas agarlyticus GJ1B (AF121273); MS115, MS116 putative β-agarases from Microscilla sp. strain PRE1 (AF339846-16, AF339846-17)
Extracellular expression and purification of RagaA7
High-level exo-production of RagaA7 was examined using B. subtilis ISW1214 and pA7AG. Supernatant from 83 ml of 72-h-cultured broth was obtained by centrifugation. The recombinant agarase RagaA7 was purified 310-fold after anion-exchange chromatography and hydroxyapatite chromatography, with a specific activity of 398 U mg−1 and a final yield of 53.8% (Table 1). SDS-PAGE and activity staining of the purified enzyme gave a single band with an apparent mass of 33 kDa (Fig. 3). This value is smaller than that deduced from the agaA7 gene sequence (calculated molecular mass, 47,009 Da). The N-terminal amino acid sequence of RagaA7 was Met-Ala-Ala-Asp-Trp-Asp-Gly-Thr-Pro-Val from amino acids 18–27, as revealed by protein sequencing. LC-MS/MS showed that the C-terminal amino acid sequence was Phe289 through Ser307 (the C-terminus) in RagaA7. Thus, the amino acid sequence of the recombinant enzyme starts with Met18 and ends with Ser307 (290 amino acid residues) with a calculated molecular mass of 32,414 Da, a value very similar to that estimated by SDS-PAGE. The difference in the molecular mass may be caused by proteolytic digestion at the C-terminus of Ser307 by the host during secretion. These results also showed that the homologous regions among related agarases, which correspond to Ala20-Val293 in AgaA7, remain intact in the purified enzyme.

SDS-PAGE and activity staining of purified RagaA7. a Polyacrylamide gel [12.5% (w/v)] stained with Coomassie Brilliant Blue (CBB). Lanes: M Protein mass markers (in kDa), P purified enzyme (0.5 µg protein). b Activity staining of the purified enzyme. After SDS-PAGE, the slab gel was overlaid onto an agar sheet containing 1.5% (w/v) agar and 50 mM MOPS (pH 7.0), followed by incubation for 30 min at 40°C. Activity was visualized as a clear zone by flooding the agar sheet with iodine solution
Effects of pH on activity and stability
The pH optimum for activity of RagaA7 was examined in various buffers. The buffers used were 50 mM Britton-Robinson universal buffers (pH 3.5–12.0), 50 mM 2-morpholinoethanesulfonic acid-NaOH buffers (MES; pH 5.5–7.0), 50 mM MOPS (pH 6.5–7.5) and 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid-NaOH buffers (HEPES; pH 7.0–8.2). Maximal activity was observed at around pH 7 in every buffer. Under the same assay conditions, PSA showed optimal pH between 6.0 and 7.0. To determine pH stability, RagaA7 was incubated at 40°C for 30 min at various pH values (3–12) in 50 mM Britton-Robinson universal buffers. The enzyme was most stable at pH 5–8 and stable at pH 3.5–9.5, retaining 50% of the original activity. The pH-activity curve of RagaA7 was broader than that of PSA, while RagaA7 exhibited pH stability in a somewhat narrower range than PSA.
Effects of temperature on activity and stability
The optimal temperature for the activity of RagaA7 was calculated to be 53°C from Arrhenius plots (Fig. 4a), the highest value yet reported for agarases (Bong et al. 1999). The optimal temperature of PSA was around 46°C. The activation energies of RagaA7 and PSA were calculated from Arrhenius plots to be 85.7 and 75.6 kJ mol−1, respectively. The thermostability of RagaA7 was examined after incubation at 50 or 55°C in 50 mM MOPS (pH 7.0) for various periods (Fig. 4b). The first order constant of irreversible thermoinactivation, k, at each temperature, was obtained by linear regression in semi-logarithmic coordinates. The enzyme half-life was calculated using the equation t1/2=ln2/k. RagaA7 was found to be stable up to 50°C. The half-life of RagaA7 (502 min) at 50°C was 30-fold greater than that of PSA (17 min). Even after heating at 80°C for 30 min, the residual activity of RagaA7 was about 40% of the original activity, whereas that of PSA was less than 10%. RagaA7 is thus much more heat-stable than PSA.

Effects of temperature on activity and stability of RagaA7 and β-agarase from Pseudomonas atlantica (PSA). a Temperature profiles (Arrhenius plots) of RagaA7 and PSA are shown by closed circles and open circles, respectively. b Thermostability of RagaA7 (at 0.2 U ml–1, 0.50 µg ml–1) and PSA (at 0.2 U ml–1, 0.62 µg ml–1). Enzymes were heated individually at the temperatures indicated. Aliquots (0.1 ml) were removed at different times and used to determine residual activity under standard enzyme assay conditions. Logarithm of residual activities (U×10−3): closed circles RagaA7 (50°C), closed triangles RagaA7 (55°C), open circles PSA (50°C), open triangles PSA (55°C)
Effects of cations and chemical reagents
The enzyme was incubated with or without 10 mM EDTA at pH 7.0 in 50 mM MOPS and at 0°C for 24 h and dialyzed extensively against distilled water. After dialysis, the specific activity was found to be unaltered by the EDTA treatment. The EDTA-treated enzyme was used to study the effects of metal ions. The activities of some agarases, such as β-agarase II from P. atlantica (Morrice et al. 1983a) and β-agarase PjaA from Pseudomonas sp. strain W7 (Ha et al. 1997), are reported to be stimulated by NaCl. However, the major metal ions found in seawater, Na+ (at 0.05–1 M), Mg2+, K+, and Ca2+ ions (each at 0.05 or 0.1 M) essentially did not affect RagaA7 activity after incubation for 5 min at pH 7.0 in 50 mM MOPS and at 50°C. Hg2+, Pb2+, and Zn2+ ions (each at 1 mM) inactivated the enzyme irreversibly within 5 min under the same conditions. Because these heavy metal ions have affinity to the SH, CO, and NH moieties of amino acids, their inhibitory effects could be due to structural alteration of the enzyme protein. The activity was not affected either by sulfhydryl inhibitors such as iodoacetoamide and p-chloromercuribenzoate or by thiol reagents such as dithiothreitol and 2-mercaptoethanol. The activity was strongly inhibited by N-bromosuccinimide (0.1 mM), suggesting that the tryptophan residue(s) in this enzyme might be important for catalysis. RagaA7 had strong resistance to EDTA up to 100 mM at 40°C for 30 min. Diethyl pyrocarbonate and 1-ethyl-3-(3-dimethyl-aminopropyl)carbonate exhibited almost no inhibition. The stability of the enzyme against several surfactants was examined after incubation for 1 h at 40°C and at pH 7.0 in 50 mM MOPS. The enzyme retained full activity even after treatment with 0.1% and 1.0% (w/v) each of SDS, Tween 20, Triton X-100, and Nonidet P-40. When concentrations of SDS varied from 0 to 2.0% (w/v), the enzyme was very stable to incubation with the surfactant at a concentration of at least up to 2.0% (Fig. 5). After incubation in 0.1% SDS, the activity increased 1.4-fold compared with that of the control (without SDS). In contrast, PSA activity was abolished completely by 0.1% SDS.

Stability of RagaA7 and PSA to SDS. RagaA7 and PSA (each added at 0.2 U ml–1) were individually preincubated for 1 h at 40°C in 50 mM MOPS buffer (pH 7.0) in the presence of SDS at various concentrations. After 1:10 dilutions, residual activities in each sample were assayed. Original activities were taken as 100%. Residual activities: closed circles RagaA7, open circles PSA
Analysis of hydrolysis products and substrate specificity
The time course of hydrolysis products from agarose was examined with 0.04 U ml–1 of RagaA7 incubated at 40°C for up to 48 h, as shown in Fig. 6a. In the initial stage, RagaA7 hydrolyzed agarose to generate many large oligosaccharides. After a 1-h incubation, the amount of large oligosaccharides decreased, with a concomitant increase in tetramers and hexamers corresponding to neoagarotetraose and neoagarohexaose. This hydrolysis pattern indicates that RagaA7 is an endo-β-agarase. After incubation for a further 24 h, the main product was neoagarotetraose with concomitant production of minor neoagarobiose, as judged by R f values on TLC (Bong et al. 1999). The quantification of reaction products after 24 h incubation was performed by gel filtration chromatography. The composition (mol%) of the products was 2.5% neoagarohexaose, 85.5% neoagarotetraose, 12.0% neoagarobiose. RagaA7 could not act on neoagarotetraose, whereas it hydrolyzed neoagarohexaose to generate neoagarotetraose with a slight amount of neoagarobiose (Fig. 6b). The results show that RagaA7 hydrolyzes agarose, neoagaro-oligosacchrides larger than neoagarohexaose, and neoagarohexaose to form neoagarotetraose as the major end product. The K m values for agar and neoagarohexaose were 3.0 and 88.8 mg ml−1, respectively. The catalytic efficiencies (k cat K m −1) for agar and neoagarohexaose were 2.9×102 and 0.29 ml mg−1 s−1, respectively. The catalytic efficiency for agar is three orders of magnitude greater than that for neoagarohexaose. RagaA7 did not degrade ι-,κ-, and λ-carrageenans, which have the same backbones as agarose with substituted sulfoxy groups (data not shown).

Thin layer chromatography (TLC) of the products of agarose hydrolysis by RagaA7. a Reactions were carried out at 40°C at pH 7.0 in 50 mM MOPS with 0.04 U ml–1 enzyme and 1.0% (w/v) agarose. At intervals, aliquots from the reaction mixture were sampled and developed by TLC. b Reactions were carried out with neoagarotetraose (NA4) or neoagarohexaose (NA6) as substrate (each at 1.0%, w/v) and enzyme (1.0 U ml–1). Products generated were analyzed by TLC as described in Materials and methods. ST Standard sugars, including d-galactose (Gal). Lanes: 1–4 reactions with NA4, 5–8 reactions with NA6
Discussion
In this study, we cloned and sequenced the gene for β-agarase from a novel species of the genus Microbulbifer (strain JAMB-A7) isolated from a sample of deep-sea sediments at a depth of 1,174 m. The recombinant enzyme RagaA7 had a molecular mass of 33 kDa and a specific activity of 398 U mg–1, with optimal pH and temperature around 7 and 50°C, respectively. RagaA7 is an endo-type β-agarase, and the final main product is neoagarotetraose. Similar agarases have been reported. A β-agarase I from P. atlantica ATCC19262, with a molecular mass of 32 kDa, has maximal activity at pH between 6 and 7, and hydrolyzes agarose to yield neoagarotetraose as the main product (Morrice et al. 1983a, 1983b). Agarase A from Vibrio sp. strain JT0107, with a high molecular mass of 107 kDa, has maximal activity at pH 8 and hydrolyzes not only agarose but also neoagarotetraose to yield neoagarobiose (Sugano et al. 1993b). The less thermostable PSA is available from several manufacturers. The properties of RagaA7 are much more beneficial than these other enzymes for use in industrial applications. For example, the high activity and thermostability at temperatures higher than the gelling temperature of agar (around 40°C) is an advantage for industrial oligosaccharide production from agar or marine algae. Moreover, the strong resistance to salts at high concentrations shows that crude marine algae in high-salt seawater can be hydrolyzed by RagaA7 to produce oligosaccharides. RagaA7 can also be used for extraction of DNA fragments from agarose gels after electrophoresis because most buffers for gene manipulation contain EDTA.
Based on the amino acid sequence similarity, β-agarases are classified into the three glycosyl hydrolase families 16, 50, and 86. For example, a β-agarase (DagA) from S. coelicolor A3(2) (Kendall and Cullum 1984), two agarases from Vibrio sp. strain JT0107 (Sugano et al. 1993a, 1994), and β-agarase I from P. atlantica T6c (Belas 1989) are members of family 16, 50, and 86, respectively. AgaA7 belongs to glycoside hydrolase family 16, based on its deduced amino acid similarity with those of enzymes in the same family.
The C-terminal half of AgaA7 showed homology not only to agarases but also to other enzymes such as cellulase and acetyl xylan esterase, and the C-terminal end appears to be a noncatalytic carbohydrate-binding module (CBM). Polysaccharide-degrading enzymes in general are known to have modular structures in which the catalytic modules are attached via linker sequences to noncatalytic modules that bind to substrates (Tomme et al.1995). Binding ligands in CBMs and their specificities are dominated by hydrophobic interactions between the sugar rings and hydrophobic and aromatic amino acid residues on the surface of binding sites (Nagy et al. 1998; Notenboom et al. 2001), and different orientations of the residues Gly, Ala, and/or Arg, which have key roles as subsites, are assumed to be responsible for the different ligand specificities of CBMs (Boraston et al. 2000; Simpson et al. 2000; Czjzek et al. 2001). The C-terminal end of AgaA7 resembles in part the 6 CBM family (http://afmb.cnrs-mrs.fr/CAZY/index.html). At present, we do not know how the CBM-like sequence contributes to the catalysis of AgaA7. We are now attempting to express an AgaA7 containing this region in E. coli and B. subtilis cells.
The inhibition by N-bromosuccinimide suggests the involvement of tryptophan residue(s) in the catalysis of RagaA7. Recently, we succeeded in crystallizing RagaA7, and X-ray crystallographic analysis is now in progress.
References
Aoki T, Araki T, Kitamikado M (1990) Purification and characterization of a novel beta-agarase from Vibrio sp. AP-2. Eur J Biochem 187:461–465
Araki C (1966) Some recent studies on the polysaccharides of agarophytes. In: Young EG, Maclachan JL (eds) Proceedings of the International Seaweed Symposium 5,1965, Pergamon Press, London, pp 3–17
Araki T, Lu Z, Morishita T (1998) Optimization of parameters for isolation of protoplasts from Gracilaria verrucosa (Rhodophyta). J Mar Biotechnol 6:193–197
Belas R (1989) Sequence analysis of the agrA gene encoding β-agarase from Pseudomonas atlantica. J Bacteriol 171:602–605
Birnboim HC, Doly J (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7:1513–1523
Bong JK, Hak JK, Soon DH, Sun HH, Dae SB, Tae HL, Jai YK (1999) Purification and characterization of β-agarase from marine bacterium Bacillus cereus ASK202. Biotechnol Lett 21:1011–1105
Boraston AB, Tomme P, Amandoron EA, Kilburn DG (2000) A novel mechanism of xylan binding by a lectin-like module from Streptomyces lividans xylanase 10A. Biochem J 350:933–941
Chang S, Cohen SN (1979) High frequency transformation of Bacillus subtilis protoplasts by plasmid DNA. Mol Gen Genet 168:111–115
Cowan ST, Steel KJ (1965) Manual for the identification of medical bacteria. Cambridge University Press, London
Czjzek M, Bolam DN, Mosbah A, Allouch J, Fontes CM, Ferreira LM, Bornet O, Zamboni V, Darbon H, Smith NL, Black GW, Henrissat B, Gilbert HJ (2001) The location of the ligand-binding site of carbohydrate-binding modules that have evolved from a common sequence is not conserved. J Biol Chem 276:48580–48587
Duckworth M, Yaphe W (1971) Structure of agar. I. Fractionation of a complex mixture of polysaccharides. Carbohydr Res 16:189–197
Ducret A, Van Oostveen I, Eng JK, Yates JR III, Aebersold R (1998) High throughput protein characterization by automated reverse-phase chromatography/electrospray tandem mass spectrometry. Protein Sci 7:706–719
Ezaki T, Hashimoto Y, Yabuuchi E (1989) Fluorometric deoxyribonucleic acid-deoxyribonucleic acid hybridization in microdilution wells as an alternative to membrane filter hybridization in which radioisotopes are used to determine genetic relatedness among bacterial strains. Int J Syst Bacteriol 39:224–229
Ha JC, Kim GT, Kim SK, Oh TK, Yu JH, Kong IS (1997) β-Agarase from Pseudomonas sp. W7: purification of the recombinant enzyme from Escherichia coli and the effects of salt on its activity. Biotechnol Appl Biochem 26:1–6
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. Mol Gen Genet 166:557–580
Kendall K, Cullum J (1984) Cloning and expression of an extracellular-agarase from Streptomyces coelicolor A3(2) in Streptomyces lividans 66. Gene 29:315–321
Kobayashi R, Takisada M, Suzuki T, Kirimura K, Usami S (1997) Neoagarobiose as a novel moisturizer with whitening effect. Biosci Biotechnol Biochem 61:162–163
Komagata K, Suzuki K (1983) Lipids and cell-wall analysis in bacteria systematics. Methods Microbiol 19:161–203
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Leifson E (1963) Determination of carbohydrate metabolism of marine bacteria. J Bacteriol 85:1183–1184
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Morrice LM, McLean MW, Williamson FB, Long WF (1983a) β-Agarases I and II from Pseudomonas atlantica. Purifications and some properties. Eur J Biochem 135:553–558
Morrice LM, McLean MW, Long WF, Williamson FB (1983b) β-Agarases I and II from Pseudomonas atlantica. Substrate specificities. Eur J Biochem 137:149–154
Naganuma T, Coury DA, Poline-Fuller M, Gibor A, Horikoshi K (1993) Characterization of agarolytic Microscilla isolates and their extracellular agarases. Syst Appl Microbiol 16:183–190
Nagy T, Simpson P, Williamson MP, Hazlewoo GP, Gilbert HJ, Orosz L (1998) All three surface tryptophans in Type IIa cellulose binding domains play a pivotal role in binding both soluble and insoluble ligands. FEBS Lett 429:312–316
Notenboom V, Boraston AB, Kilburn DG, Rose DR (2001) Crystal structures of the family 9 carbohydrate-binding module from Thermotoga maritima xylanase 10A in native and ligand-bound forms. Biochemistry 40:6248–6256
Potin P, Richard C, Rochas C, Kloareg B (1993) Purification and characterization of the α-agarase from Alteromonas agarlyticus (Cataldi) comb. nov., strain GJ1B. Eur J Biochem 214:599–607
Saito H, Miura K (1963) Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim Biophys Acta 72:619–629
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–885
Simpson PJ, Xie HF, Bolam DN, Gilbert HJ, Williamson MP (2000) The structural basis for the ligand specificity of family 2 carbohydrate-binding modules. J Biol Chem 275:441137–441142
Stackebrandt E, Goebel, BM (1994) Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44:846–849
Sugano Y, Matsumoto T, Kodama H, Noma M (1993a) Cloning and sequencing of agaA, a unique agarase 0107 gene from a marine bacterium, Vibrio sp. strain JT0107. Appl Environ Microbiol 59:3750–3756
Sugano Y, Terada I, Noma M, Matsumoto T (1993b) Purification and characterization of a new agarase from a marine bacterium, Vibrio sp. strain JT0107. Appl Environ Microbiol 59:1549–1544
Sugano Y, Matsumoto T, Noma M (1994) Sequence analysis of the agaB gene encoding a new β-agarase from Vibrio sp. strain JT0107. Biochim Biophys Acta 1218:105–108
Sumitomo N, Ozaki K, Hitomi J, Kawaminami S, Kobayashi T, Kawai S, Ito S (1995) Application of the upstream region of a Bacillus endoglucanase gene to high-level expression of foreign genes in Bacillus subtilis. Biosci Biotechnol Biochem 59:2172–2175
Tamaoka J, Komagata K (1984) Determination of DNA base composition by reversed-phase high-performance liquid chromatography. FEMS Microbiol Lett 25:125–128
Tomme P, Warren RAJ, Gilkes NR (1995) Cellulose hydrolysis by bacteria and fungi. Adv Microbiol Physiol 37:1–81
Van der Meulen HJ, Harder W (1975) Production and characterization of the agarase of Cytoplaga flevensis. Antonie van Leeuwenhoek 41:431–447
Vera J, Alvarez R, Murano E, Slebe JC, Leon O (1998) Identification of a marine agarolytic Pseudoalteromonas isolate and characterization of its extracellular agarase. Appl Environ Microbiol 64:4378–4383
Waino M, Tindall BJ, Schumann P, Ingvorsen K (1999) Gracilibacillus gen. nov., with description of Gracilibacillus halotolerans gen. nov., sp. nov.; transfer of Bacillus dipsosauri to Gracilibacillus dipsosauri comb. nov., and Bacillus salexigens to the genus Salibacillus gen. nov., as Salibacillus salexigens comb. nov. Int J Syst Bacteriol 49:821–831
Washburn MP, Wolters D, Yates JR III (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19:242–247
Wayne LG, Brenner DJ, Colwell RR et al (1987) International Committee on Systematic Bacteriology. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37:463–464
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Yoon JH, Kim H, Kang KH, Oh TK, Park YH (2003) Transfer of Pseudomonas elongata Humm 1946 to the genus Microbulbifer as Microbulbifer elongatus comb. nov. Int J Syst Evol Microbiol 53:1357–1361
Yoshizawa Y, Ametani A, Tsunehiro J, Nomura K, Itoh M, Fukui F, Kaminogawa S (1995) Macrophage stimulation activity of the polysaccharide fraction from a marine alga (Porphyra yezoensis): structure-function relationships and improved solubility. Biosci Biotechnol Biochem 59:1933–1937
Zhong Z, Toukdarian A, Helinski D, Knauf V, Sykes S, Wilkinson JE, O’Bryne C, Shea T, DeLoughery C, Caspi R (2001) Sequence analysis of a 101-kilobase plasmid required for agar degradation by a Microscilla isolate. Appl Environ Microbiol 67:5771–5779
Acknowledgement
We are grateful to Dr. Y. Sakano of Tokyo University of Agriculture and Technology for stimulating discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ohta, Y., Hatada, Y., Nogi, Y. et al. Enzymatic properties and nucleotide and amino acid sequences of a thermostable β-agarase from a novel species of deep-sea Microbulbifer . Appl Microbiol Biotechnol 64, 505–514 (2004). https://doi.org/10.1007/s00253-004-1573-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-004-1573-y