
Abstract
Diatraea saccharalis constitutes a threat to the sugarcane productivity, and obtaining borer tolerant cultivars is an alternative method of control. Although there are studies about the relationship between the interaction of D. saccharalis with sugarcane, little is known about the molecular and genomic basis of defense mechanisms that confer tolerance to sugarcane cultivars. Here, we analyzed the transcriptional profile of two sugarcane cultivars in response to borer attack, RB867515 and SP80-3280, which are considered tolerant and sensitive to the borer attack, respectively. A sugarcane genome and transcriptome were used for read mapping. Differentially expressed transcripts and genes were identified and termed to as DETs and DEGs, according to the sugarcane database adopted. A total of 745 DETs and 416 DEGs were identified (log2|ratio| > 0.81; FDR corrected P value ≤ 0.01) after borer infestation. Following annotation of up- and down-regulated DETs and DEGs by similarity searches, the sugarcane cultivars demonstrated an up-regulation of jasmonic acid (JA), ethylene (ET), and defense protein genes, as well as a down-regulation of pathways involved in photosynthesis and energy metabolism. The expression analysis also highlighted that RB867515 cultivar is possibly more transcriptionally activated after 12 h from infestation than SP80-3280, which could imply in quicker responses by probably triggering more defense-related genes and mediating metabolic pathways to cope with borer attack.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A threat to sugarcane productivity is the presence of pests which cause economic losses in the field. The sugarcane borer (Diatraea saccharalis) is the main pest in Brazilian sugarcane growing areas. Estimates have demonstrated that each 1% of sugarcane internode infested with borer results in 0.5 to 1.1% of sugar yield loss (White et al. 2008; Rossato et al. 2013). Sugarcane breeders have focused on the selection of tolerant cultivars through conventional breeding methods aiming to control this pest and reduce its impact. However, studies have been focusing on the phenotypic characterization of sugarcane tolerance to borer (Dinardo-Miranda et al. 2012; Tomaz et al. 2017), and until now, little is known about genes that are involved in resistance mechanisms to this pest in sugarcane.
In general, the plant’s defense mechanisms against insect-feeding involve the recognition of insect elicitors, followed by induction of a cascade of signal transduction pathways which leads to transcriptional changes of defense-related genes and consequently triggering the biosynthesis of defense metabolites. Throughout evolution, plants have been acquiring induced defense mechanisms for protection against insects. Usually, the first steps upon herbivory recognition are the activation of kinase networks and biosynthesis of phytohormones (Maffei et al. 2012). As a consequence, defense mechanisms tend to interfere directly with insect growth and development, mainly via the activation of toxic proteins and metabolites, such as proteinase inhibitors and peroxidases (Govind et al. 2010). It has been proposed that most of the defense mechanisms against insect attack in plants are generally regulated by genes involved in the jasmonic acid (JA) and ethylene (ET) biosynthetic and signaling pathways (Wang and Wu 2013; Rehrig et al. 2014; Pangesti et al. 2016). JA- and ET-induced responses appear to be responsible for a large portion of the differential regulation of defense genes and regulatory elements, which can prime plants against biotic and abiotic stresses, improving tolerance.
The analysis of sugarcane transcriptome is a powerful approach to identify differentially expressed genes related to hormone biosynthesis and insect defense-related genes and to unravel the molecular mechanisms of tolerance to herbivory. Among available methods, RNA sequencing (RNA-seq) is a tool that allows the analysis of transcriptional profiles of any species of interest that present contrasting characteristics by quantifying the expression of genes activated or inactivated under certain conditions (Li et al. 2012). Despite the importance of sugarcane borer and the improvements in breeding programs related to borer tolerance, little is known about the molecular mechanisms related to resistance triggered upon Diatraea saccharalis infestation in sugarcane.
The absence of a reference genome imposed a drawback for further advances in understanding the molecular physiology of sugarcane. For long, the genome of the diploid species Sorghum bicolor had been used for this purpose because of the overall sequence collinearity and similarity with the sugarcane genome. However, because of the genomic complexity of its polyploid genome, the transcriptome analysis of sugarcane cultivars had been limited by the presence of allelic variation and a variety of transcript isoforms, which are difficult to identify using diploid genome models. Only recently, a reference transcriptome for sugarcane, named SUGIT (Sugarcane Iso-Seq transcriptome database), became available (Hoang et al. 2017). SUGIT is the first reference for complete transcripts sequences (full-length) of sugarcane and provides an excellent coverage of its transcriptome, contributing significantly for gene expression studies. More recently, a reference sequence of sugarcane monoploid genome became available (Garsmeur et al. 2018). This reference is a single tiling path (STP) which encompasses a single copy of the sugarcane gene space and contains 25,316 annotated protein-coding genes. STP and SUGIT are, respectively, a well-suited genome and transcriptome references for sugarcane gene expression studies.
Here, we analyze the transcriptional profiles of Brazilian sugarcane cultivars RB867515 and SP80-3280 in response to D. saccharalis infestation. These cultivars were chosen based on their susceptibility to the borer attack. In preliminary tests, both cultivars presented different behaviors regarding culm injury and young larvae survival on leaves (Tomaz et al. 2017). In general terms, RB867515 has relatively low survival of early-stage larvae feeding on leaves and is known to be more resistant in field conditions. Conversely, SP80-3280 is more susceptible to field and has lower mortality of early-stage larvae feeding on leaves, allowing then more larvae to penetrate the stalks. Thus, this study focused on characterizing the transcriptional changes of sugarcane and on identifying differences related to defense mechanisms possibly involved in the sugarcane tolerance to D. saccharalis.
Results and discussion
RNA-seq quality assessment
In summary, the samples obtained in this study consisted of good quality RNAs (Online Resource 1. Table S1). A reduced version of SUGIT containing 38,240 canonical sequences representatives of the transcriptome (SUGIT.UniRef80) was selected as reference for read mapping (see “Materials and methods”). Even with the complexity reduction, the number of SUGIT.UniRef80’s transcripts was superior to STP’s genes (25,316 sequences), which could represent isoform variants of genes, non-coding regions or translated regions still not annotated in the sugarcane reference genome. Besides, STP was developed based on sorghum genome, which might not be sufficient to capture all sugarcane gene fractions.
Approximately 4% of low-quality reads were removed from all RNA-seq libraries. The number of raw, filtered, and mapped reads are listed in Table 1. In general, more reads were mapped to SUGIT.Ref80 reference (69.5% average) than to STP’s (58.6% average), probably because of differences in genetic backgrounds and the number of sequences included in each reference database. Despite having an overall lower percentage of mapped reads to STP’s genes, the pairwise Pearson’s correlation coefficient values ranged between 0.98 and 0.99 among the mapping outputs of read counts generated by STAR software (Online Resource 2. Fig. S1). Likewise, pairwise correlation values between 0.97 and 0.99 were observed among the normalized read abundances generated by Kallisto software (Online Resource 2. Fig. S2). These correlation values indicate that biological replicates data have good reproducibility.
Differential expression analysis
For comparison purposes, we adopted the terminology “differentially expressed transcripts” (DETs) and “differentially expressed genes” (DEGs) (log2|ratio| > 0.81; FDR corrected P value ≤ 0.01) for the expression data obtained from SUGIT.UniRef80 and STP references, respectively. Differential expression analysis highlighted a more abundant transcriptional response for RB867515 when compared to SP80-3280, regardless of the mapping reference adopted (Fig. 1a–d). This result shows that RB867515 cultivar is more transcriptionally activated after 12 h of infestation, which could imply in quicker responses upon herbivory by probably triggering more defense-related genes and mediating metabolic pathways to cope with D. saccharalis attack. The complete list of overlapping and unique DETs and DEGs with their respective annotation and log2 fold changes (log2 FCs) is shown in Online Resource 1. Tables S2 and S3.

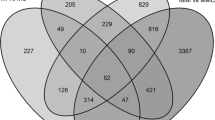
Number of differentially expressed transcripts (DETs) and genes (DEGs) identified in sugarcane in response to Diatraea saccharalis. DETs and DEGs were identified after 12 h from infestation in sugarcane cultivars RB867515 (tolerant) and SP80-3280 (susceptible) using SUGIT.UniRef80 and STP databases as mapping references, respectively. (A and B) Number of up-regulated (red) and down-regulated (blue) DETs and DEGs found (FDR corrected P value ≤ 0.01). (C and D) Venn diagram of unique and common up- and down-regulated DETs and DEGs found in the cultivars RB867515 and SP80-3280. (E) Summary of reciprocal BLAST analysis results between DETs and DEGs
A reciprocal alignment between DETs and the coding DNA sequences (CDS) of DEGs was conducted to verify sequence similarity among them. The reciprocal BLAST searches showed that 255 DEGs significantly aligned with 298 DETs (Fig. 1 (E)). Surprisingly, 447 DETs and 161 DEGs had no matches in the alignments among differentially expressed sequences. When the BLAST searches were conducted between the entire set of SUGIT.UniRef80’s transcripts and STP’s CDS, there were still 230 DETs and 81 DEGs that had no significant matches. The complete list of the BLAST searches results between DETs and DEGs is shown in Online Resource 1. Table S4. This means that different sequences are considered differentially expressed depending on the reference adopted. These alignments provide an auxiliary tool for designing confidently specific primers in genes of interest, which could be further used in gene expression analysis in sugarcane. In addition, this information reveals that sequencing and annotation gaps or different genic regions still exist between SUGIT and STP references and ensures the need of using more than one reference database to capture a broader genetic background in gene expression studies with sugarcane.
Based on common herbivory responses and stress-related defense mechanisms (Howe and Jander 2008; Pandey et al. 2017; War et al. 2018), up- and down-regulated DETs and DEGs shared between the resistant (RB867515) and susceptible (SP80-3280) cultivars were selected and compared as for the relative log2 FC (Table 2). The selected genes are involved in hemicellulose, lignin, jasmonic acid (JA), ethylene (ET), and terpenoid biosynthesis, as well as in kinase, transcription factor, peroxidase, and chitinase activity. These comparisons give insight into gene-specific regulation in each sugarcane cultivar in response to D. saccharalis infestation. Upon infestation, the expression of most of those genes increased significantly in both cultivars. However, genes involved in terpenoid and hemicellulose biosynthesis were up- and down-regulated, respectively, only in RB867515; whereas chitinase- and lignin-related genes were only up-regulated in SP80-3280.
In addition, a differential expression analysis among control samples of SP80-3280 and RB867515 was conducted to better understand their transcriptional background (SP vs. RB) (Online Resource 2. Table S17). Among the selected genes, only “cinnamoyl-CoA reductase” and “cellulose synthase-like protein E6” were differentially expressed, which means that their expression levels in the unstressed state are significantly higher in RB867515 (Table 2). Then, the difference in expression levels at the stressed conditionis attributed to how each cultivar mediates gene expression upon D. saccharalis infestation.
Functional analysis of DETs and DEGs
Sequence annotation
DETs and DEGs were functionally annotated through similarity searches to understand their possible functions in response mechanisms against D. saccharalis infestation. A total of 716 DETs (96.1%) and 403 DEGs (96.8%) were aligned to the non-redundant (nr) protein sequence database of NCBI using Blast2GO (Online Resource 2. Tables S2 and S3). Most of DETs and DEGs had significant alignments with genes from Sorghum bicolor, which is the closest-related diploid crop to sugarcane, and both share high sequence identity in their genic regions (Bundock et al. 2012).
In addition, TRAPID was used to assign conserved protein domains (PDs) to DETs and DEGs by comparing proteins encoded by their sequences with the InterPro database (Hunter et al. 2009) (Online Resource 2. Tables S5 and S6). TRAPID found 814 domains in proteins encoded by 723 DETs (97%) and 592 domains in proteins encoded by 408 DEGs (97.8%) (Table 3). TRAPID also identified 421 and 282 different gene families among the annotated DETs and DEGs, having the “Plant peroxidase” and “Oxoglutarate family” as the most abundant gene families (GFs), respectively (Table 3). In general terms, peroxidases have been implicated in physiological processes that involve scavenging of reactive oxygen species (ROS) and synthesis of lignin and phytoalexins, which are important biological processes against biotic stresses (Cosio and Dunand 2009). Enzymes belonging to the oxoglutarate family participate in a variety of plant metabolic pathways, including the synthesis of hormones, signaling molecules and secondary metabolites (Cheng et al. 2014).
KEGG pathway and GO enrichment analysis
Several stress-related pathways and biological processes were enriched in both sugarcane cultivars and a similar pattern was observed in responses after 12 h of infestation with D. saccharalis (Online Resource 2. Tables S8, S9, and S11). Pathways such as “phenylpropanoid biosynthesis,” “biosynthesis of amino acids,”, “alpha-linoleic acid metabolism,” and “linoleic acid metabolism” are known to be involved in plant stress responses and were significantly enriched and up-regulated in RB867515 and SP80-3280 (Fig. 2a and c). Pathways involved in “photosynthesis” and “carbon metabolism” were significantly enriched and down-regulated (Fig. 2b and d). Biological processes such as “response to wounding (GO:0009611),” “response to jasmonic acid (GO:0009753),” and “oxylipin metabolic process (GO:0031407)” are known to be associated to stress and plant defense were up-regulated in RB867515 and SP80-3280 (Fig. 3a and c). Only one biological process related to “photosynthesis; light harvesting in photosystem I (GO:0009768)” was observed as down-regulated in RB867515 (Fig. 3b). However, a greater number of down-regulated pathways and biological processes related to photosynthesis and carbon metabolism was observed in both sugarcane cultivars when genes showing small fold changes (0 < log2|ratio| < 0.81) were included in enrichment analysis (Online Resource 2. Figs. S3 and S4), suggesting that this transcriptional switch from an up-regulation of defense responses to a down-regulation of photosynthesis and carbon metabolism is still in a starting phase after 12 h of infestation.

Most enriched KEGG pathways identified in sugarcane cultivars in response to D. saccharalis. (A and B) Distribution of up-regulated (red) and down-regulated (blue) KEGG pathways assigned to cultivar RB867515 (tolerant). (C and D) Distribution of up-regulated (red) and down-regulated (blue) KEGG pathways assigned to cultivar SP80-3280 (susceptible). KEGG ID corresponds to reference metabolic pathways of Sorghum bicolor. The color scale represents the FDR corrected P value calculated in enrichment analysis

Most enriched biological process Gene Ontology (GO) terms identified in sugarcane in response to D. saccharalis. (A and B) Distribution of up-regulated (red) and down-regulated (blue) GO terms assigned to cultivar RB867515 (tolerant). (C and D) Distribution of up-regulated (red) and down-regulated (blue) GO terms assigned to cultivar SP80-3280 (susceptible). The color scale represents the FDR corrected P value calculated in enrichment analysis
Down-regulation of genes involved in photosynthesis has also been observed in rice under nematode (Wang et al. 2020) and bacterial pathogen mimic infection (Ranjan et al. 2015). In plants, photosynthesis and carbon metabolism might be compromised upon herbivory as a trade-off for the synthesis of defensive metabolites (Zhou et al. 2015). Down-regulation of photosynthesis-related genes is an adaptive response to biotic attack, which slow turnover of many photosynthetic proteins and allows plants to invest resources in immediate defense needs without debilitating near term losses in photosynthetic capacity (Bilgin et al. 2010). Furthermore, wound responses dependent of jasmonic acid also has been showed to promote the down-regulation of photosynthesis genes and a significant decrease in carbon assimilation (Havko et al. 2020).
As the pattern observed in differential expression analysis, RB867515 also showed a higher number of genes included in enriched pathways and biological processes than SP80-3280. RB867515 had 123 genes (considering DETs and DEGs) mapped to KEGG pathways and 186 mapped to biological processes GOs, while SP80-3280 had 70 and 68, respectively. When genes with small fold changes (0 < log2|ratio| < 0.81) are taken in consideration, the enrichment analysis also showed a different distribution of up- and down-regulated pathways and biological processes between RB867515 and SP80-3280 (Online Resource 2. Figs. S3 and S4). While SP80-3280 shows a predominant up-regulation of pathways and biological process, RB86715 shows a higher proportion of down-regulated genes involved in photosynthesis and sugar/carbon metabolism. It is also interesting to mention that the general GO term associated with “response to stress (GO:0006950)” showed opposite results for these genes with small fold changes. Meanwhile RB867515 starts showing a down-regulation of genes related to response to stress, SP80-3280 is still mediating the up-regulation of general stress-related responses against D. saccharalis after 12 h of infestation. Therefore, according to the enrichment analysis, we hypothesize that RB867515 responds faster than SP80-3280 and invests more resources in immediate defense mechanisms during D. saccharalis infestation.
Analysis of differentially expressed genes in control conditions (SP vs. RB) provide some insights that corroborate to this hypothesis. RB867515 showed an up-regulation of photosynthesis genes with enriched biological processes such as “photosynthesis, light harvesting (GO:0009765)” and “pyruvate metabolic process (GO:0006090),” while SP80-3280 showed an up-regulation of genes of oxidative metabolism with enriched biological processes such as “oxidation-reduction process (GO:0055114)” and “glutathione metabolic process (GO:0006749)” (Online Resource 2. Table S16). These observations suggest higher transcriptional activity of photosynthesis genes for RB867515 at control conditions when compared to SP80-3280.
Another possibly related point in understanding the faster response and transcriptional activation of RB867515 upon herbivory when compared to SP80-3280 is the regulation of fructose-6-phosphate 2-kinase/fructose-2,6-bisphosphatase (F2KP) (Sh09_g005060; GFHJ01038535.1). F2KP was found as a DEG (log2 FC − 0.65; FDR corrected P value 9.27e−6) and a DET (log2 FC − 0.53; FDR corrected P value 5.01e−5) in comparison between controls (Online Resource 2. Table S17), which means that it is more expressed in RB867515 at control conditions. Fructose-2,6-bisphosphate is an important traffic signal in plant metabolism, and it coordinates the photosynthetic carbon flux into sucrose and starch metabolism, allowing plants to respond effectively to external conditions as a fast regulatory system of the ever-changing photosynthetic metabolism (Nielsen et al. 2004).
Regulation of genes involved in the biosynthetic and signaling pathways of ET and JA after D. saccharalis infestation
Phytohormones are usually implicated in defense mechanisms against pests. Basically, hormones participate in signal transduction pathways, promoting cellular crosstalk communication and coordination of transcriptome changes. Analysis of DETs and DEGs revealed that the signaling and biosynthetic pathways of ET and JA were upregulated in both cultivars, suggesting the involvement of these molecules in the defense responses during infestation with D. saccharalis in sugarcane herbivory (Online Resource 1. Table S12).
In infested treatments, key enzymes of the ET biosynthetic pathway, such as 1-aminocyclopropane-1-carboxylate synthase (ACS) and 1-aminocyclopropane-1-carboxylate oxidase (ACO), were upregulated (Fig. 4). These enzymes belong to the “cysteine and methionine metabolism.” an enriched KEGG metabolic pathway upon D. saccharalis infestation in this study. ACS is an enzyme that catalysis the synthesis of 1-aminocyclopropane-1-carboxylic acid from S-adenosyl methionine (SAM), and ACO catalyzes the last step of the ET biosynthetic pathway by converting 1-aminocyclopropane-1-carboxylic acid to ethylene (Booker and DeLong 2015).

General hormonal responses implicated in sugarcane infested by D. saccharalis infestation. Genes related to the biosynthetic pathways of ethylene (ET) (left) and jasmonic acid (JA) (right). Up- and down-pointed arrows correspond to up- and down-regulation, respectively. Green and yellow arrows correspond to regulation in sugarcane cultivars RB867515 and SP80-3280, respectively
ET signaling genes were also up-regulated, and among the annotated DETs and DEGs are included: ethylene-responsive factor (ERF), reversion-to-ethylene sensitivity (RTE), ethylene response sensor 2 (ERS2).These genes were more predominantly triggered in RB867515 after D. saccharalis infestation (Online Resource 1. Table S12). In general, ERS2 belongs to a family of membrane-associated receptors responsible for recognizing ET (Lacey and Binder 2014); RTE is involved in the regulation of ET receptors and is described as a negative regulator of ET responses (Resnick et al. 2008); and ERF transcription factors are involved in positively regulating ethylene and various stress responses (Xie et al. 2019). These genes are reviewed to be responsible for triggering defenses against abiotic and insect herbivory (Nguyen et al. 2016), but little is known about their interplay in sugarcane defense mechanisms against insect herbivory.
The JA and its derivatives have been recognized as key regulators in plant defense responses. The first steps in JA biosynthesis involve enzymes that participate in the “linoleic and alpha-linoleic acid metabolism,” an enriched KEGG metabolic pathway in this study. During infestation with D. saccharalis, several DETs and DEGs belonging to the JA biosynthetic genes were identified, such as lipoxygenase (LOX), allene oxide synthase (AOS), allene oxide cyclase (AOC), and 12-oxophytodienoate reductase (12-OPR). These enzymes participate in the oxylipin biosynthetic process, and more precisely, they mediate the biosynthesis of JA and its derivatives from α-linoleic acid precursor via the octadecanoid pathway. The octadecanoid pathway has been reported to be involved in defense line mechanisms against biotic stresses (Santino et al. 2013). Here, LOX and AOS were upregulated in both cultivars, while AOC and12-OPR genes were up-regulated only in RB867515 (Fig. 4). Besides the similarities, according to the RNA-seq data, JA genes were more regulated in RB867515 in terms of expression level and number, an indication that the JA pathway is more activated in this cultivar upon herbivory (Online Resource 1. Table S12).
Genes involved in JA signaling transduction pathway, such as jasmonate ZIM domain-containing protein (TIFY/JAZ) and MYC2 transcription factor, were found to be up-regulated in this study. Belonging to the “basic-helix-loop-helix (bHLH) related transcription factor” protein domain, MYC2 acts on the first line of signaling transduction after JA perception, positively modulating JA-dependent responses. Conversely, JAZ proteins are known to function as a repressor of MYC2 activity, posing a negative effect on the JA signaling pathway (Pauwels et al. 2010). Considering DETs and DEGs, more TIFY/JAZ genes were assigned to SP80-3280, whereas more MYC2 genes were assigned to RB867515 herbivory (Online Resource 1. Table S12). This indicates that RB867515 might have better control of the interplay between JA repressors and promoters.
This hormonal regulation pattern indicates that larval herbivory by D. saccharalis triggers similar JA and ET genes in the tolerant and susceptible cultivars used in this study (Fig. 4). However, some differences in number and regulation of DETs and DEGs assigned to these pathways were observed in both cultivars, which could represent different transcript isoforms of these genes and specific gene regulation that might be associated with herbivory-related defense mechanisms (Online Resource 1. Table S12).
Other reports highlight the transcriptional changes of JA and ET related to the plant’s defense responses under caterpillar infestation. For instance, using transcriptome sequencing and analysis, similar hormonal regulation results were identified in sugarcane submitted to infection with the bacterium Acidovorax avenae subsp. avenae (Santa Brigida et al. 2016), in corn infested with the borer Ostrinia furcanalis (Yang et al. 2015; Wang et al. 2017), and in cotton submitted to Hellicoverpa armigera larvae infestation (Huang et al. 2015). Also, it has been demonstrated that JA possess a role in resistance against Phytophthora cinnamomi in maize (Allardyce et al. 2013) and against Botrytis cinera in strawberry (Jia et al. 2016).
It has been demonstrated that JA is involved in the synthesis of secondary metabolites including plant-defense proteins in certain species. For example, exogenous JA treatment induced the expression of pathogenesis-related genes (PRs) in rice (Yang et al. 2013). In sugarcane, JA-treated plants demonstrated certain resistance to D. saccharalis (Sanches et al. 2017), which could be associated with the regulation of defense proteins. Also, particular LOX isoforms can play important role in the activation of stress-induced defense responses in plants (Mariutto et al. 2011; Ogunola et al. 2017) and confer tolerance against wounding and insect attack (Wang et al. 2008). Thus, it is speculated whether different genes of LOX identified in this study, or possible isoforms, could regulate defense-related genes and impair different levels of tolerance in sugarcane.
These findings collaborate on the importance and involvement of JA and ET in the responsive mechanisms and regulation of signaling networks against biotic stresses. However, because of the polyploidy characteristic of sugarcane and the array of identified DETs and DEGs assigned to these pathways, further molecular investigation on single transcripts and genes could elucidate associations with specific phenotypic responses of sugarcane to D. saccharalis herbivory.
Regulation of defense-related proteins
Proteases and protease inhibitors
Generally, the recognition of insect elicitors by the plant’s receptors triggers signaling transduction pathways that lead to the synthesis and accumulation of defense-associated proteins. Plant peptidase inhibitors are an example of plant-defense proteins produced upon herbivory. In this study, we identified a wide array of DETs and DEGs with a protease inhibitor annotation. In summary, annotations included “Bowman-Birk type bran trypsin inhibitor and Bowman-Birk type trypsin inhibitor (BBTI),” “Bowman-Birk type wound-induced proteinase inhibitor WIP1 (BBWIP1),” “Subtilisin Chymotrypisin inhibitor-2B (SCI-2B),” “Subtilisin Chymotrypisin inhibitor-2A (SCI-2A),” and “maize proteinase inhibitor (MPI).” Interesting, DETs and DEGs assigned to BBTI, BBWIP1, and SCI-2B were up-regulated in both cultivars; DEGs assigned to SCI-2A were only up-regulated in SP80-3280; and MPI were only up-regulated in RB867515, suggesting that these genes play a role in the defense against D. saccharalis attack.
Similar regulation patterns for these protease inhibitors have been reported for sugarcane under D. saccharalis artificial attack using the same hybrid cultivar SP80-3280 (Medeiros et al. 2016). In addition to the regulation pattern, the authors verified that Bowman-Birk inhibitor genes have different levels of expression when submitted to D. saccharalis attack and to wounding, suggesting that these proteins have a specific-triggering line of defense, which might be involved in the recognition of insect-specific elicitors. Up-regulation and accumulation of protease inhibitors have also been reported in maize infested with the leaf-eater Spodoptera frugiperda (Ankala et al. 2013), maize infested with the stem-borer Ostrinia furcanalis (Yang et al. 2015), and also in sugarcane after D. saccharalis infestation (Medeiros et al. 2012).
Besides the inhibitors, we also identified differentially expressed genes with a protease annotation: “serine carboxypeptidase II-3,” “serine carboxypeptidase like-2,” “cysteine proteinase 2,” and “metalloendoproteinase 2MMP (MP).” In this study, MPs were only up-regulated by RB867515; serine carboxypeptidase like-2 and cysteine proteinases were only down-regulated in RB867515; and serine carboxypeptidase II-3 were up-regulated by both cultivars. Regulation of proteases and protease inhibitors with respective log2 fold changes is shown in Online Resource 1. Table S13.
Chitinases
Most of the up-regulated chitinase-related DETs and DEGs was mediated by SP80-3280 (Online Resource 1. Table S13), and “chitin metabolic process (GO:0006030)” was an enriched and up-regulated biological process for this cultivar (Fig. 3 (C)). It has been demonstrated that chitinases have an antifungal activity in plants, playing important role in defense mechanisms, inclusive in sugarcane (Tariq et al. 2018). Despite mostly studied for its antifungal properties, chitinases are also a protective agent against insects, attacking on chitin molecules that compose the insect’s skeleton. For long, it has been verified an augment of chitinase expression under biotic stress, including insect attack, and this pattern has caught the attention to be involved in defense mechanisms. The defensive effects of chitinase genes have been reported in some species, including resistance of tomato against Colorado potato beetle (Lawrence and Novak 2006), of transgenic corn, expressing a chitinase gene, against Spodoptera littoralis (Osman et al. 2015), and of transgenic tea (Camellia sinensis [L.] O. Kuntze), overexpressing a class I chitinase gene from potato, against blister blight (Exobasidium vexans) (Singh et al. 2015). However, the role of the sugarcane chitinase family genes remain unclear due to the highly heterozygous and aneuploidy chromosome genetic background of sugarcane (Su et al. 2015).
Peroxidases
Several up and down differentially expressed sequences had a peroxidase annotation. In total, there were 15 DETs and 7 DEGs (Online Resource 1. Table S13). Interestingly, only RB867515 showed a down-regulation of POD genes. This regulation pattern and its annotation diversity suggest that different peroxidase genes, and probably different isoforms, are mediated for defense mechanisms in sugarcane upon herbivory.
It has been demonstrated that a peroxisomal catalase, another ROS scavenger enzyme, which also belongs to a multi-gene family, is responsive to biotic stresses and is suggested to be involved in the protection of sugarcane against oxidant-related environment stimuli (Su et al. 2014). Also, it has been suggested that lipoxygenases modulate ROS burst by consuming polyunsaturated fatty acids after wounding (Roach et al. 2015; Prasad et al. 2017), a process that could be associated with an increase of POD synthesis to counter-balance the negative effects of ROSs. The large number of POD genes in sugarcane, together with the diversity of processes catalyzed by peroxidases, suggest possible functional specialization of each isoform, but assigning a precise role for each individual peroxidase gene has continued to be a major bottleneck (Cesarino et al. 2012). Since D. saccharalis infestation resulted in differential expression of PODs, a closer-detailed investigation could reveal new insights into the function and specificity of peroxidase-related DETs and DEGs found in this work.
Cell wall biosynthesis
Lignin, cellulose, and hemicellulose are plant cell wall components that might dictate plant resistance to insect injury. Lignin accumulation plays an important role in the process of plant resistance to insects and can be used as a barrier directly or through the associated hormone signal pathway to increase insect resistance of plants (Liu et al. 2018). Genes involved in the biosynthetic pathway of lignin were regulated in this study. Cinnamoyl-CoA reductase (CCR), 4-coumarate:CoA ligase (4CL), and hydroxycinnamoyltransferase (HCT) are enzymes that participate in monolignol biosynthesis and were differentially expressed after D. saccharalis infestation (Online Resource 1. Table S12).
Most of the DEGs and DETs annotated to CCR, HCT, and 4CL were up-regulated by SP80-3280, indicating that this cultivar transcriptionally tends to reinforce its structures upon D. saccharalis infestation. On the other side, this result implies that RB867515 did not invest energy on morphological barriers to prevent larvae feeding, which is advantageous because lignin biosynthesis is not an immediate line of defense.
The enzymes “cellulose synthase A catalytic subunit 6 (CESA6)” and “cellulose synthase-like protein E (CSLE6),” which are involved in cellulose and hemicellulose biosynthesis, respectively, were also found to be differentially expressed. It has been shown that reduced levels of cellulose synthesis can lead to lignin accumulation mediated in part by JA and ethylene, leading to defense responses in Arabidopsis thaliana (Cano-Delgado et al. 2003). Besides regulating few biosynthetic genes of lignin, RB867515 also showed a predominantly down-regulation of cellulose synthase E and up-regulation of cellulose synthase A, demonstrating its transcriptional versatility to mediate the biosynthesis of cell wall components upon biotic stress (Online Resource 1. Table S13).
Validation of RNA-seq by RT-qPCR
RT-qPCR analysis was performed to corroborate with RNA-seq data (Fig. 5). Three DEGs involved in JA biosynthesis (LOX), protease inhibitor (SCI), and ferredoxin energy transport (FeS), and three DETs involved in JA biosynthesis (AOS), protease inhibitor (MPI), and hemicellulose biosynthesis (CSLE) were selected for that purpose. The assayed genes exhibited the expected positive or negative fold changes in qPCR reactions (Fig. 5a and d). However, SCI had significant up-regulation in qPCR for RB867515 (Fig. 5a), and MPI and CSLE exhibited significant up-regulation for SP80-3280 in qPCR (Fig. 5d). These genes did not differentiate statistically in RNA-seq data for the considered samples (Fig. 5b and e). We attribute these differences partly to non-controlled variation and to false-positive discovery correction in RNA-seq data because the difference in read counts mapped to each gene in control and infested SP80-3280 samples leads to the regulation pattern observed in RT-qPCR, but did not represent statistical significance (FDR corrected P value ≤ 0.01) in RNA-seq. Despite the significance differences, RNA-seq and RT-qPCR data showed a high correlation for the selected genes, reaching R values of 0.96 and 0.90 for RB867515 and SP80–3280, respectively (Fig. 5c and f).

RNA-seq analysis validation by RT-qPCR using candidate genes involved in different biological pathways. Three biological replicates were used. The bars of the RT-qPCR graphs represent the relative expression mean of three technical replicates (data = mean ± SD). (A and B) Relative expression levels obtained from RT-qPCR and RNA-seq analysis for RB867515 cultivar. (D and E) Relative expression levels obtained from RT-qPCR and RNA-seq analysis for SP80-3280 cultivar. (C and F) Pearson correlation coefficient analysis between log2-transformed relative expression levels from RT-qPCR and RNA-seq results of RB867515 and SP80-3280 cultivars, respectively. *Statistical significance between control and infested treatments for RT-qPCR analysis (P value ≤ 0.05) and for RNA-seq analysis (FDR corrected P value ≤ 0.01); nsnot significant for RT-qPCR analysis (P value ≤ 0.05) and for RNA-seq analysis (FDR corrected P value ≤ 0.01). LOX lipoxygenase, AOS allene oxide synthase, SCI subtilisin-chymotrypsin inhibitor, MPI maize proteinase inhibitor, CSLE hemicellulose synthase, FeS 3Fe-4S ferredoxin
Conclusions
In summary, expression analysis highlighted a more abundant transcriptional response for tolerant cultivar RB867515 upon D. saccharalis herbivory when compared to susceptible cultivar SP80-3280, suggesting that it is more transcriptionally activated after 12 h from infestation, which may act as a tolerance mechanism to boost defense responses. Overall pathway analysis of both sugarcane cultivars showed an up-regulation of jasmonic acid (JA), ethylene (ET), and defense protein genes, as well as a down-regulation of pathways involved in photosynthesis and energy metabolism after D. saccharalis herbivory. However, RB867515 showed a higher proportion of down-regulated genes of metabolic pathways involved in photosynthesis and sugar/carbon when genes with small fold changes are considered, which suggests an immediate energy switch by probably allocating more resources to defense mechanisms in order to cope with the pest attack. In contrast to RB867515, SP80-3280 demonstrated a higher number of differentially expressed genes of metabolic pathways involved in lignin accumulation, probably to augment morphological structures upon infestation.
Materials and methods
Sugarcane cultivars, cultivation conditions, infestation, and experimental design
The sugarcane cultivars (RB867515 and SP80-3280) used in this work were obtained from RIDESA (Inter-University Network for the Development of Sugarcane Industry) (Barbosa et al. 2012). Culms from the cultivars were planted in 30 L pots filled with a mixture of 28 kg soil/manure (2:1 v/v) and grown in a greenhouse (average temperature 28.5 °C and natural light conditions) with soil humidity kept at soil water capacity. D. saccharalis larvae were obtained from a reserve colony grown in artificial diet (Hensley and Hammond 1968) with modifications (Araújo et al. 1985). The colony originated from a set of larvae collected in the field and grown in laboratory (Girón-Pérez et al. 2014). Sugarcane plants were evaluated using the leaf numbering system proposed by Kuijper (Kuijper 1915) and the leaves + 1 of individual plants were infested with 20 D. saccharalis larvae (3rd to 4th instar) after 2 months from the planting date, period in which plants were approximately 1 m high (Online Resource 2. Fig. S5).
The experiment was conducted in a randomized block design with two contrasting sugarcane cultivars (RB867515, resistant; and SP80-3280, susceptible), two treatments (control and infested), and five biological replicates. Three biological replicates of each experimental group were selected for RNA-seq analysis (Online Resource 2. Fig. S5A). The harvested vegetal material consisted of a section delimited by the bases of sheath and blade of + 1 leaf, including the internal leaves, which was collected after 12 h from the infestation. All samples were immediately frozen under liquid nitrogen after collection.
RNA extraction, RNA-seq library construction, sequencing, and bioinformatic analysis
Leaf samples were ground to fine powder under liquid nitrogen using stainless steel grind jars in the TissueLyser II (Qiagen) bead mill, and RNA was extracted using the PureLink® Plant RNA Reagent (Thermo Fisher) as described by the manufacturer. The quantification of total extracted RNA was accessed using a Qubit® 2.0 Fluorometer kit (Life technologies). The RNA integrity number (RIN) of the total RNA was analyzed using a 2100 Bioanalyzer 6000 Kit (Agilent Technologies, USA). A minimum of 10 μg of purified total RNA per sample with a RIN > 7.0 was sent out to GenOne Biotechnologies (Brazil) for constructing mRNA-seq libraries using paired-end mode (2 × 150 bp) in the Illumina HiSeq 2500 sequencer (Illumina).
The libraries underwent filtering steps to obtain clean reads, which included the following: (1) raw reads quality assessment using FASTQC version 0.11.8 (https://github.com/s-andrews/FastQC), and (2) trimming and removal of low-quality reads using AfterQC version 0.97 (Chen et al. 2017) and Trimmomatic version 0.38 (Bolger et al. 2014). After trimming and filtering, clean reads were mapped to the sugarcane monoploid genome database (STP) (Garsmeur et al. 2018) using STAR version 2.7.0a (Dobin et al. 2013) by selecting the quantification mode (“—quantMode”) and default parameters. For complementary results, we also used the SUGIT transcriptome reference source (Hoang et al. 2017) to broaden the spectrum of read mapping. It is worth mentioning that the SUGIT database was constructed using a pool of RNA from different sugarcane genotypes under different stresses and from different tissues, which enhances the probability of catching broader transcriptional-triggered response profiles in sugarcane. Before mapping, the SUGIT database (107,597 transcripts) underwent a pre-processing step involving sequence complexity reduction to obtain a unified reference (UniRef) that was comparable to STP (25,316 genes) in differential expression analysis. For that purpose, CD-hit version 4.6.8 (Li and Godzik 2006) was used to cluster transcript sequences that share 80% of identity into canonical sequences representatives of the transcriptome. The UniRef80 was selected as that one with the number of canonical sequences closest to the number of genes of Saccharum officinarum (35,525 genes) and Sorghum bicolor (34,118 genes). Then, clean reads were mapped to SUGIT.UniRef80 using Kallisto version 0.45.0 (Bray et al. 2016) by selecting the quantification algorithm (“quant”) with 1000 bootstrap samples (“--bootstrap-samples”).
For terminology purposes, we referred to differentially expressed transcripts as DETs, and differentially expressed genes as DEGs according to the respective database adopted. The integrative analysis of DEGs and DETs concomitantly broadens the spectrum of the transcriptional profile of sugarcane and gives a better grasp of the biological processes triggered upon infestation, which enhances the exploitation of resistance traits in future studies and maximizes the understanding of how sugarcane responds to D. saccharalis infestation. A schematic overview of the analysis conducted in this study is shown in Online Resource 2. Fig. S6.
DETs and DEGs were identified using the DESeq2 package version 1.6.3 (Love et al. 2014) implemented in R version 3.5.1 (R Development CoreTeam (2018) 2018). DETs and DEGs analysis were conducted using the read mapping information generated by Kallisto and STAR, respectively. A false discovery rate (FDR) corrected P value ≤ 0.01 and a log2|ratio| > 0.81 (|fold change| > 1.75) were used to select significant DEGs and DETs. In addition, Pearson’s correlation coefficient analysis was performed to compare the read abundances from the mapping outputs obtained by STAR and Kallisto software. The read abundances were normalized as transcripts per million (TPM) and then transformed to log2. This analysis included pairwise comparisons of the log2 transformation of normalized read abundances relative to all biological replicates in control and infested plants (see Online Resource 2. Figs. S1 and S2).
Characterization and functional annotation of DETs and DEGs
DETs and DEGs were functionally annotated using Blast2GO version 5.2.5 (Conesa and Götz 2008) through similarity searches using BLAST (McGinnis and Madden 2004) (Online Resource 1. Tables S2 and S3). The performed search parameters included: blastx-fast; nr database; taxonomy filter: monocots (taxa: 4447, Liliopsida); e-value: 1.0e−5; and number of Blast Hits: 5. Blast2GO was also used to perform a reciprocal BLAST analysis between DETs and DEGs using the following parameters: blastn (-task megablast); e-value, 1.0e−5; word size, 28; and HSP length cutoff, 75 (Table S4).
Conserved domains were assigned to the proteins encoded by DEGs and DETs using the online platform TRAPID: Rapid Analysis of Transcriptome Data (Van Bel et al. 2013) (Tables S5 and S6), which uses the PLAZA 4.5 database (https://bioinformatics.psb.ugent.be/plaza/) to assign functional annotations based on sequence similarity. The parameters of performed searches included the Poaceae database and an e-value of 1.0e−5.
Gene ontology (GO) terms were assigned to DETs and DEGs using the online platform PlantRegMap (Plant Transcriptional Regulatory Map) (Jin et al. 2017), which adopt the topGo package and Fisher’s exact tests to find significantly overrepresented GO terms (P value ≤ 0.01). Sequences of DETs and DEGs were mapped to Sorghum bicolor genes using the ID Mapping tool of PlantRegMap (Table S7) and four lists of non-redundant Gene IDs were submitted to GO enrichment analysis: up- and down-regulated in RB867515, up- and down-regulated in SP80-3280 (Table S8). The lists of enriched Biological Process GO terms were further summarized using REViGO (Supek et al. 2011) (Table S9).
We also used KEGG Orthology Based Annotation System (KOBAS) version 3.0 to identify enriched metabolic pathways among DETs and DEGs (Xie et al. 2011). Sequences of DETs and DEGs were mapped to S. bicolor genes using the Annotation tool of KOBAS (Table S10) and four lists of Gene IDs were submitted to pathway enrichment analysis. Enriched KEGG pathways were determined by using Fisher’s exact tests followed by Benjamini-Hochberg FDR correction method (FDR corrected P value ≤ 0.05) (Table S11).
Quantitative RT-PCR
The same three biological replicates for RNA-seq were used in RT-qPCR, but with independent RNA extraction. Total RNA was isolated using PureLink Plant RNA Reagent (Life Technologies). The extracted RNA was DNase-treated using TURBO DNase kit (Thermo Fisher) and, sequentially, the samples were submitted to heat (70 °C/15 min) as a final step for DNase deactivation. First-strand cDNA synthesis was performed from 500 ng of total RNA using RNA M-MLV Reverse transcriptase kit (Invitrogen). PCR reactions were performed in the QuantStudio3 instrument (Thermo Fisher) using the following program: 95 °C for 2 min, 40 cycles of 95 °C for 15 s, and 61 °C for 1 min, followed by melting curve. To each well was added a 10 μl final volume containing: 2 μl of diluted cDNA (1:25), 5 μl of GoTaq qPCR Master Mix (Promega), and 1.5 μl each primer (300 nM). Target gene expression was normalized using eEF-1a (eukaryotic elongation factor 1a) and eIF-4a (eukaryotic initiation factor 4a) as internal controls. Data were subjected to the 2−ΔΔCt method (Livak and Schmittgen 2001). For statistical analysis, Student’s t test (P value ≤ 0.05) was employed to compare the significance of the relative mRNA level of the selected genes between control and infested treatments. Pearson correlation test was employed to verify the correlation coefficient of log2 fold changes (log2FCs) between RT-qPCR and RNA-seq expression results. The design of primers was based on selected up- and down-regulation of DEGs and DETs in RNA-seq results with minor nucleotide correction according to read mapping. Primers are listed in Online Resource 1. Table S14.
Data availability
The RNA-Seq raw data are available at sequence read archive (SRA) (https://www.ncbi.nlm.nih.gov/sra), and this study was registered with the ID SRP243494 and BioProject ID PRJNA602327. The RNA-seq data of RB867515 (BioSample ID SAMN13892657) were registered with the following accession numbers: SRR10917298, SRR10917297, SRR10917294, SRR10917293, SRR10917292, and SRR10917291. The RNA-seq data of SP80-3280 (BioSample ID SAMN13892658) were registered with the following accession numbers: SRR10917290, SRR10917289, SRR10917288, SRR10917287, SRR10917296, and SRR10917295.
References
Allardyce JA, Rookes JE, Hussain HI, Cahill DM (2013) Transcriptional profiling of Zea mays roots reveals roles for jasmonic acid and terpenoids in resistance against Phytophthora cinnamomi. Funct Integr Genom 13:217–228. https://doi.org/10.1007/s10142-013-0314-7
Ankala A, Kelley RY, Rowe DE, Williams WP, Luthe DS (2013) Foliar herbivory triggers local and long distance defense responses in maize. Plant Sci 199–200:103–112. https://doi.org/10.1016/j.plantsci.2012.09.017
Araújo JR, Botelho PSM, Araújo SMSS, Almeida LC, Degaspari N (1985) Nova dieta artificial para criação da Diatraea saccharalis (Fabr.). Saccharum 36:45–48
Barbosa MHP, Deon M, Resende V, Antônio L, Dias S, Veríssimo De Souza Barbosa G, Augusto De Oliveira R, Peternelli LA, Daros E (2012) Genetic improvement of sugar cane for bioenergy: the Brazilian experience in network research with RIDESA. Crop Breed Appl Biotechnol 12:87–98. https://doi.org/10.1590/S1984-70332012000500010
Bilgin DD, Zavala JA, Zhu J, Clough SJ, Ort DR, Delucia EH (2010) Biotic stress globally downregulates photosynthesis genes. Plant Cell Environ 33:1597–1613. https://doi.org/10.1111/j.1365-3040.2010.02167.x
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Booker MA, DeLong A (2015) Producing the ethylene signal: regulation and diversification of ethylene biosynthetic enzymes. Plant Physiol 169:42–50. https://doi.org/10.1104/pp.15.00672
Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527. https://doi.org/10.1038/nbt.3519
Bundock PC, Casu RE, Henry RJ (2012) Enrichment of genomic DNA for polymorphism detection in a non-model highly polyploid crop plant. Plant Biotechnol J 10:657–667. https://doi.org/10.1111/j.1467-7652.2012.00707.x
Cano-Delgado A, Penfield S, Smith C, Catley M, Bevan M (2003) Reduced cellulose synthesis invokes lignification and defense responses in Arabidopsis thaliana. Plant J 34:351–362. https://doi.org/10.1046/j.1365-313X.2003.01729.x
Cesarino I, Araújo P, Sampaio Mayer JL, Paes Leme AF, Mazzafera P (2012) Enzymatic activity and proteomic profile of class III peroxidases during sugarcane stem development. Plant Physiol Biochem 55:66–76. https://doi.org/10.1016/j.plaphy.2012.03.014
Chen S, Huang T, Zhou Y, Han Y, Xu M, Gu J (2017) AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinformatics 18:91–100. https://doi.org/10.1186/s12859-017-1469-3
Cheng AX, Han XJ, Wu YF, Lou HX (2014) The function and catalysis of 2-oxoglutarate-dependent oxygenases involved in plant flavonoid biosynthesis. Int J Mol Sci 15:1080–1095. https://doi.org/10.3390/ijms15011080
Conesa A, Götz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:619832–619812. https://doi.org/10.1155/2008/619832
Cosio C, Dunand C (2009) Specific functions of individual class III peroxidase genes. J Exp Bot 60:391–408. https://doi.org/10.1093/jxb/ern318
Development CoreTeam R (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Dinardo-Miranda LL, dos Anjos IA, da Costa VP, Fracasso JV (2012) Resistance of sugarcane cultivars to Diatraea saccharalis. Pesqui Agropecu Bras 47:1–7. https://doi.org/10.1590/S0100-204X2012000100001
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Garsmeur O, Droc G, Antonise R, Grimwood J, Potier B, Aitken K, Jenkins J, Martin G, Charron C, Hervouet C, Costet L, Yahiaoui N, Healey A, Sims D, Cherukuri Y, Sreedasyam A, Kilian A, Chan A, Van Sluys MA, Swaminathan K, Town C, Bergès H, Simmons B, Glaszmann JC, Van Der Vossen E, Henry R, Schmutz J, D’Hont A (2018) A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat Commun 9:2638. https://doi.org/10.1038/s41467-018-05051-5
Girón-Pérez K, Oliveira AL, Teixeira AF, Guedes RNC, Pereira EJG (2014) Susceptibility of Brazilian populations of Diatraea saccharalis to Cry1Ab and response to selection for resistance. Crop Prot 62:124–128. https://doi.org/10.1016/j.cropro.2014.04.004
Govind G, Mittapalli O, Griebel T, Allmann S, Böcker S, Baldwin IT (2010) Unbiased transcriptional comparisons of generalist and specialist herbivores feeding on progressively defenseless Nicotiana attenuata plants. PLoS One 5:e8735. https://doi.org/10.1371/journal.pone.0008735
Havko NE, Das MR, McClain AM, Kapali G, Sharkey TD, Howe GA (2020) Insect herbivory antagonizes leaf cooling responses to elevated temperature in tomato. Proc Natl Acad Sci U S A 117:2211–2217. https://doi.org/10.1073/pnas.1913885117
Hensley SD, Hammond AH (1968) Laboratory techniques for rearing the sugar cane borer on an artificial diet. J Econ Entomol 61:1742–1743. https://doi.org/10.1093/jee/61.6.1742
Hoang NV, Furtado A, Mason PJ, Marquardt A, Kasirajan L, Thirugnanasambandam PP, Botha FC, Henry RJ (2017) A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genomics 18:395. https://doi.org/10.1186/s12864-017-3757-8
Howe GA, Jander G (2008) Plant immunity to insect herbivores. Annu Rev Plant Biol 59:41–66. https://doi.org/10.1146/annurev.arplant.59.032607.092825
Huang XZ, Chen JY, Xiao HJ, Xiao YT, Wu J, Wu JX, Zhou JJ, Zhang YJ, Guo YY (2015) Dynamic transcriptome analysis and volatile profiling of Gossypium hirsutum in response to the cotton bollworm Helicoverpa armigera. Sci Rep 5:11867. https://doi.org/10.1038/srep11867
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, Mcanulla C, McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJA, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C (2009) InterPro: the integrative protein signature database. Nucleic Acids Res 37:211–215. https://doi.org/10.1093/nar/gkn785
Jia H, Zhang C, Pervaiz T, Zhao P, Liu Z, Wang B, Wang C, Zhang L, Fang J, Qian J (2016) Jasmonic acid involves in grape fruit ripening and resistant against Botrytis cinerea. Funct Integr Genom 16:79–94. https://doi.org/10.1007/s10142-015-0468-6
Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, Gao G (2017) PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res 45:D1040–D1045. https://doi.org/10.1093/nar/gkw982
Kuijper J (1915) DeGroei van Bladschijf, Bladscheede em Stengel van het suikerriet. Arch Suikerind Ned Indië 23(1):528–556
Lacey RF, Binder BM (2014) How plants sense ethylene gas - the ethylene receptors. J Inorg Biochem 133:58–62. https://doi.org/10.1016/j.jinorgbio.2014.01.006
Lawrence SD, Novak NG (2006) Expression of poplar chitinase in tomato leads to inhibition of development in Colorado potato beetle. Biotechnol Lett 28:593–599. https://doi.org/10.1007/s10529-006-0022-7
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. https://doi.org/10.1093/bioinformatics/btl158
Li SW, Yang H, Liu YF, Liao QR, Du J, Jin DC (2012) Transcriptome and gene expression analysis of the rice leaf folder, Cnaphalocrocis medinalis. PLoS One 7:e47401. https://doi.org/10.1371/journal.pone.0047401
Liu Q, Luo L, Zheng L (2018) Lignins: biosynthesis and biological functions in plants. Int J Mol Sci 19:335. https://doi.org/10.3390/ijms19020335
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Maffei ME, Arimura GI, Mithöfer A (2012) Natural elicitors, effectors and modulators of plant responses. Nat Prod Rep 29:1288–1303. https://doi.org/10.1039/c2np20053h
Mariutto M, Duby F, Adam A, Bureau C, Fauconnier ML, Ongena M, Thonart P, Dommes J (2011) The elicitation of a systemic resistance by Pseudomonas putida BTP1 in tomato involves the stimulation of two lipoxygenase isoforms. BMC Plant Biol 11:29. https://doi.org/10.1186/1471-2229-11-29
McGinnis S, Madden TL (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32:20–25. https://doi.org/10.1093/nar/gkh435
Medeiros AH, Franco FP, Matos JL, de Castro PA, Santos-Silva LK, Henrique-Silva F, Goldman GH, Moura DS, Silva-Filho MC (2012) Sugarwin : a sugarcane insect-induced gene with antipathogenic activity. Mol Plant-Microbe Interact 25:613–624. https://doi.org/10.1094/MPMI-09-11-0254
Medeiros HA, Mingossi BF, Dias OR, Franco PF, Vicentini R, Mello OM, Moura SD, Silva-Filho CM (2016) Sugarcane serine peptidase inhibitors, serine peptidases, and Clp protease system subunits associated with sugarcane borer (Diatraea saccharalis) Herbivory and wounding. Int J Mol Sci 17:1444. https://doi.org/10.3390/ijms17091444
Nguyen D, Rieu I, Mariani C, van Dam NM (2016) How plants handle multiple stresses: hormonal interactions underlying responses to abiotic stress and insect herbivory. Plant Mol Biol 91:727–740. https://doi.org/10.1007/s11103-016-0481-8
Nielsen TH, Rung JH, Villadsen D (2004) Fructose-2,6-bisphosphate: a traffic signal in plant metabolism. Trends Plant Sci 9:556–563. https://doi.org/10.1016/j.tplants.2004.09.004
Ogunola OF, Hawkins LK, Mylroie E, Kolomiets MV, Borrego E, Tang JD, Williams WP, Warburton ML (2017) Characterization of the maize lipoxygenase gene family in relation to aflatoxin accumulation resistance. PLoS One 12:1–19. https://doi.org/10.1371/journal.pone.0181265
Osman GH, Assem SK, Alreedy RM, El-Ghareeb DK, Basry MA, Rastogi A, Kalaji HM (2015) Development of insect resistant maize plants expressing a chitinase gene from the cotton leaf worm, Spodoptera littoralis. Sci Rep 5:18067. https://doi.org/10.1038/srep18067
Pandey SP, Srivastava S, Goel R, Lakhwani D, Singh P, Asif MH, Sane AP (2017) Simulated herbivory in chickpea causes rapid changes in defense pathways and hormonal transcription networks of JA/ethylene/GA/auxin within minutes of wounding. Sci Rep 7:1–14. https://doi.org/10.1038/srep44729
Pangesti N, Reichelt M, van de Mortel JE, Kapsomenou E, Gershenzon J, van Loon JJA, Dicke M, Pineda A (2016) Jasmonic acid and ethylene signaling pathways regulate glucosinolate levels in plants during rhizobacteria-induced systemic resistance against a leaf-chewing herbivore. J Chem Ecol 42:1212–1225. https://doi.org/10.1007/s10886-016-0787-7
Pauwels L, Barbero GF, Geerinck J, Tilleman S, Grunewald W, Pérez AC, Chico JM, Vanden BR, Sewell J, Gil E, García-Casado G, Witters E, Inzé D, Long JA, De Jaeger G, Solano R, Goossens A (2010) NINJA connects the co-repressor TOPLESS to jasmonate signalling. Nature 464:788–791. https://doi.org/10.1038/nature08854
Prasad A, Sedlářová M, Kale RS, Pospíšil P (2017) Lipoxygenase in singlet oxygen generation as a response to wounding: in vivo imaging in Arabidopsis thaliana. Sci Rep 7:1–10. https://doi.org/10.1038/s41598-017-09758-1
Ranjan A, Vadassery J, Patel HK, Pandey A, Palaparthi R, Mithöfer A, Sonti RV (2015) Upregulation of jasmonate biosynthesis and jasmonate-responsive genes in rice leaves in response to a bacterial pathogen mimic. Funct Integr Genom 15:363–373. https://doi.org/10.1007/s10142-014-0426-8
Rehrig EM, Appel HM, Jones AD, Schultz JC (2014) Roles for jasmonate- and ethylene-induced transcription factors in the ability of Arabidopsis to respond differentially to damage caused by two insect herbivores. Front Plant Sci 5:1–14. https://doi.org/10.3389/fpls.2014.00407
Resnick JS, Rivarola M, Chang C (2008) Involvement of RTE1 in conformational changes promoting ETR1 ethylene receptor signaling in Arabidopsis. Plant J 56:423–431. https://doi.org/10.1111/j.1365-313X.2008.03615.x
Roach T, Colville L, Beckett RP, Minibayeva FV, Havaux M, Kranner I (2015) A proposed interplay between peroxidase, amine oxidase and lipoxygenase in the wounding-induced oxidative burst in Pisum sativum seedlings. Phytochemistry 112:130–138. https://doi.org/10.1016/j.phytochem.2014.06.003
Rossato JADS, Costa GHG, Madaleno LL, Mutton MJR, Higley LG, Fernandes OA (2013) Characterization and impact of the sugarcane borer on sugarcane yield and quality. Agron J 105:643–648. https://doi.org/10.2134/agronj2012.0309
Sanches PA, Santos F, Peñaflor MFGV, Bento JMS (2017) Direct and indirect resistance of sugarcane to Diatraea saccharalis induced by jasmonic acid. Bull Entomol Res 107:828–838. https://doi.org/10.1017/S0007485317000372
Santa Brigida AB, Rojas CA, Grativol C, De Armas EM, Entenza JOP, Thiebaut F, Lima MDF, Farrinelli L, Hemerly AS, Lifschitz S, Ferreira PCG (2016) Sugarcane transcriptome analysis in response to infection caused by Acidovorax avenae subsp. avenae. PLoS One 11:e0166473. https://doi.org/10.1371/journal.pone.0166473
Santino A, Taurino M, De Domenico S, Bonsegna S, Poltronieri P, Pastor V, Flors V (2013) Jasmonate signaling in plant development and defense response to multiple (a)biotic stresses. Plant Cell Rep 32:1085–1098. https://doi.org/10.1007/s00299-013-1441-2
Singh HR, Deka M, Das S (2015) Enhanced resistance to blister blight in transgenic tea (Camellia sinensis [L.] O. Kuntze) by overexpression of class I chitinase gene from potato (Solanum tuberosum). Funct Integr Genom 15:461–480. https://doi.org/10.1007/s10142-015-0436-1
Su Y, Guo J, Ling H, Chen S, Wang S, Xu L, Allan AC, Que Y (2014) Isolation of a novel peroxisomal catalase gene from sugarcane, which is responsive to biotic and abiotic stresses. PLoS One 9:e84426. https://doi.org/10.1371/journal.pone.0084426
Su Y, Xu L, Wang S, Wang Z, Yang Y, Chen Y, Que Y (2015) Identification, phylogeny, and transcript of chitinase family genes in sugarcane. Sci Rep 5:10708. https://doi.org/10.1038/srep10708
Supek F, Bošnjak M, Škunca N, Šmuc T (2011) Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. https://doi.org/10.1371/journal.pone.0021800
Tariq M, Khan A, Tabassum B, Toufiq N, Bhatti MU, Riaz S, Nasir IA, Husnain T (2018) Antifungal activity of chitinase II against colletotrichum falcatum went. causing red rot disease in transgenic sugarcane. Turkish J Biol 42:45–53. https://doi.org/10.3906/biy-1709-17
Tomaz AC, Coutinho AE, Soares BO, Peternelli LA, Pereira EJG, Barbosa MHP (2017) Assessing resistance of sugarcane varieties to sugarcane borer Diatraea saccharalis Fab. (Lepidoptera: Crambidae). Bull Entomol Res 108:547–555. https://doi.org/10.1017/S0007485317001183
Van Bel M, Proost S, Van Neste C, Deforce D, Van de Peer Y, Vandepoele K (2013) TRAPID: an efficient online tool for the functional and comparative analysis of de novo RNA-Seq transcriptomes. Genome Biol 14:R134. https://doi.org/10.1186/gb-2013-14-12-r134
Wang L, Wu J (2013) The essential role of jasmonic acid in plant-herbivore interactions - using the wild tobacco Nicotiana attenuata as a model. J Genet Genomics 40:597–606. https://doi.org/10.1016/j.jgg.2013.10.001
Wang R, Shen W, Liu L, Jiang L, Liu Y, Su N, Wan J (2008) A novel lipoxygenase gene from developing rice seeds confers dual position specificity and responds to wounding and insect attack. Plant Mol Biol 66:401–414. https://doi.org/10.1007/s11103-007-9278-0
Wang H, Li S, Teng S, Liang H, Xin H, Gao H, Huang D, Lang Z (2017) Transcriptome profiling revealed novel transcriptional regulators in maize responses to Ostrinia furnacalis and jasmonic acid. PLoS One 12:e0177739. https://doi.org/10.1371/journal.pone.0177739
Wang HL, Yang SH, Lv M, Ding SW, Li JY, Xu CL, Xie H (2020) RNA-Seq revealed that infection with white tip nematodes could downregulate rice photosynthetic genes. Funct Integr Genomics 20:367–381. https://doi.org/10.1007/s10142-019-00717-9
War RA, Taggar GK, Hussain B, Taggar MS, Nair RM, Sharma HC (2018) Plant defense against herbivory and insect adaptations. AoB Plants 10:ply037. https://doi.org/10.1093/aobpla/ply037
White WH, Viator RP, Dufrene EO, Dalley CD, Richard EP, Tew TL (2008) Re-evaluation of sugarcane borer (Lepidoptera: Crambidae) bioeconomics in Louisiana. Crop Prot 27:1256–1261. https://doi.org/10.1016/j.cropro.2008.03.011
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:316–322. https://doi.org/10.1093/nar/gkr483
Xie Z, Nolan TM, Jiang H, Yin Y (2019) AP2/ERF transcription factor regulatory networks in hormone and abiotic stress responses in Arabidopsis. Front Plant Sci 10:1–17. https://doi.org/10.3389/fpls.2019.00228
Yang D-L, Yang Y, He Z (2013) Roles of plant hormones and their interplay in rice immunity. Mol Plant 6:675–685. https://doi.org/10.1093/MP/SST056
Yang F, Zhang Y, Huang Q, Yin G, Pennerman KK, Yu J, Liu Z, Li D, Guo A (2015) Analysis of key genes of jasmonic acid mediated signal pathway for defense against insect damages by comparative transcriptome sequencing. Sci Rep 5:16500. https://doi.org/10.1038/srep16500
Zhou S, Lou Y-R, Tzin V, Jander G (2015) Alteration of plant primary metabolism in response to insect herbivory. Plant Physiol 169:1488–1498. https://doi.org/10.1104/pp.15.01405
Acknowledgments
The authors would like to thank the Núcleo de Análise de Biomoléculas (NuBioMol) and Sugarcane Breeding Program, Universidade Federal de Viçosa, MG, Brazil, for providing the facilities necessary for execution of the experiments.
Funding
This work was funded by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG).
Author information
Authors and Affiliations
Contributions
USM and PMPV analyzed data and wrote the manuscript. PMPV, CEV, ACT, and MHPB planned and designed the research. USM, PMPV, CEV, ACT, and MF conducted the experiments. LAP and MHPB coordinated the research. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Code availability
Not applicable.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
de Mello, U.S., Vidigal, P.M.P., Vital, C.E. et al. An overview of the transcriptional responses of two tolerant and susceptible sugarcane cultivars to borer (Diatraea saccharalis) infestation. Funct Integr Genomics 20, 839–855 (2020). https://doi.org/10.1007/s10142-020-00755-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-020-00755-8