
Abstract
Henoch–Schönlein purpura (HSP) is a systemic disorder characterized by leukocytoclastic vasculitis involving the capillaries and the deposition of IgA immune complexes. Renal involvement is the principal cause of morbidity and mortality in children with HSP. Thus, it is important to clarify the onset mechanism of Henoch–Schönlein purpura nephritis (HSPN) and to identify the most appropriate treatment. We herein review the pathogenesis and treatment of HSPN. As to the pathogenesis, several studies suggest that galactose-deficient IgA1 is recognized by anti-glycan antibodies, leading to the formation of circulating immune complexes and their mesangial deposition, thereby inducing renal injury. Aggressive therapies for the treatment of severe HSPN, including multiple drug combination therapy and plasmapheresis, have been shown to be effective in ameliorating proteinuria and histological severity. Nevertheless, detailed investigations of the pathogenesis of HSPN and double-blind randomized control studies on children with HSPN are still necessary.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Henoch–Schönlein purpura (HSP) was first recognized by Heberden in 1801 and first described as an association between purpura and arthritis by Schönlein in 1837. Henoch added descriptions of gastrointestinal involvement in 1874 and renal involvement in 1899. HSP is a small vessel vasculitis, the major manifestations of which include arthritis, non-thrombocytopenic purpura, abdominal pain, and renal disease. HSP is one of the most common vasculitides of childhood and is considered to be self-limiting. One manifestation of HSP that can continue to cause lifelong problems is renal involvement [1, 2]. Approximately 40% of pediatric patients develop nephritis within 4–6 weeks of the initial presentation. Most children with Henoch–Schönlein purpura nephritis (HSPN) present with only hematuria and/or low-grade proteinuria, or both, and have a good chance of recovery. However, patients with massive proteinuria at onset frequently have a progressive course. In specialized centers, the proportion of children with HSPN who progress to renal failure or end-stage renal disease varies from 1 to 17% [2–8].
It is therefore important to clarify the onset mechanism of and to ascertain the most appropriate treatment for HSPN. Herein we review the literature on the pathogenesis and treatment of HSPN.
Incidence of disease
Gardner-Medwin et al. [6] examined the frequency of and ethnic variations in childhood vasculitides in the West Midlands region of the United Kingdom. Their survey was completed using monthly questionnaires sent to consultants and a single questionnaire sent to family doctors, along with a review of case notes with diagnostic codes for vasculitis. The annual incidence of HSP in the study was 22.1 cases per 100,000 children, which was higher than previous estimates of 13.5–18.0 cases per 100,000 children [7, 8]. The authors postulated that a higher incidence of HSP may lead to increases in the incidences of renal disease and the need for renal medical treatment. Stewart et al. [7] evaluated a total of 270 patients with HSP from a total childhood population of 155,000 over a 13-year period and showed that the incidence of HSPN was 2.7 cases per 100,000 children. The mean incidence of HSPN in Asian children, however, has been reported to be 4.9 cases per 100,000 children per year, and over a 22-year period in Japan, Kawasaki et al. [9] reported that the mean number of HSPN cases per 100,000 children per year was 3.6 ±1.0 (Fig. 1).

Number of patients diagnosed with HSPN per 100,000 children each year. The mean number of cases of HSPN per 100,000 children per year was 3.5 ± 1.2 between 1987 and 1997, and 3.6 ± 0.8 between 1998 and 2008, with no significant difference between the groups
Pathogenesis
The pathogenesis of HSP remains unknown; however, HSP is generally believed to be an immune complex-mediated disease characterized by the presence of polymeric IgA1 (pIgA1)-containing immune complexes predominantly in the dermal, gastrointestinal and glomerular capillaries [10]. The pathognomonic granular IgA and C3 deposits in the mesangium are indistinguishable from those seen in IgA nephropathy, and similar immunohistologic findings have been observed in the kidneys of patients with liver cirrhosis, dermatitis herpetiformis, celiac disease, and chronic inflammatory disease of the lung.
Structure of IgA
IgA exists as a heterogeneous molecule, and two subclasses (IgA1 and IgA2) have been determined that are differentiated by the presence or absence of a 13-amino-acid hinge region [11, 12]. This region, exclusively present in IgA1, has many O-linked glycosylation sites and is a target for at least two families of IgA1 bacterial proteases, the expression of which has been linked to pathogenicity. In addition, IgA2 is primarily polymeric, and has a secretory component synthesized by glandular epithelial cells [8, 9]. In HSPN, mesangial deposits predominantly contain pIgA1, with a bridging J protein lacking the secretory component [13, 14].
Role of IgA-containing circulating immune complexes in HSPN pathogenesis
Although the pathogenic mechanisms of HSPN have not been fully elucidated, perturbations in the immune system, elevations in the serum levels of IgA1, IgA1-containing circulating immune complexes, circulating IgA-antineutrophil cytoplasmic antibodies (ANCA), and IgA-rheumatoid factors have been documented for patients with HSP [13, 15–18]. Coppo et al. [19] also reported elevated serum levels of IgA and IgA-containing immune complexes in patients with HSPN. In addition, it was reported that all HSP patients have IgA1-containing circulating immune complexes of small molecular mass, but only those with nephritis have additional large-molecular-mass IgA1-IgG-containing circulating immune complexes [20]. Furthermore, using GalNAc-specific lectin from Helix aspersa, Lau et al. reported that the serum levels of galactose-deficient IgA1 (Gd-IgA1) were higher in children with HSPN than in healthy controls and patients with C1q nephropathy [21]. However, the median levels of serum Gd-IgA1 in children with HSP without nephritis did not significantly differ from those in healthy controls. These data corroborate a potential pathogenic role for Gd-IgA1 in HSPN.
Mechanism of mesangial IgA deposition in HSPN pathogenesis
Both increased IgA synthesis and diminished clearance have been implicated in the pathogenesis of IgA immune complex deposition. Increased polymeric IgA production by the mucosal immune system in response to a mucosally presented antigen, such as bacteria, viruses, or fungi, has been hypothesized as a potential mechanism for the development of HSP [9]. Hyper-reactivity of both B and T cells in response to specific antigenic stimuli in vitro has been reported in patients with HSP, resulting in increased polymeric IgA production, including Gd-IgA in mucosal and tonsillar cells [18]. Gd-IgA1, in particular, is currently assumed to have a pivotal role in the pathogenesis of HSPN [21].
The mechanisms of renal injury by the Gd-IgA1 immunocomplex in HSPN are as follows. (1) The Gd-IgA1 immunocomplex in the mesangial areas activates a complement pathway, such as the alternative or lectin pathways [22, 23]. Deposition of C3 and properdin without C1q or C4 is typical, suggesting alternate pathway activation. Despite the demonstration of complement components in skin and renal biopsies, questions remain regarding the role of the complement system in the pathogenesis of HSP [20]. Hisano et al. [24] found that complement activation through the lectin pathway may contribute to the development of advanced glomerular injuries and prolonged urinary abnormalities in patients with HSPN. (2) The Gd-IgA1 immunocomplex in the mesangial areas activates mesangial cells, which results in the proliferation of cells, such as macrophages and lymphocytes, and the production of inflammatory and profibrogenic cytokines and chemokines, suggesting a pivotal role in mesangial cell proliferation, matrix expansion, and inflammatory cell recruitment [25]. Kawasaki et al. [26] reported that the accumulation of macrophages in the glomeruli was a predictor of poor prognosis in HSPN patients.
On the other hand, there have been several reports of endothelial cell dysfunction in HSPN [27, 28]. Fujieda et al. [27] showed that the endothelial cells are damaged in HSPN, and high IgA antiendothelial cell antibody titers and elevated serum thrombomodulin values may be clinically useful markers of renal involvement in patients with HSPN. Kawasaki et al. [28] reported that serum E-selectin concentrations at the time of the first biopsy in patients with HSPN were higher than those at the time of the second biopsy and could be used to evaluate the glomerular endothelial dysfunction in HSPN.
Other possible pathogenic mechanisms of HSPN
Masuda et al. [29] showed that nephritis-associated plasmin receptor (NAPlr), a group A streptococcal antigen, may also have a pathogenetic role in a subset of patients with HSPN. Among 33 children with biopsy-proven HSPN, 30% had segmental or global mesangial NAPlr antigen depositions, compared to 3% in children with non-HSPN glomerular diseases (half of these children had IgAN). The exact pathophysiologic mechanism, if any, and the relationship between NAPlr and HSPN require further investigation.
A study by Davin et al. [30] compared 22 children with HSPN with 16 children with IgAN. In their cohort, elevated plasma IgE levels were more commonly found in patients with HSPN (77 vs. 44%), leading them to hypothesize that the IgA-containing immune complexes could enhance local IgE production via the stimulation of dermal and intestinal mast cells. Deposition of the IgA immune complexes was further enhanced by subsequent increases in local capillary permeability [30]. Notwithstanding the higher incidence of elevated plasma IgE levels in patients with HSPN, the pathogenetic roles of IgE remains unclear, as mast cells are not usually found in the mesangium [31].
Eosinophil activation has also been proposed to play a role in the pathogenesis of HSPN [32, 33]. Namgoong et al. analyzed serum eosinophil cationic protein (ECP) levels in HSP patients and IgA nephropathy patients in order to identify the relationship between eosinophil activation in HSP and IgA nephropathy. The levels of ECP were significantly higher in HSP patients (mean 9.7 ± 1.8 μg/l) than in a control group (mean 4.6 ± 0.7 μg/l). The classification of HSP patients into two groups, one with normal urine and one with abnormal urine, revealed that the latter group showed higher levels of ECP. However, ECP levels were not significantly higher in IgA nephropathy patients than in a control group. These results suggest that eosinophil activation may be involved in the pathogenesis of HSP, but not in IgA nephropathy [32]. Kawasaki et al. [33] reported that patients with HSPN also had higher serum concentrations of ECP and interleukin-5. These studies suggested that ECP might have a role in the initiation of nephritis in patients with HSP.
Recently, the renal expression of alpha-smooth muscle actin (α-SMA) and c-Met, which is the receptor for hepatocyte growth factor, has also been associated with the progression of renal injury in patients with HSPN [34]. Kawasaki et al. studied 35 patients in Japan with biopsy-proven HSPN who were categorized into three groups: (1) nephritis with an ISKDC histological grade (the classification system used by the International Study of Kidney Disease in Children) of II or less, (2) those with ISKDC grade III or greater and a good prognosis, and (3) those with ISKDC grade III or greater and a poor prognosis. All patients except those in group 1 underwent repeated biopsies during the course of the follow-up. The authors found that the mean scores for glomerular and interstitial α-SMA staining, and glomerular c-Met staining, at the first biopsy were higher in HSPN patients with crescents. At the second biopsy, α-SMA expression was also higher in patients with a poor prognosis (Fig. 2). Although the mechanism underlying the phenotypic changes in mesangial cells and increased expression of α-SMA in patients with HSPN is unclear, α-SMA is the predominant actin isoform within vascular smooth muscle, and these observations suggest that increased renal α-SMA expression may be an early histological indicator of the progression of HSPN. Similarly, increased expression of α-SMA in the tubulointerstitial area, but not in glomeruli, was associated with poor prognosis in patients with IgAN.

Renal expression of alpha-smooth muscle actin and c-Met in children with Henoch–Schönlein purpura nephritis. The mean scores for glomerular and interstitial α-SMA staining in groups 2 and 3 were significantly higher than those in group 1. At the first renal biopsy, none of the patients in group 1 and all patients in groups 2 and 3 clearly expressed c-Met in the glomeruli. At the first renal biopsy, AI and Cl in groups 2 (P < 0.01) and 3 (P < 0.01) were higher than those in group 1
Diagnostic investigation
There is no specific serologic test available for the diagnosis of HSP. Elevated levels of IgA, IgA-rheumatoid factor, and IgA-containing immune complexes have been detected in patients with HSP [35], yet despite a proposed correlation between serum IgA level and clinical features, no such a relationship has been consistently demonstrated. Diagnosis is made instead on the basis of clinical suspicion, and laboratory tests are mainly directed toward excluding other diagnostic possibilities and assessing the extent of renal involvement. Renal biopsy is particularly useful in distinguishing HSP from other disorders, and in assessing prognosis and indicating the need for treatment for patients with urinary protein excretion of >0.5 g/day at 1–2 months after the onset of HSPN.
Course and clinicopathological correlations
Although HSP is generally a benign, self-limiting disorder, there may be episodic and recurrent bouts of rash, arthralgia, gastrointestinal symptoms, and hematuria for several months or even years after the initial onset.
In patients with focal and segmental proliferative glomerular lesions, the overall mortality is <10% at 5 and 10 years after onset [35–37]. In a large series of patients seen by Meadow et al. [37] 2 years or more after diagnosis, 55% were entirely normal, 22% had residual urinary abnormalities but normal GFR, 10% had both abnormal urine sediment and reduced GFR, and 8% had a severe reduction in GFR, were receiving dialysis, or had died of renal failure. The occurrence of acute nephritic syndrome at onset, persistent nephrotic syndrome, and older age were indicators of a poor prognosis. All renal deaths occurred in patients with clinical and histological findings of crescentic glomerulonephritis. In the group of patients who recovered or improved clinically, repeated biopsies also revealed a lessening in the severity of glomerular alterations. Hypercellularity diminished or disappeared, and focal lesions decreased in terms of the number and extent of glomerular involvement. Furthermore, IgA deposits diminished to a considerable extent or even disappeared in a few patients. Capillary wall deposits also disappeared in association with clinical improvement [35].
In a long-term follow-up of 78 patients, with an average observation period of 23 years after the onset of HSPN in childhood, Goldstein et al. noted that 44% of patients presenting with nephrotic syndrome or acute nephritis had persisting hypertension or a progressive decline in GFR, whereas 82% of those who presenting with hematuria alone were normal. Sixteen of 44 full-term pregnancies were also complicated by proteinuria and/or hypertension, even in the absence of active renal disease [36]. Subsequent deterioration in clinical status was observed in approximately 20–25% of patients, even after initial and apparently full recovery, indicating the need for the long-term follow-up of patients with HSP [36].
Treatment of HSPN
The extrarenal manifestations of HSPN are managed by appropriate symptomatic measures. Severe skin lesions may require oral corticosteroids [13, 38–40], which may also improve abdominal pain and protein-losing enteropathy [38]. Severe gastrointestinal complications may occasionally require surgical intervention [39].
As for the treatment of HSPN, there have been many reports dealing with the use of corticosteroids and multiple combined agents, including immunosuppressive drugs. The action and side effects of these drugs, as well as their efficacy in the treatment of HSPN, are described below.
Action of each drug in multiple drug combination therapies including prednisolone, immunosuppressive drugs, warfarin, and dipyridamole
Corticosteroids
Corticosteroids inhibit T cell proliferation, T cell-dependent immunity, and cytokine gene transcription (including interleukin-1, IL-2, IL-6, interferon-gamma and tumor necrosis factor-α genes). Although no individual cytokine can totally reverse the inhibitory effects of corticosteroids upon mitogen-stimulated T cell proliferation, a combination of cytokines is effective in restoring T cell proliferation [41].
Immunosuppressive drugs
Cyclophosphamide
Cyclophosphamide is a potent alkylating agent that inhibits lymphocyte proliferation, leading to the repression of B- and T-lymphocyte function as well as to a reduction in their numbers [42]. Cyclophosphamide has also been used with success in the treatment of several other forms of crescentic glomerulonephritis, particularly those characterized by altered autoimmunity, including Wegener’s granulomatosis and systemic lupus erythematosus. However, some side effects, such as myelosuppression, hemorrhagic cystitis, and interstitial pneumonia have been observed in association with cyclophosphamide.
Azathioprine
Azathioprine (AZP) is the 1-methyl-4-nitro-5-imidazolyl derivative of 6-mercaptopurine. Azathioprine, a purine analog, functions as a purine antagonist and thereby inhibits cellular proliferation [41]. Again, some side effects, such as myelosuppression and liver dysfunction, have been reported in association with the use of AZP.
Mizoribine
Mizoribine (MZB) (p-INN: 4-carbamoyl-1-β-d-ribofuranosyl imidazolium-5-olate) is an antibiotic agent produced by the soil fungus Eupenicillium brefeldianum [43]. In the early 1980s, Japanese studies began to appear that demonstrated the inhibitory effects of MZB on T- and B-lymphocyte activity in vitro and examined the pharmacokinetics and immunosuppressive effects of MZB in vivo [44]. In view of its inhibition of purine synthesis and its relative lack of toxicity, MZB has been used increasingly in Japan during the last decade in place of azathioprine as part of immunosuppressive drug regimens: it was first approved by the Ministry of Health and Welfare of Japan as a drug indicated for the prevention of rejection in renal transplantation in 1984, and since then the indication has been extended to include lupus nephritis (1990), rheumatoid arthritis (1992), and primary nephrotic syndrome (1995) [45, 46].
Cyclosporine A
Cyclosporine A (CyA) has more recently been used for the treatment of a wide range of autoimmune diseases. CyA is a cyclic lipophilic undecapeptide that has immunosuppressive effects as well as a very selective inhibitory effect on T-helper cell function that operates by blocking the transcription of genes for specific cytokines such as interleukin-2 and interferon-gamma. Initially used in transplantation patients to control tissue rejection [47].
The efficacy of steroids
Most patients with HSPN have either no clinical renal involvement or have microhematuria, mild proteinuria, and normal renal function. These patients do not require steroid therapy, and the disease is usually managed symptomatically. Niaudet and Habib [48] reported that MPT is effective for patients with the risk of progression of nephropathy. Another study appeared to confirm that the early administration of prednisone is useful in preventing the development of HSPN [49]. In an uncontrolled study, Kawasaki et al. performed a long-term observation of the clinical manifestations and prognosis of 56 patients undergoing methylprednisolone and urokinase pulse therapy (MUPT). The mean urinary protein excretion after 6 months of treatment was found to have decreased significantly compared with the “pre-MUPT” level. Hypercoagulant status after the completion of urokinase pulse therapy was also improved compared with the “pre-MUPT” status (Table 1). First renal biopsies were performed in all patients, and second biopsies were performed in 27 patients. The activity index decreased significantly from 4.1 ± 1.9 at the first biopsy to 2.5 ± 1.7 at the second biopsy, whereas there were no differences in chronicity index between the first and second biopsies. No patients showed any renal insufficiency and the renal survival rate was 100% for the decade. These results suggested that MUPT is effective for those patients at risk of nephropathy progression, particularly if started early in the course of the disease before the crescents become fibrous. Urokinase is a plasminogen activator derived from fresh human urine that first attracted attention as a therapeutic agent for thrombotic diseases such as cardiovascular diseases or cerebral thrombosis. The rationale for such treatment was as follows: (1) stronger defibrinating activity was observed with urokinase administration than with anti-coagulant drug administration, (2) specific accumulations of urokinase were seen in the kidney and liver despite a very short turnover rate, and (3) adverse effects were very rare, even when urokinase was administered for a long period [50].
The efficacy of multiple-drug therapy
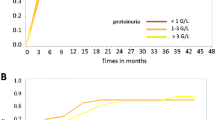
A prospective study of 12 patients with HSP who presented with rapidly progressive glomerulonephritis suggested benefits could be derived from intensive multiple-drug therapy [51]. Clinical improvement associated with combined corticosteroid and azathioprine therapy was also suggested by another study of 21 children with severe HSPN [52]. Iijima et al. [53] showed that multiple combined therapy including prednisolone, cyclophosphamide, heparin/warfarin, and dipyridamole was effective in the treatment of histologically severe HSN. In addition, Flynn et al. [54] report that treating children with HSPN with high-dose corticosteroids plus oral cyclophosphamide is safe and, as in nephrotic syndrome, appears to significantly reduce proteinuria. This study, however, was not a controlled study. Therefore, Kawasaki et al. evaluated the efficacy of methylprednisolone and urokinase pulse therapy combined with cyclophosphamide for patients with HSPN. They studied 37 patients who had been diagnosed with HSPN of at least ISKDC grade IVb. Of them, 20 (group A) were treated with methylprednisolone and urokinase pulse therapy, and 17 (group B) were treated with methylprednisolone and urokinase pulse therapy combined with cyclophosphamide. Methylprednisolone and urokinase pulse therapy combined with cyclophosphamide, but not methylprednisolone and urokinase pulse therapy alone, was found to significantly reduce urinary protein excretion (Fig. 3) and prevent any increase in crescentic or sclerosed glomeruli in HSPN patients with at least ISKDC grade IV HSPN. At the most recent follow-up, no patients among those treated with methylprednisolone and urokinase pulse therapy combined with cyclophosphamide were observed to have persistent nephropathy or renal insufficiency [55]. Shin et al. [56] suggests that CyA therapy is effective in reducing proteinuria, which is a known risk factor for the development of renal insufficiency in HSPN and may lead to a regression in renal pathology in patients with nephrotic-range proteinuria.

Comparison of proteinuria between group A and group B up to the most recent follow-up. The mean urinary protein excretions for group A and group B were reduced from 154 ± 73 and 181 ± 85 mg/m2/h to 29 ± 19 (P < 0.001) and 10 ± 5 mg/m2/h (P < 0.001), respectively, at the 6-month follow-up. The mean urinary protein excretion in group B was lower than that in group A from 2 months after the initiation of treatment to the most recent follow-up
However, there are some problems, such as anemia, leukopenia, alopecia, hemorrhagic cystitis, carcinogenesis, and hypogonadism, associated with the use of the above immunosuppressive drugs [46]. Thus, Kawasaki et al. evaluated whether methylprednisolone and urokinase pulse therapy combined with mizoribine, which has only mild side effects and is comparatively safe (MUPM), was effective in children with severe HSPN. They studied 12 patients who had been diagnosed with HSPN of at least ISKDC grade III. All patients were treated with MUPM, and clinical features, pathological findings, and prognoses were investigated prospectively. Ten patients (responders; nine with ISKDC grade IIIb and one with grade IVb) were treated with MUPM, whereas MUPM was discontinued due to a lack of response in two patients (nonresponders; both with grade IVb). Among the responders, urinary protein excretion had decreased significantly from 99.7 ± 37.8 to 25.9 ± 33.4 mg/m2 per h after 3 months of therapy. The acute index and tubulointerstitial scores decreased significantly from 5.8 ± 1.5 and 3.8 ± 0.6 at the first biopsy to 2.3 ± 1.3 and 1.0 ± 0.8 at the second biopsy, respectively. At the most recent follow-up, eight of the responders had normal urine, and two had minor urinary abnormalities. The nonresponders demonstrated continued high levels of urinary protein excretion after 3 months of therapy, and MUPM was discontinued. These results suggest that MUPM is effective at ameliorating the proteinuria and histological severity of HSPN in patients with <50% crescents, but is not so effective for HSPN in patients with >50% crescents [57].
Plasmapheresis
There have been a number of reports on plasmapheresis (PP) for HSPN in childhood [58, 59]. Hattori et al. retrospectively evaluated the clinical courses of nine children with a rapidly progressive type of HSPN who were treated with PP as the sole therapy. All patients had nephrotic-range proteinuria (4.9 ± 2.5 g/m2/day, mean ± SD) and a decreased glomerular filtration rate (GFR) (46.5 ± 9.5 ml/min/1.73 m2) at the time of the initiation of PP. Biopsy specimens taken before PP showed large crescents involving more than 50% of the glomerular circumference in 56.8 ± 6.9% of the glomeruli examined. The mean interval between disease onset and initiation of PP was 39.1 ± 22.1 days. The PP regimen consisted of thrice-weekly treatment for 2 weeks, then weekly treatment for 6 weeks. All patients responded promptly to PP with improvement in renal function, reduction of proteinuria, and subsidence of purpuric rash and abdominal pain. Six of nine patients showed further improvements without any other treatment; four recovered completely, and two had only microscopic hematuria at the latest observation (follow-up period, 9.6 ± 4.3 years). The remaining three patients showed a rebound increase in proteinuria after the completion of PP; two of whom progressed to end-stage renal failure at 14.1 and 1.8 years, respectively, after disease onset. These results suggests that PP as the sole therapy is effective at improving the prognoses of patients with rapidly progressive HSP nephritis, particularly if instituted early in the course of the disease [58].
On the other hand, Kawasaki et al. reviewed the cases of six Japanese children with rapidly progressive HSPN who received multiple drug therapy combined with PP. After five courses of PP, multiple drug therapy (including methylprednisolone and urokinase pulse therapy, oral prednisolone, cyclophosphamide, dipyridamole, and warfarin) was given. At presentation, urine protein excretion and histological indices of the mean activity and chronicity were 245 ±101 mg/m2 per h, 6.6 ±1.2, and 1.5 ±1.3, respectively. After 6 months of therapy, urinary protein excretion had decreased significantly (P < 0.001). The activity index was also decreased significantly at the second renal biopsy performed at a mean interval of 4.3 months after the first biopsy (2.8 ± 1.4, P < 0.05), whereas there was no change in the chronicity index. At the most recent observation, all patients showed clinical improvement. Two patients had normal urine, three had proteinuria of <20 mg/m2 per h, one had proteinuria of >20 mg/m2 per h, and none showed any renal insufficiency. Although this case series was examined without controls, this treatment protocol may be of benefit to children with rapidly progressive HSPN [59].
The benefits of the abovementioned treatments for treating HSPN deserve to be assessed further in larger randomized controlled trials.
Other types of treatment
The use of intravenous immunoglobulin (IVIg) for the treatment of HSP is anecdotal, and has been advocated as being effective for abdominal pain and other gastrointestinal symptoms [60].
Some studies have reported that tonsillectomy was effective for patients with severe HSPN [61, 62]. Kawasaki et al. report an 11-year-old boy with HSPN accompanied by recurrent purpura and persistent nephropathy despite conventional therapy such as prednisolone, methylprednisolone pulse therapy and mizoribine. The patient was treated with tonsillectomy plus methylprednisolone pulse therapy. This treatment decreased proteinuria, induced the disappearance of microscopic hematuria, and improved renal pathological findings. This case suggests that tonsillectomy plus methylprednisolone pulse is an effective and useful therapy for some children with recurrent purpura and persistent nephropathy [62].
Transplantation
HSPN may recur after transplantation, and rates of recurrence are increased in recipients of living-related transplantations [63, 64]. Meulders et al. [63] reported the actuarial risks for renal recurrence and for graft loss due to recurrence were 35 and 11%, respectively, at 5 years after transplantation. Recurrence appeared to be associated with shorter duration of the original disease episode and occurrence despite delays of more than 1 year between the disappearance of purpura and transplantation, and was not prevented by a triple immunosuppressive regimen that included CyA.
Finally, our recommendations for the treatment of HSPN in our hospital are shown in Table 2. We try to perform renal biopsy at 1–2 months after the onset of HSPN for patients with a urinary protein excretion of more than 0.5 g/day, and provide aggressive therapy according to the severity of pathological lesions. We believe these treatments are effective at improving the prognosis for HSPN.
Conclusion
We have reviewed the pathogenesis of and treatment for HSPN, including multiple drug combination therapies. Further detailed investigation of HSPN pathogenesis and treatment is necessary to identify the most appropriate treatment.
References
Mills JA, Michel BA, Bloch DA, Calabrese LH, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch–Schonlein purpura. Arthritis Rheum. 1990;33(8):1114–21.
Saulsbury FT. Epidemiology of Henoch-Schonlein purpura. Cleve Clin J Med. 2002;69:187–9.
Bunchman TE, Mauer SM, Sibley RK, Vernier RL. Anaphylactoid purpura: characteristics of 16 patients who progressed to renal failure. Pediatr Nephrol. 1988;2:393–7.
Counahan R, Winterborn MH, White RH, Heaton JM, Meadow SR, Bluett NH, Swetschin H, Cameron JS, Chantler C. Prognosis of Henoch–Schönlein nephritis in children. Br Med J. 1977;2:11–4.
Yoshikawa N, Ito H, Yoshiya K, Nakahara C, Yoshiara S, Hasegawa O, Matsuyama S, Matsuo T. Henoch–Schoenlein nephritis and IgA nephropathy in children: a comparison of clinical course. Clin Nephrol. 1987;27:233–7.
Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch–Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197–202.
Stewart M, Savage JM, Bell B, McCord B. Long term renal prognosis of Henoch–Schönlein purpura in unselected childhood population. Eur J Pediatr. 1988;147:113–5.
Nielsen HE. Epidemiology of Schonlein–Henoch purpura. Acta Paediatr Scand. 1988;77:125–31.
Kawasaki Y, Suyama K, Yugeta E, Katayose M, Suzuki S, Sakuma H, et al. The incidence and severity of Henoch–Schönlein purpura nephritis over a 22-year period in Fukushima Prefecture, Japan. Int Urol Nephrol. 2010;42:1023–9.
Vogler C, Eliason SC, Wood EG. Glomerular membranopathy in children with IgA nephropathy and Henoch–Schoenlein purpura. Pediatr Dev Pathol. 1999;2:227–35.
Kerr MA. The structure and function of human IgA. Biochem J. 1990;271:285–96.
Yoo EM, Morrison SL. IgA: an immune glycoprotein. Clin Immunol. 2005;116:3–10.
Jones CL, Powell HR, Kincaid-Smith P, Roberton DM. Polymeric IgA and immune complex concentrations in IgA-related renal disease. Kidney Int. 1990;38:323–31.
Allen AC, Willis FR, Beattie TJ, Feehally J. Abnormal IgA glycosylation in Henoch–Schoenlein purpura restricted to patients with clinical nephritis. Nephrol Dial Transplant. 1998;13:930–4.
Egmond M, Damen CA, Spriel AB, Vidarsson G, Garderen E, Winkel JGJ. IgA and IgA Fc receptor. Trends Immunol. 2001;22:205–11.
Moja P, Quesnel A, Resseguier V, Lambert C, Freycon F, Berthoux F, et al. Is there IgA from gut mucosal origin in the serum of children with Henoch–Schoenlein purpura? Clin Immunol Immunopathol. 1998;86:290–7.
Silva FG. IgA nephropathy and Henoch–Schoenlein syndrome. In: Jennette JC, Olson JL, Schwartz MM, Silva FG, editors. Heptinstall’s pathology of the kidney. 15th ed. Philadelphia: Lippincott-Raven; 1998. p. 479–540.
Allen A, Harper S, Feehally J. Origin and structure of pathogenic IgA in IgA nephropathy. Biochem Soc Trans. 1997;25:486–90.
Coppo R, Basolo B, Bulzomi MR, Roccatello D, Carbonara AO, Barbiano di Belgiojoso G, et al. IgA1 and IgA2 immune complexes in primary IgA nephropathy and Henoch–Schoenlein purpura nephritis. Clin Exp Immunol. 1984;57:583–90.
Levinsky RJ, Barratt TM. IgA immune complexes in Henoch–Schoenlein purpura. Lancet. 1979;24:1100–3.
Leu KK, Wyatt RJ, Moldoveanu Z, Tomana M, Julian BA, Hogg RJ, et al. Serum levels of galactose-deficient IgA in children with IgA nephropathy and Henoch–Schoenlein purpura. Pediatr Nephrol. 2007;22:2067–72.
Oortwijn BD, Eijgenraam JW, Rastaldi MP, Roos A, Daha MR, van Kooten C. The role of secretary IgA and complement in IgA nephropathy. Semin Nephrol. 2008;28:58–65.
Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M. Complement activation through the lectin pathway in patients with Henoch–Schönlein purpura nephritis. Am J Kidney Dis. 2000;35:401–7.
Hisano S, Matsushita M, Fujita T, Iwasaki H. Activation of the lectin complement pathway in Henoch–Schönlein purpura nephritis. Am J Kidney Dis. 2005;45:295–302.
Moura IC, Benhamou M, Launay P, Vrtovsnik F, Blank U, Monteiro RC. The glomerular response to IgA deposition in IgA nephropathy. Semin Nephrol. 2008;28:88–95.
Kawasaki Y, Suzuki J, Nemoto K, Nozawa R, Suzuki S, Suzuki H. Clinical and pathological features of children with Henoch–Schoenlein purpura nephritis: risk factors associated with poor prognosis. Clin Nephrol. 2003;60:153–60.
Fujieda M, Oishi N, Naruse K, Hashizume M, Nishiya K, Kurashige T, Ito K. Soluble thrombomodulin and antibodies to bovine glomerular endothelial cells in patients with Henoch–Schoenlein purpura. Arch Dis Child. 1998;78:240–4.
Kawasaki Y, Suzuki J, Nozawa R, Sakai N, Tannji M, Isome M, et al. FB21, a monoclonal antibody that reacts with a sialic-acid-dependent carbohydrate epitope, is a marker for glomerular endothelial cell injury. Am J Kidney Dis. 2004;44:239–49.
Masuda M, Nakanishi K, Yoshizawa N, Iijima K, Yoshikawa N. Group A streptococcal antigen in the glomeruli of children with Henoch–Schönlein nephritis. Am J Kidney Dis. 2003;41:366–70.
Davin JC, Pierard G, Dechenne C, Grossman D, Nagy J, Quacoe M, et al. Possible pathogenic role of IgE in Henoch–Schönlein purpura. Pediatr Nephrol. 1994;8:169–71.
Ehara T, Shigematsu H. Mast cells in the kidney. Nephrology (Carlton). 2003;8:130–8.
Namgoong MK, Lim BK, Kim JS. Eosinophil cationic protein in Henoch–Schönlein purpura and in IgA nephropathy. Pediatr Nephrol. 1997;11:703–6.
Kawasaki Y, Hosoya M, Suzuki H. Possible pathologenic role of interleukin-5 and eosino cationic protein in Henoch–Schönlein purpura nephritis. Pediatr Int. 2005;47:51251–7.
Kawasaki Y, Imaizumi T, Matsuura H, Ohara S, Takano K, Suyama K, et al. Renal expression of alpha-smooth muscle actin and c-Met in children with Henoch–Schönlein purpura nephritis. Pediatr Nephrol. 2008;23:913–9.
Algoet C, Proesmans W. Renal biopsy 2–9 years after Henoch–Schönlein purpura. Pediatr Nephrol. 2003;18:471–3.
Goldstein AR, White RH, Akuse R, Chantler C. Long-term follow-up of childhood Henoch–Schoenlein nephritis. Lancet. 1992;339:280–2.
Meadow SR, Glasgow EF, White RH, Moncrieff MW, Cameron JS, Ogg CS. Schoenlein–Henoch nephritis. Q J Med. 1972;41:241–5.
Reif S, Jain A, Santiago J, Rossi T. Protein-losing enteropathy as a manifestation of Henoch–Schoenlein purpura. Acta Paediatr Scand. 1991;80:482–5.
Cull DL, Rosario V, Lally KP, Ratner I, Mahour GH. Surgical implications of Henoch–Schoenlein purpura. J Pediatr Surg. 1990;25:741–3.
Kawasaki Y, Suzuki H. Henoch–Schoenlein nephritis. In: Geary DF, Schaefer F, editors. Comprehensive pediatric nephrology. Mosby: Elsevier; 2008. p. 343–51.
Suthanthiran M, Strom TB. Immunoregulatory drugs: mechanistic basis for use in organ transplantation. Pediatr Nephrol. 1997;11:651–7.
Taube D, Brown Z, Williams DG. Long-term impairment of suppressor-cell function by cyclophosphamide in minimal-change nephropathy and its association with therapeutic response. Lancet. 1981;1:235–8.
Mizuno K, Tsujino M, Takada M, Hayashi M, Atsumi K. Studies on bredinin. I. Isolation, characterization and biological properties. J Antibiot. 1974;27:775–82.
Ichikawa Y, Ihara H, Takahara S. The immunosuppressive mode of action of mizoribine. Transplantation. 1984;38:262–7.
Ishikawa H. Mizoribine and mycophenolate mofetil. Curr Med Chem. 1999;6:575–97.
Kawasaki Y. Mizoribine: a new approach in the treatment of renal disease. Clin Dev Immunol. 2009;681482.
Diasio RB, Lo Buglio AF. Immunomodulators: immunosuppressive agents and immunostimulants. In: Hardman JG, editor. Goodman and Gilman’s: The pharmacological basis of therapeutics. 9th ed, vol. 52. New York: McGraw-Hill; 1996. p. 1291–308.
Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schoenlein–Henoch purpura nephritis. Pediatr Nephrol. 1998;12:238–43.
Mollica F, Li Volti S, Garozzo R, Russo G. Effectiveness of early prednisone treatment in preventing the development of nephropathy in anaphylactoid purpura. Eur J Pediatr. 1992;151:140–4.
Kawasaki Y, Suzuki J, Nozawa R, Suzuki S, Suzuki H. Efficacy of methylprednisolone and urokinase pulse therapy for severe Henoch–Schoenlein nephritis. Pediatrics. 2003;111:785–9.
Oner A, Tinaztepe K, Erdogan O. The effect of triple therapy on rapidly progressive type of Henoch–Schönlein nephritis. Pediatr Nephrol. 1995;9(1):6–10.
Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch–Schoenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol. 1998;49:9–14.
Iijima K, Ito-Kariya S, Nakamura H, Yoshikawa N. Multiple combined therapy for severe Henoch–Schoenlein nephritis in children. Pediatr Nephrol. 1998;12:244–8.
Flynn JT, Smoyer WE, Bunchman TE, Kershaw DB, Sedman A. Treatment of Henoch–Schoenlein purpura glomerulonephritis in children with high-dose corticosteroids plus oral cyclophosphamide. Am J Nephrol. 2001;21:128–33.
Kawasaki Y, Suzuki J, Suzuki H. Efficacy of methylprednisolone and urokinase pulse therapy combined with or without cyclophosphamide in severe Henoch–Schoenlein nephritis: a clinical and histopathological study. Nephrol Dial Transplant. 2004;19:858–64.
Shin JI, Park JM, Shin YH, Kim JH, Lee JS, Jeong HJ. Henoch–Schönlein purpura nephritis with nephrotic-range proteinuria: histological regression possibly associated with cyclosporin A and steroid treatment. Scand J Rheumatol. 2005;34:392–5.
Kawasaki Y, Suyama K, Hashimoto K, Hosoya M. Methylprednisolone pulse plus mizoribine in children with Henoch–Schoenlein purpura nephritis. Clin Rheumatol. 2011;30:529–35.
Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Schoenlein–Henoch purpura nephritis in children. Am J Kidney Dis. 1999;33:427–33.
Kawasaki Y, Suzuki J, Murai M, Takahashi A, Isome M, Nozawa R, et al. Plasmapheresis therapy for rapidly progressive Henoch–Schoenlein nephritis. Pediatr Nephrol. 2004;19:920–3.
Heldrich FJ, Minkin S, Gatdula CI. Intravenous immunoglobulin in Henoch–Schoenlein purpura: a case study. Md Med J. 1993;42:577–9.
Suguyama H, Watanabe N, Onoda T, Kikumoto Y, Yamamoto M, Maeta M, et al. Successful treatment of progressive Henoch–Schoenlein purpura nephritis with tonsillectomy and steroid pulse therapy. Intern Med. 2005;44:611–5.
Kawasaki Y, Suyama K, Matsumoto A, Takano K, Hashimoto K, Suzuki S, et al. Efficacy of tonsillectomy plus methylprednisolone pulse therapy for a child with Henoch–Schoenlein purpura nephritis. Tohoku J Exp Med. 2007;211:291–5.
Meulders Q, Pirson Y, Cosyns JP, Squifflet JP, de Strihou C. Course of Henoch–Schoenlein nephritis after renal transplantation: report on ten patients and review of the literature. Transplantation. 1994;58:1179–86.
Ramos EL. Recurrent diseases in the renal allograft. J Am Soc Nephrol. 1991;2:109–21.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Kawasaki, Y. The pathogenesis and treatment of pediatric Henoch–Schönlein purpura nephritis. Clin Exp Nephrol 15, 648–657 (2011). https://doi.org/10.1007/s10157-011-0478-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-011-0478-1