
Abstract
White-tip nematode, Aphelenchoides besseyi is a kind of widely distributed migratory parasitic nematode that can infect plant shoots. Transcriptome sequencing of plant parasitic nematodes and their host plants is helpful for understanding their interaction relationship. This study first reported expression patterns of defense-related genes in rice, and rice transcriptomes at different periods after infection with A. besseyi. The result showed that the defense response pathways of rice changed obviously in the early stage of A. besseyi infection, including upregulated salicylic acid and jasmonate pathways and a downregulated ethylene pathway. Transcriptome analysis results suggested that A. besseyi infection was associated with the downregulation of multiple genes related to photosynthesis with possible suppression of the photosynthetic activity. It suggested that the photosynthesis system of rice could be suppressed by infections of migratory nematodes, including A. besseyi and Hirschmanniella oryzae, but was stimulated by that of a sedentary nematode, Meloidogyne graminicola, by comparing our study with the reported transcriptome. OS09G0417800 (OsWRKY62) might play an important role in the interaction of migratory nematodes and rice. It also indicated that the infection strategy of both A. besseyi and the reported migratory nematode H. oryzae was similar to that of the fungal pathogen Magnaporthe grisea. These results provided an interesting starting point to elucidate the mechanism of the interaction between rice and A. besseyi, as well as the host and migratory plant nematodes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plant parasitic nematodes are important plant pathogens. The annual global loss of crops damaged by plant parasitic nematodes worldwide was 157 billion US dollars (Abad et al. 2008). During interactions between hosts and plant nematodes, plant hosts have a series of self-recognition and self-defense mechanisms to resist the invasion of plant nematodes, while plant nematodes have various parasitic strategies to respond to the recognition and defense mechanisms of hosts (Kyndt et al. 2012b). It has been generally recognized that nematodes can evade or destroy the hosts’ defense mechanism by secreting effector proteins. In recent years, transcriptome sequencing has been increasingly used to study the interaction of plant nematodes and host plants. Over 500 effectors from more than ten plant nematodes have been predicted and identified by RNA-Seq (Jacob et al. 2008; Haegeman et al. 2011, 2013; Akker et al. 2014; Bauters et al. 2014; Thorpe et al. 2014; Wang et al. 2014; Petitot et al. 2016; Barnes et al. 2018). To respond to pathogen infection, the expressions of pathogen-related genes (PR genes) in plant can be induced through the plant self-regulations of hormones. At present, there are many studies that have focused on plant defense pathways mediated by the hormones salicylic acid (SA), jasmonic acid (JA), and ethylene (ET) (Haegeman et al. 2011; Nahar et al. 2011; Kyndt et al. 2012b). Through transcriptome sequencing of host plants infected by nematodes, the expression patterns of defense-related genes in the infected plants could be revealed, which is helpful to understand the parasitic mechanism of plant nematodes and the defense strategies of plants, thereby better understanding their interaction mode. Therefore, RNA-Seq has also been increasingly used in recent studies of host plants infected by nematodes. Swiecicka et al. (2009) reported the transcriptome of tomato infected by Globodera rostochiensis, and found that the genes of the infected host were enriched in hormone regulation, defense response, cell cycle and cytoskeletal regulation, cell wall conformation, and transcription factors. Kyndt et al. (2012a, b) found that rice root knot nematodes, Meloidogyne graminicola could suppress host plant defense reactions and stimulate the metabolism in galls to enhance root nutrition transportation, but rice root rot nematodes, Hirschmanniella oryzae, could stimulate the host defense response and suppress host metabolism by comparing the transcriptomes of rice infected with the two nematodes. Mota et al. (2018) found 52 resistance genes belonging to the nucleotide-binding site and the leucine-rich repeat (NBS-LRR) family by comparing the transcriptomes of two different resistant wild peanut relatives infected by M. arenaria. These studies focused on the defense responses and transcriptome of plant roots infected with plant nematodes. There have been few reports about the defense response and transcriptome of plant shoots infected by plant nematodes. So far, only transcriptomes of pinus spp. infected with Bursaphelenchus xylophilus have been reported (Santos et al. 2012).
Rice is one of the most important food crops. Global economic losses of rice caused by plant nematodes are over 16 billion US dollars, and Aphelenchoides besseyi contributes to most of this loss (Lilley et al. 2011). Aphelenchoides besseyi is a migratory plant nematode that causes white tip and spikelet symptoms in rice. It can cause economic losses of up to 50% due to severe damage (Lin et al. 2004; Karssen and Groza 2018; Wang et al. 2018). At present, some of the most studied PR genes in rice are OsPR1a involved in the general defense pathway, OsPR1b and OsPAL involved in the SA pathway, JiOsPR10 involved in the JA pathway, and OsERF70 involved in the ET pathway (Kyndt et al. 2012b; Nahar et al. 2011; Karmakar et al. 2016). In this study, we tried to understand the rice defense mechanism and physiological metabolic activity in response to A. besseyi infection by investigating symptoms of the infected rice, examining the expression patterns of five selected PR genes (OsPR1a, OsPR1b, OsPAL, JiOsPR10, and OsERF70) after the rice was infected with A. besseyi, and sequencing the transcriptome of rice plants infected with the nematodes at different times, to provide a basis to reveal the parasitic strategy of A. besseyi and to understand the interactions of rice and A. besseyi. This study also provided valuable data for understanding the interaction mechanism of plants and migratory parasitic nematodes.
Materials and methods
Plant material and nematode
Rice, Oryza sativa L. cv. “Nipponbare” was provided by Prof. Guohui Zhou from Laboratory of Plant Virus, South China Agricultural University. Healthy rice seeds were germinated for 12 h at 37 °C and then 48 h at 30 °C after surface disinfection. Seeds with radicles of 0.5–1.0 cm long were selected to sown in sterile soil and further grown in a growth chamber at 30 °C with the conditions of watering once a week under a 16 h:8 h light:dark regime, with 150 μmol/m−2/s−1 light density, and 70–75% relative humidity.
Aphelenchoides besseyi used in this study was collected from rice (O. sativa) in Luhe Town, Nanjing, Jiangsu Province, China. Nematodes were isolated, identified, preserved, and cultured by Laboratory of Plant Nematology, South China Agricultural University. The preservation and cultivation methods of nematodes were carried out as the described method by Cheng et al. (2013).
Inoculation
Nematodes were inoculated at the two-leaf stage of rice. A nematode suspension containing 2000 nematodes/ml was prepared, and a 50-μl suspension containing 100 nematodes was dropped into a cotton ball. The cotton ball was placed into a rice sheath and fixed with a preservative membrane. Control treatments were healthy plants that were inoculated by a cotton ball soaked with 50 μl sterilized water. The inoculated rice plants were grown in the growth chamber at 30 °C with the conditions of watering once a week under a 16 h:8 h light:dark regime, 150 μmol/m−2/s−1 light density, and 70–75% relative humidity.
Rice symptom observation and nematode isolation after A. besseyi infection
Inoculated rice leaves were collected at 0.5, 1, 3, 5, 7, and 10 days (DAI) after rice was inoculated with A. besseyi. Samples were stained with 3% acid fuchsin, destained with acid glycerol, made into slides, and observed and photographed with microscopes as described by Wang et al. (2016). At the same time, shoots were collected and gently washed by tap water at each time point (DAI). Then, nematodes were separated from the shoots by the Bayman funnel method (Wang et al. 2016) and counted under microscope. Three biological replicates, each composed of five plants for each inoculation period, were taken.
Defense-related genes expression patterns of rice infected with A. besseyi
Shoots of DAI 0.5, DAI 1, DAI 3, DAI 5, DAI 7, and DAI 10 rice plants were collected for RNA extraction. RNA extraction was conducted using the RNAprep Pure Plant Kit (Tiangen, China). The extracted RNA was diluted to 100 ng/μl using RNase-free water as a template for cDNA reverse transcription after examination by electrophoresis for integrity and Nanodrop™ spectrophotometer for purity. Reverse transcription was performed following the instructions of the HiScript™ Q RT SuperMix for qPCR (+gDNA wiper) kit (Vazyme, China). The relative expression levels of target defense-related genes in rice inoculated with nematodes at different times were detected by qPCR using the reverse transcribed cDNA as a template. The target genes that were selected and the primers that were used are shown in Table 1. Each DAI treatment included three biological replicates composed of three rice plants. All PCRs were performed in two technical replicates. qPCRs were performed in the CFX-96 (Bio-Rad), and data were analyzed using Bio-Rad CFX 96 Manager and REST 384 software (Pfaffl et al. 2002).
RNA-Seq and data analysis of rice tissues infected with A. besseyi
For RNA extraction, shoots were taken from DAI 1, DAI 3, and DAI 7 rice plants. Three biological replicates, each containing a pool five individual plants were taken. RNA degradation and contamination were monitored on 1% agarose gels. RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA). Afterwards, the Illumina HiSeq™ X TEN platform was used for RNA-Seq analysis. Qualified RNA was used for cDNA library construction and then RNA-Seq, which were performed by Novogene Bioinformatics Technology Co., Ltd. -Tianjin, Beijing, China.
Raw data (raw reads) of fastq format were firstly processed through in-house perl scripts. Clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low-quality reads from raw data. At the same time, Q20, Q30, and GC content in the clean data were calculated. All the downstream analyses were based on the clean data with high quality (Novogene). Thereafter, clean sequence reads were mapped to the available Japonica rice Nipponbare genome (ftp://ftp.ensemblgenomes.org/pub/plants/release-37/fasta/oryza _sativa/dna/) using Hisat 2 v2.0.4 software (Kim et al. 2015). Splice variants mapped were treated as a single gene. Cufflinks v2.1.1 (Trapnell et al. 2014) was used to construct and identify both known and novel transcripts from the alignment results. HTSeq v 0.9.1 was used to count the read numbers mapped to each gene. The expected number of fragments per kilobase of transcript sequence per millions base pairs sequenced (FPKM), was used to estimate gene expression levels (Anders et al. 2015). As described by Anders and Huber (2010), the DESeq R (1.18.0) package was used to analyze the differential expression between the treated and control rice transcripts. The resulting p values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. Differentially expressed genes (DEGs) were defined as genes that had absolute values of log2 fold change (Log2FC) ≥ 1 and false discovery rate (FDR) ≤ 0.05.
Gene Ontology (GO) enrichment analysis of DEGs was implemented by the GOseq R package (Young et al. 2010). KOBAS 3.0 (Mao et al. 2005) was used to test the statistical enrichment of differential expression genes in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Mapman (Thim et al. 2004) was used to visualize the expression of genes onto the biotic stress pathway (http://MapMan.gabipd. org).
DEGs obtained in this study were analyzed and compared with those retrieved from reported transcriptome data of rice infected with M. oryzae and H. oryzae (Kyndt et al. 2012a) by using the website http://rice.hzau.edu.cn/cgi-bin/rice2/id_mapping_rs2 and software TBtools v0.66853 (Chen et al. 2018).
Validation of RNA-Seq results by qPCR
To verify the accuracy of RNA-Seq results, 22 DEGs with potentially important functions were randomly selected for qPCR. RNA from DAI 1, DAI 3, and DAI 7 rice infected with the nematodes was extracted according to the method described in “Rice symptom observation and nematodes isolation after A. besseyi infection”, and cDNA was synthesized by reverse transcription. Primers (Table S2) were designed according to EST sequences of transcripts, and OsUBQ5 was selected as the reference gene. Template extraction, detection, and data analysis were performed as described in “Rice symptom observation and nematodes isolation after A. besseyi infection”. Three biological replicates with two technical replicates each were taken.
Statistical analysis
All data in this study were subjected to an analysis of variance (ANOVA), and multiple comparisons of means were conducted by Duncan’s Multiple Range Test at p = 0.05 using SAS (Release 8.01).
Results
Symptoms of rice and the number of nematodes at different time points after A. besseyi infection
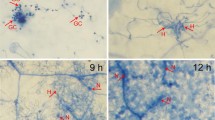
Slight deformation and discoloration on the leaf tips were observed in 15.56% of the rice plants, and nematode invading leaf tissue were observed at DAI 3 (Fig. 1). Obvious leaf deformation and white tip were observed (Fig. 1A), and symptoms were present in 33.33%, 43.33%, and 65.56% of the rice plants on DAI5, DAI7, and DAI 10, respectively, which were significantly higher than that on DAI 3 (p < 0.05). There was no significant difference between the rates of symptomatic rice plants at DAI5 and DAI7, but both were significantly lower than at DAI 10 (p < 0.05).

Rice disease symptoms and symptomatic rates after Aphelenchoides besseyi infection. A. Symptoms on leaves of infected rice. B. Nematodes were observed in the rice plant tissue at DAI3 and were visualized using acid fusion staining. C. Rate of symptomatic plants; DAI: days after inoculation. CK: healthy rice plants. Bars indicate standard errors of mean data (n = 3), and different letters indicate significant differences (p < 0.05) between treatments
No nematodes were separated from rice plants treated at DAI 0.5 and DAI1. From DAI3, nematodes were separated from rice inoculated with A. besseyi, and ten nematodes were separated at DAI 3. Separated nematode numbers increased with the extension of the nematode infection time. Separated nematode numbers had no significant difference (p > 0.05) between DAI5 and DAI7, but both were significantly higher than that at DAI 3 and less than that at DAI 10 (p < 0.05) (Table 2).
Expression patterns of rice defense–related genes after A. besseyi infection
Expression levels of five selected defense genes OsPR1a, OsPR1b, OsPAL, JiOsPR10, and OsERF70, were detected by qPCR and RNA-seq. The relative expression levels of five defense-related genes in rice changed significantly after the inoculation of A. besseyi (p < 0.05), which indicated that rice defense was induced and defense pathways were mediated by the hormones SA, JA, and ET responding to nematode infection in different ways. According to the qPCR results, OsERF70 involved in the ET pathway was significantly downregulated at DAI 1, DAI 3, DAI 5, DAI 7, and DAI 10; JiOsPR10 involved in the JA pathway was significantly upregulated at DAI 0.5, DAI1, DAI3, DAI5, and DAI7, but not at DAI 10; OsPAL and OsPR1b involved in the SA pathway were significantly upregulated at DAI 1, DAI3, and DAI5, and OsPAL was significantly upregulated while OsPR1b was significantly downregulated at DAI 7 and DAI 10; and the general defense-related gene OsPR1a was significantly upregulated at DAI 3, DAI 5, DAI 7, and DAI 10, but not at DAI0.5 and DAI1 (Fig. 2A). Compared with RNA-seq results at DAI1, DAI3, and DAI7, the five defense-related genes expressed with similar patterns. Among these five genes, OsERF70 involved in the ET pathway was significantly downregulated at DAI 3; JiOsPR10 involved in the JA pathway was significantly upregulated at DAI1 and DAI3; OsPAL and OsPR1b involved in the SA pathway were significantly upregulated at DAI 1, and OsPR1b was significantly downregulated at DAI 7; and the general defense-related gene OsPR1a was significantly upregulated at DAI 3 and DAI 7, but not at DAI1. Some differences were between qPCR and RNA-seq, and that might be due to different samples used for detection.

Expression of five defense-related genes in rice tissue at different time after inoculation with Aphelenchoide besseyi. A. Bars represent the mean expression levels, and SE was from three biologicals and two technical replicates, with each containing a pool of three plants. Data are shown as relative expression levels of treated tissue in comparison with the control tissue. Gene expression levels were normalized using the internal reference gene OsUBQ5. Bars indicate standard errors of mean data (n = 3). The asterisk (*) means a significant differential expression (p < 0.05) between inoculated and healthy rice plants according to Duncan’s multiple range test. B. Bars represent the FPKM ratio (treated and untreated) from three biologicals, with each containing a pool of five plants. Differentially expressed genes (FDR < 0.05, Log2FC ≥ 1 or Log2FC ≤ −1) were labeled with the asterisk (*). DAI: Days after inoculation
RNA sequencing and data analysis
Transcriptome sequencing of rice shoots inoculated with A. besseyi (DAI1, DAI3, and DAI7) and healthy rice shoots (CK1, CK3, and CK7) was performed in the Illumina HiSeq ™ X ten platform. Obtained raw reads numbers ranged from 59,125,350–68,752,185 and clean reads numbers ranged from 58,001,991–68,500,163. Clean reads mapped to the rice reference genome ranged from 93.26 to 94.28% (Table S2).
Identification of DEGs in rice inoculated with A. besseyi at different time points after inoculation
A total of 2037 DEGs were identified in rice plants inoculated with A. besseyi at three time points after inoculation; comparisons and statistical analysis between transcriptome data of inoculated rice and healthy rice were based on FDR < 0.05 and Log2FC ≥ 1 or Log2FC ≤ −1 (Fig. 3), which included 260 DEGs (185 upregulated and 75 downregulated) at DAI1, 147 DEGs (81 upregulated and 66 downregulated) at DAI3, and 1630 DEGs (459 upregulated and 1171 downregulated) at DAI7. Eleven DEGs overlapped at DAI 1, DAI 3, and DAI 7. There were 182, 92, and 1546 unique DEGs at DAI1, DAI3, and DAI7, respectively (Fig. 3B).

Differentially expressed genes were retrieved from RNA-Seq of rice infected with Aphelenchoides besseyi at different time points after inoculation (1 day, 3 days, and 7 days). A Overall, 2037 differentially expressed genes were identified in the infected rice compared to healthy rice; B Venn diagram illustrating the intersections of differentially expressed genes at three time points after inoculation compared to healthy rice tissues. DAI1, DAI3, and DAI7: rice plants at 1 day, 3 days, and 7 days after inoculation, respectively. CK: healthy rice tissues
GO enrichment analysis of the DEGs in rice infected with A. besseyi
The GO enrichment analysis was performed on the DEGs obtained from DAI1, DAI3, and DAI7. At DAI1, in the cell component category, significantly upregulated GO terms were involved in the “anchored component of plasma membrane” and “anchored component of membrane” pathways (Table S3) (p value < 0.05). At DAI 3 and DAI7, 62 and 158 significantly enriched GO terms were annotated (p value < 0.05), respectively, and were mainly classified into three categories (biological process, cell composition, and molecular function) (Table S4, S5; Fig. 4). In the biological process category, enriched and upregulated GO terms were involved in chitin metabolic and catabolic processes, aminoglycan catabolic processes, and glucosamine-containing compound catalysis and metabolic processes at DAI3; enriched and upregulated GO terms were involved in processing and the metabolic process of ribosomal RNA (rRNA) and non-coding RNA (ncRNA) at DAI7; enriched and downregulated GO terms were involved in photosynthesis at both DAI3 and DAI7. In the cell composition category, enriched and downregulated GO terms were involved in photosynthesis, including chloroplast, thylakoid, and plastid, at DAI3 and DAI7. In the molecular functional category, two enriched and upregulated GO terms were involved in chitin-related pathway, and two enriched and upregulated GO terms were involved in peroxidase activity–related pathway at DAI3; enriched GO terms were from pathways related to “chlorophyll binding,” “small non-coding RNA (snoRNA) binding,” “transmembrane receptor protein activity,” “hydrolyzing O-glycosyl compounds,” and “translation elongation factor activities” at DAI7.

Gene Ontology (GO) enrichment analysis of differentially expressed genes of rice tissue infected with Aphelenchoides besseyi at different time points after inoculation. DAI3 and DAI7: rice plants at 3 days and 7 days after inoculation, respectively. CK: healthy rice tissues
Plants infected with A. besseyi show downregulation of several photosynthetic genes
KEGG pathway analysis of DEGs annotated from DAI1, DAI3, and DAI7 rice plants was performed. A total of seven enriched pathways (p value < 0.05) were annotated, including three at DAI 3 and four at DAI 7 (Table S6). No KEGG pathway was annotated at DAI1. Photosynthesis antenna protein and photosynthesis pathways were annotated at both DAI 3 and DAI 7. In the photosynthesis antenna pathway, two and 11 downregulated DEGs associated with putative light harvesting chlorophyll protein (LHC) genes were annotated at DAI 3 and DAI 7, respectively. Among those, in DAI3 and DAI7 plants, one and three DEGs were annotated to be involved in photosynthesis system I(PSI), while one and eight were involved in photosynthesis system II(PSII), respectively. In the photosynthesis pathway, ten and 21 significantly downregulated DEGs of were annotated at DAI3 and DAI7, respectively (Table 3). KEGG pathway analysis results suggested that photosynthesis and photosynthetic antenna protein pathways of rice infected with A. besseyi were obviously suppressed.
DEGs related to the interaction of rice and A. besseyi
By comparing the RNA-Seq data of rice plants inoculated with A. bessieyi and that of healthy rice plants, the DEGs of 21 kinase responsive genes and 31 transcription factor genes were identified (FDR < 0.05, Log2FC ≥ 1, or Log2FC ≤ − 1). Identified differentially expressed kinases and transcription factors appeared to be involved in the interaction between rice and A. besseyi.
Ten of 21 differentially expressed kinase responsive genes contained NBS-LRR. At DAI3, only OS02G0156600 (receptor-like protein 3) was upregulated. At DAI7, four DEGs, including OS02G0156600, were upregulated and six were downregulated. Among these 21 differentially expressed kinase responsive genes, OS01G0741200 (lysM domain receptor-like protein 3) of the lysM domain receptor-like kinase (LysM-RLK) family was upregulated at DAI3 and downregulated at DAI7 (Table S7).
Thirty-one differentially expressed transcription factor genes were identified as belonging to 15 families. Among those, three were upregulated at DAI 1, five were upregulated and one was downregulated at DAI 3, and eight were upregulated and 20 were downregulated at DAI7. Five, four, six, and four of the 31 DEGs belonged to WRKY, Basic helix-loop-helix (bHLH), MYB, and bZIP families, respectively. Among the five DEGs of the WRKY family, OS09G0417800 (OsWRKY62) was upregulated at DAI1, DAI3, and DAI7; OS02G0770500 (OsWRKY47) and OS11G0117400 (OsWRKY67) were upregulated at DAI3 and DAI7; and OS01G0182700 (OsWRKY13) and OS02G0698800 (OsWRKY14) were downregulated at DAI7. Among the four DEGs of the bHLH family, OS01G0952800 was upregulated at DAI1; OS01G0575200 (OsbHLH041) was significantly upregulated at DAI1 and DAI7; OS01G0159800 (OsbHLH107) was upregulated at DAI3 and DAI7; and OS02G0315600 (OsbHLH144) was downregulated at DAI7. The six DEGs of the MYB family were annotated only at DAI7, in which OS01G0975300(OsMYB59) and OS02G0168200 were upregulated while the other four were significantly downregulated. OS01G0859300 (bZIP transcription factor ABI5 homolog) in the four DEGs of the bZIP family were upregulated at DAI3 and DAI7 (Table S8).
A Mapman analysis was performed to investigate DEGs (FDR < 0.05, Log2FC ≥ 1 or Log2FC ≤ − 1) involved in the interactions between rice and A. besseyi (Table S9-S11, Fig.5). The results showed that the numbers of DEGs and their related pathways increased with the extension of the nematode infection time. At DAI1, eight DEGs involved in seven pathways were annotated, including an upregulated gene OS02G0756200 (protein exordium) and a downregulated gene OS02G0466400 (inositol-tetrakisphosphate 1-kinase 4-like), which were involved in signaling pathway. At DAI3, ten DEGs involved in ten pathways were annotated, including OS01G0326300 (peroxidase 1) of the antioxidant pathway. At DAI7, 104 DEGs involved in 18 pathways were annotated, including 20 downregulated genes of cell wall-related pathways whose functions were cell wall synthesis, degradation, and deformation.

Mapman visualization of defense response differentially expressed genes (FDR < 0.05, Log2FC ≥ 1 or Log2FC ≤ −1) in rice at three infection periods of Aphelenchoides besseyi. The ranges of the upregulated and downregulated genes are shown in red and blue colors, respectively. The Log2FC are shown in the scale bar. DAI1, DAI3, and DAI7: rice plants at 1 day, 3 days, and 7 days after infection, respectively
The PR gene OS01G0516200 was downregulated at DAI1, DAI3, and DAI7, and the downregulated range at DAI7 was greater than that at DAI1 and DAI3. Among all three time points, upregulated genes of the SA and JA pathways and downregulated genes of the ET pathway were annotated and were involved in hormone signaling. Moreover, an upregulated gene OS01G0797600 (ethylene- responsive transcription factor 8) involved in the ET pathway was annotated at DAI7. Other DEGs were also annotated including those involved in auxins and brassinosteroid pathways of hormone signaling.
Comparative analysis of the transcriptomes of rice infected with three nematode species
Aphelenchoides besseyi and H. oryzae were both migratory nematodes, and had similar infection strategies, which were different from sedentary nematode M. graminicola. DEGs (FDR < 0.05, Log2FC ≥ 1 or Log2FC ≤ −1) retrieved from RNA sequencing in this study were compared with the reported DEGs of rice plants infected with H. oryzae and M. graminicola (Kyndt et al. 2012a). At DAI3, three overlapping DEGs were identified between rice plants infected with A. besseyi and H. oryzae: OS01G0326300 (Peroxidase 1), OS03G0664800 (N-acetyltransferase p20), and OS04G0288100 (germin-like protein 8–14); one overlapping DEG were identified between rice plants infected with A. besseyi and M. oryzae: OS11G0702100 (xylanase inhibitor protein 1-like); no overlapping DEGs were identified between rice plants infected with H. oryzae and M. graminicola. At DAI7, 11 overlapping DEGs were identified between rice plants infected with A. besseyi and H. oryzae; 62 overlapping DEGs were identified between rice plants infected with A. besseyi and M. graminicola; five overlapping DEGs were identified between rice plants infected with H. oryzae and M. graminicola (Fig. 6). The number of overlapping DEGs between rice plants infected with A. besseyi and H. oryzae was much fewer than that between rice plants infected with A. besseyi and M. graminicola, which was because the DEG numbers differed in 1630 genes of A. besseyi, and 995 genes of M. graminicola, but only 114 genes of H. oryzae. Four overlapping DEGs identified in rice plants infected with three different nematodes at DAI7 were OS06G0101600 (plastocyanin, chloroplastic-like), OS03G0778100 (photosystem I reaction center subunit III, chloroplastic), OS07G0141400 (oxygen-evolving enhancer protein 2, chloroplastic) and OS01G0600900 (chlorophyll a-b binding protein 2, chloroplastic-like), and those genes were all involved in photosynthesis. These four genes were downregulated in rice infected with A. besseyi and H. oryzae, but were upregulated in rice infected with M. graminicola.

Graphic representation of all intersections illustrating the differentially expressed genes retrieved from the transcriptome of rice plants infected with Aphelenchoides besseyi, Hirschmanniella oryzae (Kyndt et al. 2012a), and Meloidogyne graminicola (Kyndt et al. 2012a). The black bars indicate the intersections among the transcriptomes of rice plants infected with A. besseyi, H. oryzae, and M. gramminicola. The set size represents the number of DEGs at each condition (genotype/DAI) and the black dots, their intersections. Ab: A. besseyi. Ho: H. oryzae. Mg: M. graminicola. DAI3 and DAI7: rice plants at 3 days and 7 days after infection, respectively
OS09G0417800 (OsWRKY62) was identified to be an overlapping and upregulated DEG between rice plants infected with A. besseyi and H. oryzae at both DAI3 and DAI7. OS07G0539900 (glucan endo-1,3-beta-glucosidase 3) was identified to be an overlapping DEG among rice plants infected with A. besseyi at DAI3, H.oryzae at DAI3, and M. graminicola at DAI7. OS01G0946700 (glucan endo-1,3-beta- glucosidase GV), OS10G0515900 (cytochrome P450 89A2), and OS01G0159800 (putative transcription factor bHLH107) were identified in rice plants infected with H. oryzae at DAI3, H. oryzae at DAI7, and M. graminicola at DAI7, respectively, while these DEGs were identified to be overlapping genes in rice plants infected with A. besseyi at both DAI3 and DAI7. OS06G0726200 (chitinase 1-like), OS02G0756200 (protein EXORDIUM) and OS01G0974200 (metallothionein-like protein 2B), and OS04G0308600 were identified in rice plants infected with A. besseyi at DAI3, A. besseyi at DAI7, and H. oryzae at DAI3, respectively, while these DEGs were identified to be overlapping genes in rice plants infected with M. graminicola at both DAI3 and DAI7; and OS02G0756200 (chitinase 1-like) was identified to be upregulated in rice plants infected with A. besseyi at DAI3, and M. graminicola at both DAI3 and DAI7. No overlapping DEGs were identified between rice plants infected with A. besseyi and M. graminicola, or between rice plants infected with H. oryzae and M. graminicola, at both DAI3 and DAI7 (Fig. 6).
Validation of RNA-Seq results by qPCR
Seventeen DEGs of putative kinase responsive genes, transcription factors, and photosynthesis-related genes were selected for qPCR. qPCR results revealed that most of the selected DEGs had similar expression patterns but different expression levels compared with the RNA-Seq results (Table 4). OS00G0117400 (OsWRKY67) and OS02G0156600 (receptor-like protein 3) were significantly upregulated at DAI1 by qPCR (p < 0.05), while they were upregulated but not significantly by RNA-Seq. OS11G0242800 was significantly downregulated at DAI1 and DAI3 by qPCR (p < 0.05), while there was no significant change by RNA-Seq. Overall, qPCR basically validated the results that were revealed by RNA-Seq analysis.
Discussion
This study demonstrated that rice plant defense pathways changed significantly after A. bessey infection, including the upregulated SA and JA pathways and a downregulated ET pathway, by detecting the expression patterns of defense-related genes via RNA-Seq. It had been reported that the defense responses of rice differed between the infections of migratory and sedentary nematodes in the early stage. After H. oryzae infection, the SA, JA, and ET pathways were upregulated. After M. graminicola infection, the SA and ET pathways were downregulated, and genes involved in the JA pathway were both upregulated and downregulated. Therefore, migratory nematodes induced plant defense–related pathways, while sedentary nematodes suppressed those in different modes (Kyndt et al. 2012a, b). This study obtained the same result that the SA and JA pathways of rice defense responses were upregulated after A. besseyi infection. In addition, A. besseyi also belonged to the migratory nematodes. However, the change in the ET pathway in rice infected with A. besseyi was different from that infected with H. oryzae, which might be due to the different parasite parts that were infected: A. besseyi infected rice shoots and H. oryzae infected roots. Further studies should be carried out.
Plant kinases played a crucial role in the signal recognition and transmission of plant defense to pathogen attack. Kinase receptors could recognize signaling molecules, then transport signals to down streams, and activate transcription factor expression to regulate biochemical programs in response to plant-pathogen interactions (Wuriyanghan et al. 2007). There were more than 1500 kinase responsive genes in rice. Some of those were resistance genes, that contained the NBS-LRR domain and could induce self-defense by recognizing pathogen avirulence genes (Tariq et al. 2018). In our study, four upregulated and six downregulated DEGs containing the NBS-LRR domain were identified in rice plants infected with A. besseyi and were predicted to be involved directly or indirectly in the interaction of rice and A. besseyi. OS02G0156600 was significantly upregulated at DAI3 and DAI7, which suggests it played an important role in plant-nematode interactions and deserves further study. Proteins containing the lysine motif (LysM domain) were receptors of pathogen signals in plants (Liu et al. 2012). LYP4 and LYP6 were homologous proteins containing the LysM domain of rice. They played a dual role in the perception of peptidoglycan and chitin in the innate immunity of rice. These two LYP genes could selectively combined with peptidoglycan or chitin in vitro. Silencing either of these two LYP genes could specifically disturb the defense response that is induced by peptidoglycan or chitin in rice (Zhu et al. 2018). In our study, a LysM gene, OS01G0741200 (lysM domain receptor-like protein 3) was identified, and was upregulated significantly in the early infection stage. This LysM gene might be involved in chitin-related pathways. At the same time, it was also found that the upregulated and enriched GO term was involved in chitin-related pathways in the early infection stage. It had been reported that host chitin-related pathways were upregulated by infections of cyst nematodes and root knot nematodes (Bagnaresi et al. 2013; Kong et al. 2015). It was suggested that the kinase responsive gene OS01G0741200 might play an important role in pathogen recognition and in the activation of chitin-induced defense response against the attack of A. besseyi. Interestingly, OS02G0756200 (chitinase 1-like) was identified to be upregulated in rice plants infected with migratory nematode A. besseyi at DAI3, and sedentary nematode M. graminicola at both DAI3 and DAI7, but not a DEG in plants infected with another migratory nematode H. oryzae. Chitin-induced defense response of rice infected with A. besseyi may have some similarity with that infected M. graminicola.
Rice could counter various biotic or abiotic stresses by regulating transcription factor genes. WRKY transcription factor genes, which contain W-box domains, played an important role in pathogen perception and signal amplification. They could regulate the expression of disease-resistant genes and, afterward, induce plant defense pathways directly or indirectly (Jimmy and Babu 2015; Jiang et al. 2017). OsWRKY47 and OsWRKY67 are positive transcription factors regulating the rice defense response, and both were related to rice resistance to rice blast disease (Wei et al. 2013; Vo et al. 2018). In this study, the transcription factors OsWRKY47 and OsWRKY67 were identified to be significantly upregulated at the early and late stages of nematode infection. This result suggested that the defense mechanism of rice responding to migratory nematode A. besseyi might be similar to that of the rice blast pathogen. Kyndt et al. (2012b) reported that the mechanism of rice response to the infection of another migratory plant nematode, H. oryzae, was similar to that of the rice blast pathogen. Thus, this defense response mechanism might generally exist in the interaction of host plants and migratory plant nematodes. OsWRKY62 was a negative regulator of the defense response in rice, and its overexpression suppressed the expression of defense genes (Peng et al. 2008). However, OsWRKY62 was present and was significantly upregulated in rice infected with A. besseyi and in rice infected with H. oryzae at different stages, which suggested that migratory nematodes could suppress the host defense response by upregulating this gene.
Pathogens had an impact on photosynthesis, and often leaded to a decrease in photosynthesis of chlorotic and necrotic areas and to decrease in photosynthetic assimilate production (Berger et al. 2007; Göhre et al. 2012). The decrease in photosynthesis in response to infection might be an active process as part of the plant defense program to limit carbon source availability for the pathogen or result from a prioritization of metabolic processes in favor or defense reaction (Bolton 2009). Recently, studies have shown that the chloroplast was a key component of early immune responses in plants. Chloroplasts could switch off photosynthesis, navigate to the site of infection, and participate in the defense responses against pathogens infection. Most pathogens could secrete effectors to target the chloroplast and inhibit or destroy plant photosynthesis (De Torres Zabala et al. 2015; Toufexi et al. 2019). So far, no study has been reported that nematodes could secrete effectors to target the chloroplast. But it had been reported that the migratory nematodes H. oryzae and B. xylophilus could inhibit the photosynthesis of hosts (Kyndt et al. 2012a; Zhang et al. 2014). Our results showed rice photosynthesis-related pathways were inhibited downregulated after A. besseyi infection, including photosynthesis antenna proteins and photosynthesis-related components of the chloroplast, thylakoid, and plastid. When comparing the RNA-Seq data of rice infected with A. besseyi, H. oryzae, and M. graminicola, four overlapping DEGs involved in photosynthesis were identified. These four DEGs were downregulated in rice infected with the migratory nematodes A. besseyi and H. oryzae, but were upregulated in rice infected with sedentary nematode M. graminicola. In addition, host photosynthesis of infecting sites was also found to be upregulated in response to the attack of another sedentary nematode H. schachtii (Kyndt et al. 2012a; Szakasits et al. 2009). Therefore, it suggested that plant nematodes could also affect photosynthesis of host plants as a plant pathogen, and different parasitic strategies of plant nematodes affected the photosynthesis of the host in different ways.
This study first reported rice defense-related gene expression patterns and rice transcriptomes at different stages after A. besseyi infection. The result showed that the defense response pathways of rice changed obviously in the early stage of A. besseyi infection, which included upregulated SA and JA pathways and a downregulated ET pathway. It also indicated the infection strategy of both A. besseyi and the migratory nematode H. oryzae was similar to that of fungal pathogen Magnaporthe grisea. Moreover, OS09G0417800 (OsWRKY62) was supposed to play an important role in the interaction of migratory nematodes and rice. Furthermore, it suggested that the photosynthesis of the host may be suppressed by infections of migratory plant nematodes. However, further studies were required to verify the results. The results of this study provide an interesting starting point to elucidate the mechanism of the interaction of rice and rice white tip nematodes, as well as the host and migratory plant nematodes. To verify the findings of this paper, functional analysis was required. Techniques of molecular biology including yeast two hybridation, transgenic plants or mutants should be considered in future studies.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. Sequences were deposited in SRA (BioProject Accession: PRJNA541104).
References
Abad P, Gouzy J, Aury JM, Castagnone-Sereno P, Danchin EGJ, Deleury E, Perfus-Barbeoch L, Anthouard V, Artiguenave F, Blok VC, Caillaud MC, Coutinho PM, Dasilva C, Luca FD, Deau F, Esquibet M, Flutre T, Goldstone JV, Hamamouch N, Hewezi T, Jaillon O, Jubin C, Leonetti P, Magliano M, Maier TR, Markov GV, McVeigh P, Pesole G, Poulain J, Robinson-Rechavi M, Sallet E, Se’gurens B, Steinbach D, Tytgat T, Ugarte E, Ghelder CV, Veronico P, Baum TJ, Blaxter M, Bleve-Zacheo T, Davis EL, Ewbank JJ, Favery B, Grenier E, Henrissat B, Jones JT, Laudet V, Maule AG, Quesneville H, Rosso MN, Schiex T, Smant G, Weissenbach J, Wincker P (2008) Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat Biotechnol 26
Akker SED, Lilley CJ, Canchin EGJ, Rancurel C, Cock PJA, Urwin PE, Jones JT (2014) The transcriptome of Nacobbus aberrans reveals insights into the evolution of sedentary endoparasitism in plant-parasitic nematodes. Genome Biol Evol 6(9):2181–2194
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106
Anders S, Pyl PT, Huber W (2015) HTSeq. a Python framework to work with high-throughput sequencing data. Bioinformatics 31(2):166–169
Bagnaresi P, Sala T, Irdani T, Scotto C, Lamontanara A, Beretta M, Rotino GL, Sestili S, Cattivelli L, Sabatini E (2013) Solanum torvum responses to the root-knot nematode Meloidogyne incognita. BMC Genomics 14:540
Barnes SN, Wram CL, Mitchum MG, Baum TJ (2018) The plant-parasitic cyst nematode effector GLAND4 is a DNA-binding protein. Mol Plant Pathol 19(10):2263–2276
Bauters L, Haegeman A, Kyndt T, Gheysen G (2014) Analysis of the transcriptome of Hirschmanniella oryzae to explore potential survival strategies and host–nematode interactions. Mol Plant Pathol 15(4):352–363
Berger S, Sinha AK et al (2007) Plant physiology meets phytopathology: plant primary metabolism and plant–pathogen interactions. J Exp Bot 58(15/16):4019–4026
Bolton MD (2009) Primary metabolism and plant defense—fuel for the fire. Mol Plant-Microbe Interact 22:487–497
Cheng X, Xiang Y, Xie H, Xu CL, Xie TF, Zhang C, Li Y (2013) Molecular characterization and functions of fatty acid and retinoid binding protein gene (Ab-far-1) in Aphelenchoides besseyi. PLoS One 8:e66011
Chen CJ, Xia R, Chen H, He YH (2018) TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. bioRix. https://doi.org/10.1101/289660
De Torres Zabala M, Littlejohn G, Jayaraman S, Studholme D, Bailey T, Lawson T, Tillich M, Licht D, Bölter B, Delfino L, Truman W, Mansfield J, Smirnoff N, Grant M (2015) Chloroplasts play a central role in plant defence and are targeted by pathogen effectors. Nat Plants 1:15074
Göhre V, Jones AME, Sklenář J, Robatzek S, Weber APM (2012) Molecular crosstalk between PAMP-triggered immunity and photosynthesis. MPMI 25(8)
Haegeman A, Joseph S, Gheysen G (2011) Analysis of the transcriptome of root lesion nematode Pratylenchus coffeae generated by 454 sequencing technology. Mol Biochem Parasitol 178:7–14
Haegeman A, Bauters L, Kyndt T, Rahman MM, Gheysen G (2013) Identification of candidate effector genes in the transcriptome of the rice root knot nematode Meloidogyne graminicola. Mol Plant Pathol 14(4):379–390
Jacob J, Mitreva M, Vanholme B, Gheysen G (2008) Exploring the transcriptome of the burrowing nematode Radopholus similis. Mol Gen Genomics 280:1–17
Jiang J, Ma S, Ye N, Jiang M, Cao J, Zhang J (2017) WRKY transcription factors in plant responses to stresses. J Integr Plant Biol 59:86–101
Jimmy JL, Babu S (2015) Role of OsWRKY transcription factors in rice disease resistance. Trop Plant Pathol 40:355–361
Karmakar S, Molla KA, Chanda PK, Sarkar SN, Datta SK, Datta K (2016) Green tissue-specific co-expression of chitinase and oxalate oxidase 4 genes in rice for enhanced resistance against sheath blight. Planta 243:115–130
Karssen G, Groza M (2018) First report of the plant-parasitic nematode Aphelenchoides besseyi (Nematoda: Aphelenchoididae) on rice in Romania. Bull OEPP/EPPO Bull 48(2):254–255
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4)
Kong LA, Wu DQ, Huang WK, Peng H, Wang GF, Cui JK, Liu SM, Li ZG, Yang J, Peng DL (2015) Large-scale identification of wheat genes resistant to cereal cyst nematode Heterodera avenae using comparative transcriptomic analysis. BMC Genomics 16:801
Kyndt T, Denil S, Haegeman A, Trooskens G, Bauters L, Van CW, De MT, Gheysen G (2012a) Transcriptional reprogramming by root knot and migratory nematode infection in rice. New Phytol 196(2012a):887–900
Kyndt T, Hofte M, Gheysen L (2012b) Comparing the defence-related gene expression changes upon migratory and sedentary nematode attack in rice. Plant Biol:73–82
Lilley CJ, Kyndt T, Gheysen G (2011) Nematode resistant GM crops in industrialised and developing countries. In: Jones JT, Gheysen G, Fenoll C (eds) Genomics and molecular genetics of plant–nematode interactions. Springer, Heidelberg López-Martínez, N., Colinas-L 517–541
Lin MS, Ding XF, Wang ZM, Zhou FM, Lin N (2004) Description of Aphelenchoides besseyi from abnormal rice with ‘small grains and erect panicles’ symptom in China. Rice Sci 12(4):289–294
Liu B, Li JF, Ao Y, Wang J, Wang HB (2012) Lysin motif-containing proteins LYP4 and LYP6 play dual roles in peptidoglycan and chitin perception in rice innate immunity. Plant Cell 24(8):3406–3419
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21(19):3787–3793
Mota APZ, Vidigal B, Danchin EGJ, Togawa RC, Leal-Bertioli SCM, Bertioli DJ, Araujo ACG, Brasileiro ACM, Guimaraes PM (2018) Comparative root transcriptome of wild Arachis reveals NBS-LRR genes related to nematode resistance. BMC Plant Biol 18:159
Nahar K, Kyndt T, Vleesschauwer DD, Höfte M, Gheysen G (2011) The Jasmonate pathway is a key player in systematically induced defense against root nematodes in rice. Plant Physiol 157:305–316
Peng Y, Bartley LE, Chen XW, Dardick C, Chern M, Ruan R (2008) OsWRKY62 is a negative regulator of basal and Xa21-mediated defense against Xanthomonas oryzae pv. oryzae in Rice. Mol Plant 3(1):446–458
Petitot AS, Dereeper A, Agbessi M, Da CS (2016) Dual RNA-Seq reveals Meloidogyne graminicola transcriptome and candidate effectors during the interaction with rice plants. Mol Plant Pathol 17(6):860–874
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:36
Santos CS, Pinheiro M, Silva AI, Egas C, Vasconcelos MW (2012) Searching for resistance genes to Bursaphelenchus xylophilus using high throughput screening. BMC Genomics 13:599
Swiecicka M, Filipecki M, Lont D, Van VJ, Goverse A, Bakker J, Helder J (2009) Dynamics in the tomato root transcriptome on infection with the potato cyst nematode Globodera rostochiensis. Mol Plant Pathol 10(4):487–500
Szakasits D, Heinen P, Wieczorek K, Hofmann J, Wagner F, Kreil DP (2009) The transcriptome of syncytia induced by the cyst nematode Heterodera schachtii in Arabidopsis roots. Plant J 57:771–784
Tariq R, Wang CL, Qin TF, Xu F, Tang Y, Gao Y, Ji Z, Zhao K (2018) Comparative transcriptome profiling of rice near-isogenic line carrying Xa23 under infection of Xanthomonas oryzae pv. Oryzae. Int J Mol Sci 19:717
Thim O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37(6):914–939
Thorpe P, Mantelin S, Cock PJA, Blok VC, Coke MC, Akker SEV, Guzeeva DE, Lilley CJ, Smant G, Reid AJ, Wright KM, Urwin PE (2014) Genomic characterisation of the effector complement of the potato cyst nematode Globodera pallida. BMC Genomics 15:923
Toufexi A, Duggan C, Pandey WP, Savage Z, Segretin ME, Yuen LH, Gaboriau D, Leary A, Khandare V, Ward AD, Botchway SW, Bateman BC, Pan I, Schattat MH, Sparkes I, Bozkurt T (2019). Chloroplasts navigate towards the pathogen interface to counteract infection by the Irish potato famine pathogen. BioRxiv. https://doi.org/10.1101/516443
Trapnell CL, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2014) Differential gene and transcript expression analysis of RNA-Seq experiments with TopHat and cufflinks. Nat Protoc 2513
Vo KTX, Kim CY, Hoang TV, Lee SK, Shirsekar G, Seo YS, Lee SW, Wang GL, Jeon JS (2018) OsWRKY67 plays a positive role in basal and XA21-mediated resistance in rice. Front Plant Sci 8:2220
Wang F, Li DL, Wang ZY, Dong A, Liu L, Wang B, Chen Q, Liu X (2014) Transcriptomic analysis of the rice white tip nematode, Aphelenchoides besseyi (Nematoda: Aphelenchoididae). PLoS One 9(3):1–10
Wang DW, Peng XF, Xie H, Xu CL, Cheng DQ, Li JY, Wu WJ, Wang K (2016) Arabidopsis thaliana as a suitable model host to research on interactions between plant and foliar nematodes, parasites of plant shoot. Sci Rep 6:38286
Wang DW, Xu CL, Ding SW, Huang X, Cheng X, Zhang C, Chen C, Xie H (2018) Identification and function of FAR protein family genes from a transcriptome analysis of Aphelenchoides besseyi. Bioinformatics:1–8
Wei T, Qu B, Li JB, Zhao Y, Guo D, Zhu Y, Chen Z, Gu H, Li C, Qin G, Qu LJ (2013) Transcriptional profiling of Rice early response to Magnaporthe oryzae identified OsWRKYs as important regulators in rice blast resistance. PLoS One 8(3):e59720
Wuriyanghan H, Chen LJ, Dong Y, Lei G, Jia FX, Zhang JS, Chen SY (2007) Rice receptor-like kinase OsSI-RLK2 inhibits internode elongation. Chin Sci Bull 19(52):2657–2663
Young MD, Wakefiled MJ, Smyth GK, Oshlack A (2010) Method gene ontology analysis for RNA-Seq: accounting for selection bias. Genome Biol 11:R14
Zhang H, Huan XF, Xu HC (2014) Changes of photosynthesis and antioxidant enzyme activities at different stage naturally infected by Bursaphelenchus xylophilus. J Environ Entomol 36(2):139–144 (In Chinese)
Zhu Q, Zhang XL, Nadir S, Doku HA, Chen LJ, Li DX (2018) Cloning and overexpression strain construction of a LysM domain-containing gene OsEMSA1 in Rice. Mol Plant Breed (In Chinese) 1(16):22–30. https://doi.org/10.13271/j.mpb.016.000022
Funding
This work was funded by National Natural Science Foundation of China (No.31871939 and 31371920) and Doctoral innovative talents (domestic training) Cultivation Project of South China Agricultural University in 2019 (No. CX2019N048).
Author information
Authors and Affiliations
Contributions
HX and HLW designed the experiments; HLW, SHY, ML, SWD, and JYL performed the experiments; HLW, SWD, JYL, and CLX analyzed the data; HLW, HX, and CLX wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
Animals were treated in strict accordance with the Animal Ethics Procedures and Guidelines of the People’s Republic of China. All animal procedures were approved by the Animal Ethics Committee of the South China Agricultural University.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Table S1-S2
(DOC 50 kb)
Table S3-S5
(XLSX 142 kb)
Table S6
(XLSX 11 kb)
Table S7
(XLSX 15 kb)
Table S8
(XLS 42 kb)
Table S9-S11
(XLS 178 kb)
Rights and permissions
About this article

Cite this article
Wang, HL., Yang, SH., Lv, M. et al. RNA-Seq revealed that infection with white tip nematodes could downregulate rice photosynthetic genes. Funct Integr Genomics 20, 367–381 (2020). https://doi.org/10.1007/s10142-019-00717-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-019-00717-9