
Abstract
Plant defensins are small (45 to 54 amino acids) positively charged antimicrobial peptides produced by the plant species, which can inhibit the growth of a broad range of fungi at micro-molar concentrations. These basic peptides share a common characteristic three-dimensional folding pattern with one α-helix and three β-sheets that are stabilized by eight disulfide-linked cysteine residues. Instead of using two single-gene constructs, it is beneficial when two effective genes are made into a single fusion gene with one promoter and terminator. In this approach, we have linked two plant defensins namely Trigonella foenum-graecum defensin 2 (Tfgd2) and Raphanus sativus antifungal protein 2 (RsAFP2) genes by a linker peptide sequence (occurring in the seeds of Impatiens balsamina) and made into a single-fusion gene construct. We used pET-32a+ vector system to express Tfgd2-RsAFP2 fusion gene with hexahistidine tag in Escherichia coli BL21 (DE3) pLysS cells. Induction of these cells with 1 mM IPTG achieved expression of the fusion protein. The solubilized His6-tagged recombinant fusion protein was purified by immobilized-metal (Ni2+) affinity column chromatography. The final yield of the fusion protein was 500 ng/μL. This method produced biologically active recombinant His6-tagged fusion protein, which exhibited potent antifungal action towards the plant pathogenic fungi (Botrytis cinerea, Fusarium moniliforme, Fusarium oxysporum, Phaeoisariopsis personata and Rhizoctonia solani along with an oomycete pathogen Phytophthora parasitica var nicotianae) at lower concentrations under in vitro conditions. This strategy of combining activity of two defensin genes into a single-fusion gene will definitely be a promising utility for biotechnological applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Defensins are superfamily of antimicrobial peptides (AMPs), which are found in all living organisms. These are most studied among the plant AMPs (Thomma et al. 2002; Lay and Anderson 2005; Carvalho and Gomes 2009; Stotz et al. 2009) and are involved in innate immunity. Innate immunity is a much more widespread ancient defense strategy involving the production of AMPs (Boman 1995).
Plant defensins are typically highly basic proteins of small molecular weight (<10 kDa) with an even number of cysteine residues (typically 4, 6, or 8) that stabilize the protein structure through formation of disulfide bridges and provide structural and thermodynamic stability to the protein (Hancock and Lehrer 1998; Lay and Anderson 2005; Benko-Iseppon et al. 2010). They have been isolated from all parts of the plants including seeds, pods, fruits, flower parts, leaves, tubers, roots, and stems (Thomma et al. 2002; Lay and Anderson 2005; Wong et al. 2007; Carvalho and Gomes 2009). Most of the plant defensins isolated are seed derived and have been characterized at the molecular, biochemical and structural levels (Terras et al. 1993; Osborn et al. 1995; Thomma et al. 2002; Lay and Anderson 2005; Song et al. 2005).
They are structurally related to insect defensins (Broekaert et al. 1995) and even more to the antifungal insect peptide drosomycin (Landon et al. 1997). Among defensins of different plant species, homology in amino acid sequences is rather low except for the conservatively located cysteine residues. At the same time, similarity in primary structure among families is considerably higher than it is between defensins of plants belonging to different families (Odintsova et al. 2007). Despite differences in the amino acid sequence, there is a similarity in three-dimensional structures of these proteins represented by one α-helix and three β-strands.
Plant defensins are secretory proteins which are produced as precursors with an endoplasmic reticulum signal sequence and a mature defensin domain (Broekaert et al 1997). Majority of these proteins display antimicrobial activity against a broad range of fungi (Broekaert et al. 1995; Thomma et al. 2002; Lay and Anderson 2005; Wong et al. 2007; Vijayan et al. 2008). Apart from antimicrobial activity, plant defensins have also been shown to have other biological activities like an α-amylase inhibitor (Bloch and Richardson 1991; Pelegrini et al. 2008), protease inhibitor (Wijaya et al. 2000; Melo et al. 2002; Zhang and Falla 2009), 12 protein translation inhibitor (Mendez et al. 1990, 1996; Chen et al. 2005), growth inhibitor of roots of parasitic plants (Zelicourt et al. 2007), ion channel blockers (Kushmerick et al. 1998; Spelbrink et al. 2004) and mediation of metal tolerance (Mirouze et al. 2006). These diverse biological activities displayed by the plant defensins are believed to be caused by high variation in primary sequences (Thomma et al. 2002; Lay and Anderson 2005; Carvalho and Gomes 2009).
The exact mechanism of action of plant defensins is not completely understood, but there is evidence that plant defensins interact with sphingolipids on fungal cell membrane and integrate into the phospholipid bilayer of the membrane, resulting in the permeabilization and membrane destabilization (Thevissen et al. 2003, 2004). It was recently demonstrated that plant defensins can also be internalized into the cytoplasm and be involved in specific interactions with intracellular targets (Lobo et al. 2007; Van der Weerden et al. 2008). With these proteins, fungal inhibition occurs through the mechanism of ion efflux.
Most of the bacterially expressed plant defensins were shown to be active as antifungal peptides (Xu and Reddy 1997; Park et al. 2002; Olli and Kirti 2006; Hui et al. 2007; Olli et al. 2007; Solis et al. 2007; Vijayan et al. 2008).
Tfgd2 and RsAFP2 are potent antifungal proteins which belong to the family of small AMPs. Antifungal activity of Tfgd2 was tested in vitro against Rhizoctonia solani and Fusarium moniliforme fungal pathogens (Olli et al. 2007). Similarly, RsAFP2 was also reported as an efficient antifungal protein (Terras et al. 1995; Parashina et al. 2000; Li et al. 2011).
The present study deals with recombinant expression of Tfgd2 and RsAFP2 defensins (linked with each other by a linker peptide sequence from the seeds of Impatiens balsamina) as a single-fusion protein in Escherichia coli systems, with the aim to study their combinational role in exhibiting antifungal activity against Botrytis cinerea, F. moniliforme, Fusarium oxysporum, Phaeoisariopsis personata and R. solani fungal pathogens along with an oomycete pathogen, Phytophthora parasitica var nicotianae under in vitro conditions.
Materials and methods
Construction of fusion gene (Tfgd2-RsAFP2) expression vector
Fusion gene (Tfgd2-RsAFP2; GenBank accession number KF498667) construct is made up of Tfgd2 (Trigonella foenum-graecum defensin 2; GenBank accession number AY227192) and RsAFP2 (Raphanus sativus antifungal protein 2; GenBank accession number U18556) genes, which are linked with each other by a linker peptide sequence, which is fourth internal propeptide from naturally occurring IbAMP polyprotein precursor from the seeds of I. balsamina. The complete open reading frame sequence coding for the Tfgd2 was amplified by PCR with Tfgd2-specific forward primer (5′GGGGGTACCATGGAGAAGAAATCACTAGCT 3′) and reverse primer along with linker peptide sequence (5′GTCTTGTGGCGTCGCCACCTCGTCTGCTGCGTTGCTACATCTTTTAGTACACCAGCA 3′) using cDNA sequence of Tfgd2 as a template with the following conditions. Initial denaturation was performed at 94 °C for 4 min followed by 29 cycles at 94 °C for 1 min (denaturation), 55 °C for 45 s (annealing) and 72 °C for 2 min (extension). Final extension was carried out at 72 °C for 10 min resulted in the amplification of 255 bp PCR product of Tfgd2 along with linker peptide. In a similar way, RsAFP2 was amplified by PCR with RsAFP2 forward primer along with linker peptide sequence (5′AGCAACGCAGCAGACGAGGTGGCGACGCCACAAGACATGGCTAAGTTTGCTTCTATC 3′) and reverse primer (5′TTAACAAGGGAAATAACAGATACA 3′) using cDNA sequence of RsAFP2 as a template with the following conditions. Initial denaturation was performed at 94 °C for 4 min followed by 29 cycles at 94 °C for 1 min (denaturation), 59.7 °C for 45 s (annealing) and 72 °C for 2 min (extension). Final extension was carried out at 72 °C for 10 min resulted in the amplification of 279 bp PCR product of RsAFP2 along with linker peptide. Followed by another PCR using first and second PCR amplified products as templates with Tfgd2 forward and RsAFP2 reverse primers using the following cycling conditions; 94 °C for 4 min initial denaturation followed by 29 cycles at 94 °C for 1 min (denaturation), 54.5 °C for 50 s annealing and 72 °C for 2 min (extension). Final extension was conducted at 72 °C for 15 min resulted in the amplification of 500 bp fusion gene with Tfgd2 and RsAFP2 genes with intervening linker peptide sequence.
This 500-bp fusion gene was cloned in pTZ57R/T vector and clones were screened by double digestion with KpnI and BamHI enzymes. Fusion gene positive clones were finally confirmed by commercial sequencing. Later, this gene was released from pTZ57R/T vector by double digestion with KpnI and BamHI enzymes and cloned at the same sites of the T7 polymerase expression vector pET-32a+ vector (Novagen, USA) and transformed into BL21 (DE3) pLysS E. coli cells.
Overexpression and purification of His6-tagged recombinant fusion protein (Tfgd2-RsAFP2) in E. coli
E. coli BL21 (DE3) pLysS cells carrying the fusion gene were cultured overnight at 37 °C in 5 mL of Luria-Bertani (LB) medium under vigorous agitation (200 rpm). This pre-inoculum suspension was then used to inoculate 500 mL of fresh LB selection medium, which was agitated till O.D.600 reached 0.6–0.8. Thereafter, an aliquot of noninduced cells was collected and reserved as a control. Expression of His6-tagged fusion protein was induced in the remaining portion of cells by addition of IPTG to a final concentration of 1 mM. The cells were cultivated at 37 °C for 4 h in the induction medium (i.e., LB selective medium plus IPTG) and afterwards induced cells were pelleted by centrifugation at 5,000 rpm for 10 min at 4 °C.
The pellet was initially washed with fresh 1× PBS buffer. Following that, the first lysis (prolysis) buffer (300 mM NaCl, 50 mM NaHPO4, 10 mM imidazole, 15 % glycerol and 0.1 % SDS) was added and mixed homogeneously. Subsequently, cells were lysed by sonication at 6× pulse for 5 min at 4 °C (every 20-s pulse, a 10-s interval was given). After, obtained lysate was centrifuged at 12,000 rpm for 20 min to remove the debris. The supernatant was mixed with the second lysis buffer (300 mM NaCl, 50 mM NaHPO4 and 10 mM imidazole) along with 2 mL Ni-NTA (Qiagen, Germany) and the volume was made up to 50 mL/L culture of cells and mixed on a rocker at 60 rpm for 1 h at 4 °C. The supernatant mixture was loaded on to the column (Sigma-Aldrich, USA) and the flow-through was collected at the rate of 0.5 mL/min, which was maintained till the end. Finally, the column was washed with 10 vol. of wash buffer (300 mM NaCl, 50 mM NaHPO4, 20 mM imidazole and 15 % glycerol). The recombinant fusion protein was eluted with elution buffer (300 mM NaCl, 50 mM NaHPO4 and 250 mM imidazole), according to the manufacturer's instructions (Qiagen, Germany). Purified protein concentrations were measured according to Bradford's (1976) method. Later, protein samples were resolved on 12 % SDS-polyacrylamide gels according to the method of Laemlli (1970). After electrophoresis, the gel was stained with Coomassie brilliant blue.
Fungal growth inhibition bioassays
The purified recombinant fusion protein (Tfgd2-RsAFP2) was analyzed for its ability to inhibit the in vitro growth of plant pathogenic fungi along with an oomycete pathogen P. parasitica var nicotianae. The fungal pathogens used for the tests were B. cinerea, F. moniliforme, F. oxysporum, R. solani and P. personata. These were obtained from the Directorate of Oilseeds Research, Hyderabad, India; Central Tobacco Research Institute, Rajahmundry, India; and University of Agricultural Sciences, Dharwad and Bangalore, India. Antifungal activity of recombinant fusion protein was tested by both microspectrophotometry and in vitro plate assay methods. Different concentrations of purified fusion protein were prepared by diluting 10 μL of purified protein and pipetted into a 96-well microtitre plate well containing 90 μL of the test fungal spore suspension (∼2.5 × 104 spores/mL) in potato dextrose broth and placed in an incubator at 28 °C. Antifungal activity of individual protein concentration was tested and this experiment was conducted in three replicates. Germination and growth of fungal spores were observed microscopically and optical density (OD) was measured with microplate reader at a wavelength of 595 nm after inoculation for 30 min and 48 h. Control samples were analyzed similarly except that the protein was not added. Growth inhibition values lower than 10 % was not considered as significant (growth inhibition is defined as the ratio of corrected absorbance of the control at 595 nm minus the corrected absorbance of the test sample at 595 nm divided by the corrected absorbance of the control at 595 nm). The corrected absorbance is defined as the absorbance at 48 h minus that at 30 min. IC50 is defined as the concentration of protein, at which 50 % growth inhibition was reached.
Percent inhibition of fungal growth on the plate has been estimated as area of mycelial growth in the absence of antifungal protein minus area of mycelial growth in the presence of antifungal protein/area of mycelial growth in the absence of antifungal protein × 100.
A graph plotting the percent inhibition of fungal growth against the concentration of protein tested was used to determine the IC50 value of fusion protein for different pathogens, i.e., the concentration of protein used to produce 50 % growth inhibition.
For the in vitro plate assay, a piece of agar containing fungal mycelium was inoculated at the center of the potato dextrose agar (Himedia, India) plate with 25 mL of the medium and incubated at 28 °C. When the mycelia reached 6 cm in diameter, four sterile Whatman no. 1 filter paper discs (1 cm diameter) were placed on the plate at equal distance from the center. Purified recombinant fusion protein was added at various concentrations (ranging from 10 to 40 μg/μL) at the center of the discs on the plate. The elution buffer was used as a control. The plates were incubated at 28 °C and were observed periodically till the mycelial growth covered the control discs.
For P. personata conidia germination assay, conidia were collected from infected leaves of diseased plants and were diluted to 50,000 conidia/mL. Different concentrations of purified recombinant fusion protein were taken into cavity slides. As a control, elution buffer was taken as mentioned earlier. Equal concentrations of conidia (50,000 conidia/mL) were added to both control and recombinant protein containing cavity slides. They were observed under microscope after 48 h to check conidia germination
Results
Cloning of fusion gene (Tfgd2-RsAFP2) coding sequence
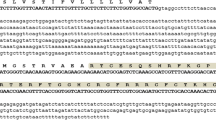
Tfgd2 and RsAFP2 genes were amplified separately by using their specific forward and reverse primers in two individual PCR reactions as represented in Fig. 1a. Finally, third PCR was conducted by using the mixture of the first two PCR reactions amplified products as templates along with Tfgd2 forward and RsAFP2 reverse primers, resulted in the amplification of 500 bp fusion gene (GenBank accession number KF498667) containing Tfgd2 and RsAFP2 genes with intervening linker peptide sequence as represented in Fig. 1b. This gene was cloned in pTZ57R/T-vector by T/A cloning and confirmed by double digestion with KpnI and BamHI enzymes (Supplementary Fig. 1). Finally, gene sequence of the fusion gene was confirmed by commercial sequencing (Fig. 2).

Construction of fusion gene (Tfgd2-RsAFP2) construct. a PCR amplification of Tfgd2 and RsAFP2 genes. Red arrow in lanes A and B represent Tfgd2 amplification; blue arrow in lanes C and D represents RsAFP2 amplification, Lane M represents 1 kb ladder. b PCR amplification of the fusion gene. Lanes E and F represent fusion gene amplification; Lane M represents Lambda EcoRI+HindIII double-digest marker

Nucleotide and deduced amino acid sequences of the fusion gene (Tfgd2-RsAFP2). Sequences of Tfgd2 and RsAFP2 genes were represented in black with intervening linker peptide sequence indicated in red. Starting codons for both the genes were represented in green. There is no terminating codon for Tfgd2 and RsAFP2 terminating codon was represented in purple
Furthermore, this gene was released from pTZ57R/T-vector by KpnI and BamHI double digestion and cloned at the same sites in pET-32a+ expression vector, which contains an inbuilt His6-tag sequence, resulted into a His6-tagged recombinant fusion protein coding sequence (pET/Tfgd2-RsAFP2 construct; Supplementary Fig. 2). The expected size of the His6-tagged recombinant fusion protein is 35 kDa, calculated from deduced amino acid sequence (ExPASY Protein Parameters Tools Analysis, Wilkins et al. 1999).
Bacterial expression and purification of His6-tagged recombinant fusion protein (Tfgd2-RsAFP2)
The hexahistidine (His6)-tagged recombinant fusion protein made up of two defensins namely Tfgd2 and RsAFP2 with intervening 12 amino acid linker peptide was overexpressed in E. coli heterologous system by using pET-32a+ expression system. This overexpressed fusion protein was purified by immobilized-metal (Ni2+) affinity chromatography (IMAC).
To obtain information about the solubility of His6-tagged recombinant fusion protein produced in E. coli, this was expressed in BL21 (DE3) pLysS E. coli cells carrying the pET-32a+-fusion-gene construct with 1 mM IPTG induction. Total cell protein fraction was retrieved from both noninduced and induced bacterial cells lysed under native conditions. The majority of the probable His6-tagged recombinant fusion protein was present within the soluble protein fraction. Then, soluble protein fractions from induced protein fractions were separated by centrifugation. Equal amounts of different protein fractions like 0 h without IPTG induction and 1, 2, 3 and 10 h with 1 mM IPTG induction were analyzed on 12 % SDS-PAGE (Fig. 3). A protein presenting a mass around the predicted molecular weight of the His6-tagged recombinant fusion protein, 35 kDa was present in high amounts within the induced total protein fraction as compared with the noninduced total protein fraction (Fig. 3). By silver staining of the protein gels, it was confirmed that low quantity of the His6-tagged recombinant fusion protein was present in flow through and wash but most of the protein was found in the elute (Supplementary Fig. 3, lanes D and E). Based on these findings, large-scale expression and purification of His6-tagged recombinant fusion protein was conducted by incubation of E. coli culture at 37° C for 4 h of expression with 1 mM IPTG induction. This method resulted in the production of substantial quantities of soluble His6-tagged recombinant fusion protein, which was purified by immobilized metal affinity chromatography. Two of the IMAC eluates revealed that obtained protein was 100 % pure as evident by the presence of one single band with SDS-PAGE analysis (Supplementary Fig. 3). This purified protein was confirmed by western blotting using anti his-tagged rabbit antibodies (Fig. 4). This approach yielded high quality, pure, soluble and denatured His6-tagged recombinant fusion protein (500 ng/μL) from 500 mL of induced E. coli culture.

Induction of recombinant fusion protein (Tfgd2-RsAFP2) with 1 mM IPTG at different time intervals. BL21 (DE3) pLysS cells transformed with pET-32a+vector harboring fusion gene were grown up to 0.6–0.8 OD and induced with 1 mM IPTG. After harvesting cells, total protein was extracted with PBS and analyzed on 12 % SDS-PAGE. Lane A, no detectable protein in 0 h; lanes B, C, D and E, concentration of protein was increased gradually in subsequent intervals of time with IPTG induction that led to expression of 35 kDa fusion protein. Lanes A, 0 h without IPTG induction; B, 1 h with IPTG induction; C, 2 h with IPTG induction; D, 3 h with IPTG induction; E, 10 h with IPTG induction; and M, medium range protein marker

Western blot of recombinant fusion protein (Tfgd2-RsAFP2) induced at different time intervals with anti His-tagged rabbit antibody and detected with Penta His-HRP antibody (Qiagen,GmbH). Lanes A, 0 h without IPTG induction; B, after 1 h of IPTG induction; C, after 2 h of IPTG induction; D, after 3 h of induction with IPTG; and E, positive control for anti His-tag antibodies
In vitro antifungal activity of His6-tagged recombinant fusion protein (Tfgd2-RsAFP2)
The antifungal activity of the purified His6-tagged recombinant fusion protein was determined by in vitro growth inhibition of the following fungi viz., B. cinerea, F. moniliforme, F. oxysporum, P. personata and R. solani along with an oomycete pathogen P. parasitica var. nicotianae. The tested concentrations of His6-tagged recombinant fusion protein were 10, 25 and 40 μg/mL respectively. Elution buffer was used as a negative control. The highest concentration checked to obtain the maximum inhibitory effect of the recombinant fusion protein against the above said fungi was 40 μg/mL. Most of the tested pathogens appeared to be more sensitive with increased concentrations of the fusion protein (Figs. 5 and 6; Supplementary Fig. 4).

In vitro antifungal assay of recombinant fusion protein showing inhibition of the mycelial growth of different plant pathogenic fungi after 48 h of inoculation. a B. cinerea, b F. moniliforme, c F. oxysporum; discs C and 1–3, negative control (elution buffer)—10, 25 and 40 μg/mL concentrations of recombinant his6-tagged fusion protein, respectively. d R. solani; discs C and 1–3, negative control (elution buffer)—10, 40 and 25 μg/mL of fusion protein concentrations

P. personata conidia germination pattern between control and test sample with recombinant fusion protein after 48 h of inoculation. A marked difference is noticeable in the germination pattern of P. personata conidia in control (elution buffer) and test (40 μg/mL of fusion protein) cavity slides. Maximum number of conidia were germinated in the control sample; where as in the case of test sample with fusion protein, their germination was completely arrested, indicating the effective antifungal nature of the fusion protein
The growth of fungal pathogens was inhibited at very lower concentration of the fusion protein (10 μg/mL) and moderately higher concentrations (40 μg/mL) were needed to inhibit the growth of other pathogens. Antifungal activity of the fusion protein was identified at IC50 value less than 10 μg/mL with fungal pathogens like B. cinerea, F. moniliforme, F. oxysporum and P. personata (Table 1). With R. solani, its IC50 value was determined as 20 μg/mL and at that concentration, it showed detrimental effect on the spread of R. solani mycelium and efficiently arrested its growth (Fig. 5d). Out of all the tested fungal pathogens, P. personata appeared to be the most sensitive with this protein. There were different zones of inhibition in the tested fungal pathogens depending upon concentration of the fusion protein used in Petri plate fungal assay but at 40 μg/mL concentration, a clear zone of inhibition was observed in all the tested fungal pathogens, i.e., B. cinerea, F. moniliforme, F. oxysporum, R. solani (Fig. 5) and in an oomycete pathogen P. parasitica var nicotianae (Supplementary Fig. 4). These results prove that fusion protein directly inhibits the growth of tested pathogens in a dose and concentration dependent manner.
Discussion
Plant defensins are highly potent AMPs produced by the plant to protect itself against the pathogen attack. Hence, characterization of efficient defensins from different sources is an emerging tool in biotechnology to protect plants against a broad range of phytopathogens (Osusky et al. 2000; Punja 2001).
Many defensins have been purified from the plants in a native condition. The demonstration of antifungal activity of the defenisn protein predicted from a gene sequence requires its in vivo (cell system or in planta) expression and purification from a heterologous systems (Campos et al. 2008; Kovaleva et al. 2011; De Beer and Vivier 2011).
In this work, we report the successful expression, purification and antifungal activity of the His6-tagged recombinant fusion protein (Tfgd2-RsAFP2) obtained by linking two individual defensins, i.e., Tfgd2 and RsAFP2 respectively with linker peptide. E. coli system has been used to produce numerous amounts of recombinant proteins because of easy handling, inexpensive media and large-scale production (Makrides 1996). Therefore, we opted E. coli expressing system to successfully produce high amounts of active recombinant fusion protein.
Xu and Reddy found that bacteria failed to produce Arabidopsis PR-5 protein's preprotein because its N terminus affected E. coli growth. Similarly, under the same conditions when the Trichosanthes kirilowii defensin (TDEF1) gene with its signal peptide-coding region was inserted into pET-32a+vector, recombinant protein was not induced. Therefore, the partial TDEF1 cDNA corresponding to the mature peptide was inserted into the expression vector and TDEF1 was produced as the recombinant protein in E. coli without the N-terminal signal. However, the antifungal activity of the expressed protein was very low, requiring a dose up to 250 μg/mL of TDEF1 to have an effect on F. oxysporum (Hui et al. 2007). In another case, Tephrosia villosa defensin 1 (TvD1) expressed in E. coli along with its signal peptide exerted significant antifungal activity after purification (Vijayan et al. 2008). Based on these studies, we have expressed our fusion gene (Tfgd2-RsAFP2) with its signal peptide in E. coli and purified efficiently as recombinant fusion protein to determine its antifungal activity against selected plant pathogens.
To express the fusion protein in E. coli, the 500-bp fusion gene encoding sequence from pTZ57R/T plasmid was isolated and cloned into pET-32a+expression vector and was transformed into E. coli BL21 (DE3) pLysS cells. The conditions herein established to overexpress this protein in E. coli (induction with 1 mM IPTG and incubation at 37 °C for 4 h) led to expression of the protein in soluble form.
The antimicrobial activity of the recombinant fusion protein was tested in vitro by using several plant pathogens. This to our knowledge is a first report of expressing antifungal recombinant fusion protein (made up of two defensins) in bacteria and not reported earlier.
The antifungal activity assay has shown that 40 μg/mL concentration of recombinant fusion protein was sufficient to form an inhibitory zone for all the tested fungi under in vitro conditions. It was 100 and 150 μg/mL for Tfgd1 and Tfgd2 defensins respectively from T. foenum-graecum against R. solani and F. moniliforme fungi (Olli and Kirti 2006; Olli et al. 2007). Some PR proteins like chitinases from sorghum and wheat expressed in E. coli required higher concentrations like 300 μg/mL to inhibit the growth of different fungal species, viz., Alternaria sp., Fusarium sp., B. cinerea, R. solani, Colletotrichum falcatum, Pestalotia theae and Sarocladium oryzae (Kirubakaran and Sakthivel 2007; Singh et al. 2007). TvD1 defensin from Tephrosia villosa showed inhibitory zone for P. parasitica f. sp. nicotiana, F. moniliforme, B. cinerea and R. solani pathogens at 100 μg/mL concentration (Vijayan et al. 2008). A defensin from seeds of Phaseolus vulgaris (L.), PvD1 exhibited antifungal activity and caused membrane permeabilization in the filamentous fungi like F. oxysporum, F. solani, F. lateritium and in yeast strains like Candida parapsilosis, Pichia membranifaciens, Candida tropicalis, Candida albicans, Kluyveromyces marxiannus and Saccharomyces cerevisiae at a concentration of 100 μg/mL (Mello et al. 2011).
To investigate the effect of recombinant fusion protein on fungal conidia germination, we have tested this protein with P. personata fungal pathogen. The conidia of P. personata failed to germinate at 40 μg/mL concentration of the fusion protein, whereas Tfgd1 and Tfgd2 defensin proteins required 100,150 μg/mL concentrations to inhibit the germination of conidia (Olli and Kirti 2006; Olli et al. 2007).
However, 50 % inhibitory effect was observed in the case of B. cinerea, F. moniliforme, F. oxysporum and P. personata fungal pathogens at <10 μg/mL concentration of the fusion protein, whereas with R. solani the same effect was observed at 20 μg/mL concentration and at higher concentrations inhibition of mycelial growth was distinctly observed. Antifungal action of this protein was intensely associated with targeted fungus. The concentration at which the fungal growth was inhibited by the fusion protein was not constant and varied with fungal pathogens under consideration.
As fusion protein (Tfgd2-RsAFP2) displays potent antimicrobial activity at lower concentrations (10 and 20 μg/mL) against economically important plant pathogenic fungi and oomycetes, it represents a novel antifungal protein with promising utility for biotechnological applications.
References
Benko-Iseppon AM, Galdino SL, Calsa T Jr, Kido EA, Tossi A, Belarmino LC, Crovella S (2010) Overview on plant antimicrobial peptides. Curr Protein Pept Sci 11:181–188
Bloch CJ, Richardson MA (1991) A new family of small (5 kDa) protein inhibitors of insect α-amylases from seeds of sorghum (Sorghum bicolor (L.) Moench) have sequence homologies with wheat γ-purothionins. FEBS Lett 279:101–104
Boman HG (1995) Peptide antibiotics and their role in innate immunity. Annu Rev Immunol 13:61–92
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Broekaert WF, Terras FR, Cammue BP, Osborn RW (1995) Plant defensins: novel antimicrobial peptides as components of the host defense system. Plant Physiol 108:1353–1358
Broekaert WF, Cammune BPA, De Bolle MFC, Thevissen K, De Samblanx GW, Osborn RW (1997) Antimicrobial peptides from plants. Crit Rev Plant Sci 16:297–323
Campos MDA, Silva MS, Magalhaes CP, Ribeiro SG, Sarto RPD, Vieira EA, de Sa MFG (2008) Expression in Escherichia coli, purification, refolding and anti-fungal activity of an osmotin from Solanum nigrum. Microb Cell Factories 7:7
Carvalho AO, Gomes VM (2009) Plant defensins-prospects for the biological functions and biotechnological properties. Peptides 30:1007–1020
Chen GH, Hsu MP, Tan CH, Sung HY, Kuo CG, Fan MJ, Chen HM, Chen S, Chen CS (2005) Cloning and characterization of a plant defensin VaD1 from azuki bean. J Agric Food Chem 53:982–988
De Beer A, Vivier MA (2011) Four plant defensins from an indigenous South African Brassicaceae species display divergent activities two test pathogens despite high sequence similarity in the encoding genes. BMC Res Notes 4:459–477
Hancock REW, Lehrer R (1998) Cationic peptides: a new source of antibiotics. Trends Biotechnol 16:82–88
Hui LD, Liang JG, Tao ZY, Min AT (2007) Bacterial expression of a Trichosanthes kirilowii defensin (TDEF1) and its antifungal activity on Fusarium oxysporum. Appl Microbiol Biotechnol 74:146–151
Kirubakaran SI, Sakthivel N (2007) Cloning and overexpression of antifungal barley chitinase gene in Escherichia coli. Protein Expr Purif 52:159–166
Kovaleva V, Krynytskyy H, Gout I, Gout R (2011) Recombinant expression, affinity purification and functional characterization of Scots pine defensin1. Appl Microbiol Biotechnol 89:1093–1101
Kushmerick C, Castro MS, Cruz JS, Bloch CJ, Beiraio PSL (1998) Functional and structural features of γ-zeathionins, a new class of sodium channel blockers. FEBS Lett 440:302–306
Laemlli UK (1970) Clevage of structural proteins during the assembly of head of bacteriophage T4. Nature 227:680–685
Landon C, Sodano P, Hetru C, Hoffmann J, Ptak M (1997) Solution structure of drosomycin, the first inducible antifungal protein from insects. Prot Sci 6:1878–1884
Lay FT, Anderson MA (2005) Defensins—components of the innate immune system in plants. Curr Protein Pept Sci 6:85–101
Li Z, Zhou M, Zhang Z, Ren L, Du L, Zhang B, Xu H, Xin Z (2011) Expression of a radish defensin in transgenic wheat confers increased resistance to Fusarium graminearum and Rhizoctonia cerealis. Funct Integr Genomics 11:63–70
Lobo DS, Pereira IB, Fragel-Madeira L, Medeiros LN, Cabral LM, Faria J, Bellio M, Campos RC, Linden R, Kurtenbach E (2007) Antifungal Pisum sativum defensin1 interacts with Neurospora crassa cyclin F related to the cell cycle. Biochemistry 46:987–996
Makrides SC (1996) Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 60:512–538
Mello EO, Ribeiro SF, Carvalho AO, Santos IS, Da Cunha M, Santa-Catarina C, Gomes VM (2011) Antifungal activity of PvD1 defensin involves plasma membrane permeabilization, inhibition of medium acidification, and induction of ROS in fungi cells. Curr Microbiol 62:1209–1217
Melo FR, Rigden DJ, Franco OL, Mello LV, Ary MB, Grossi- De- Sa MF, Bloch C (2002) Inhibition of trypsin by cowpea thionin: characterization, molecular modeling, and docking. Proteins 48:311–319
Mendez E, Moreno A, Colilla F, Pelaez R, Limas GG, Mendez R, Soriano F, de Salinas M, Haro C (1990) Primary structure and inhibition of protein synthesis in eukaryotic cell-free system of a novel thionin, gamma-thionin, from barley endosperm. Eur J Biochem 194:533–539
Mendez E, Rocher A, Calero M, Girbes T, Citores L, Soriano F (1996) Primary structure of ω-hordothionin, a member of a novel family of thionins from barley endosperm, and its inhibition of protein synthesis in eukaryotic and prokaryotic cell-free systems. Eur J Biochem 239:67–73
Mirouze M, Sels J, Richard O, Czernic P, Loubet S, Jacquier A, Franciois I, Cammue BPA, Lebrun M, Berthomieu P, Marques L (2006) A putative novel role for plant defensins: a defensin from the zinc hyper-accumulating plant, Arabidopsis halleri, confers zinc tolerance. Plant J 47:329–342
Odintsova TI, TsA E, Musolyamov AK, Odintsova MS, Pukhalsky VA, Grishin EV (2007) Seed defensins from T. kiharae and related species: genome localization of defensing encoding genes. Biochimie 89:605–612
Olli S, Kirti PB (2006) Cloning, characterization and antifungal activity of defensin Tfgd1 from Trigonella foenum-graecum L. J Biochem Mol Biol 39:278–283
Olli S, Guruprasad L, Kirti PB (2007) Characterization of defensin (Tfgd2) from Trigonella foenum-graecum. Curr Sci 93:365–369
Osborn RW, De Samblanx GW, Thevissen K, Goderis I, Torrekens S, Van Leuven F, Attenborough S, Rees SB, Broekaert WF (1995) Isolation and characterisation of plant defensins from seeds of Asteraceae, Fabaceae, Hippocastanaceae and Saxifragaceae. FEBS Lett 368:257–262
Osusky M, Zhou G, Osuska L, Hancock RE, Kay WW, Misra S (2000) Transgenic plants expressing cationic peptide chimeras exhibit broad-spectrum resistance to phytopathogens. Nat Biotechnol 18:1162–1166
Parashina EV, Serdobinskii LA, Kalle EG, Lavorova NV, Avetisov VA, Lunin VG, Naroditskii BS (2000) Genetic engineering of oilseed rape and tomato plants expressing a radish defensin gene. Russ J Plant Physiol 47:417–423
Park HC, Kang YH, Chun HJ, Koo JC, Cheong YH, Kim CY, Kim MC, Chung WS, Kim JC, Yoo JH, Koo YD, Koo SC, Lim CO, Lee SY, Cho MJ (2002) Characterization of a stamen-specific cDNA encoding a novel plant defensin in Chinese cabbage. Plant Mol Biol 50:59–69
Pelegrini PB, Lay FT, Murad AM, Anderson MA, Franco OL (2008) Novel insights on the mechanism of action of α-amylase inhibitors from the plant defensin family. Proteins 73:719–729
Punja ZK (2001) Genetic engineering of plants to enhance resistance to fungal pathogens—a review of progress and future prospects. Can J Plant Pathol 23:216–235
Singh A, Kirubakaran SI, Sakthivel N (2007) Heterologous expression of a new antifungal chitinase from wheat. Protein Expr Purif 56:100–109
Solis J, Medrano GG, Ghislain M (2007) Inhibitory effect of a defensin gene from the Andean crop maca (Lepidium meyenii) against Phytophthora infestans. J Plant Physiol 164:1071–1082
Song X, Wang J, Wu F, Li X, Teng M, Gong W (2005) cDNA cloning, functional expression and antifungal activities of a dimeric plant defensin SPE10 from Pachyrrhizus erosus seeds. Plant Mol Biol 57:13–20
Spelbrink RG, Dilmac N, Allen A, Smith TJ, Shah DM, Hockerman GH (2004) Differential antifungal and calcium channel-blocking activity among structurally related plant defensins. Plant Physiol 135:2055–2067
Stotz HU, Thomson JG, Wang Y (2009) Plant defensins: defense, development and application. Plant Signal Behav 4:1010–1012
Terras FRG, Torrekens S, Van Leuven F, Osborn RW, Vanderleyden J, Cammue BPA, Broekaert WF (1993) A new family of basic cysteine-rich plant antifungal proteins from Brassicaceae species. FEBS Lett 316:233–240
Terras FR, Eggermont K, Kovaleva V, Raikhel NV, Osborn RW, Kester A, Rees SB, Torrekens S, van Leuven F, Vanderleyden J (1995) Small cysteine-rich antifungal proteins from radish: their role in host defense. Plant Cell 7:573–588
Thevissen K, Ferket KK, Francois IE, Cammue BP (2003) Interactions of antifungal plant defensins with fungal membrane components. Peptides 24:1705–1712
Thevissen K, Warnecke DC, Francois IE, Leipelt M, Heinz E, Ott C, Zahringer U, Thomma BP, Ferket KK, Cammue BP (2004) Defensins from insects and plants interact with fungal glucosylceramides. J Biol Chem 279:3900–3905
Thomma BPHJ, Cammue BPA, Thevissen K (2002) Plant defensins. Planta 216:193–202
Van der Weerden NL, Lay FT, Anderson MA (2008) The plant defensin, NaD1, enters the cytoplasm of Fusarium oxysporum hyphae. J Biol Chem 283:14445–14452
Vijayan S, Guruprasad L, Kirti PB (2008) Prokaryotic expression of a constitutively expressed Tephrosia villosa defensin and its potent antifungal activity. Appl Microbiol Biotechnol 80:1023–1032
Wijaya R, Neumann GM, Condron R, Hughes AB, Poly GM (2000) Defense proteins from seed of Cassia fistula include a lipid transfer protein homologue and a protease inhibitory plant defensin. Plant Sci 159:243–255
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552
Wong JH, Xia L, Ng TB (2007) A review of defensins of diverse origins. Curr Protein Pept Sci 8:446–459
Xu H, Reddy ASN (1997) Cloning and expression of a PR5- like protein from Arabidopsis: inhibition of fungal growth by bacterially expressed protein. Plant Mol Biol 34:949–959
Zelicourt A, Letousey P, Thoiron S, Campion C, Simoneau P, Elmorjani K, Marion D, Simier P, Delavault P (2007) Ha-DEF1, a sunflower defensin, induces cell death in Orobanche parasitic plants. Planta 226:591–600
Zhang L, Falla TJ (2009) Host defense peptides for use as potential therapeutics. Curr Opin Investig Drugs 10:164–171
Acknowledgments
This work was supported by a research grant from the Andhra Pradesh-Netherlands Biotechnology Program administered by the Institute of Public Enterprise, Osmania University Campus, Hyderabad. The authors thank the Head, Department of Plant Sciences for facilities provided by the UGC-SAP, DST-FIST, COSIST, etc.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 426 kb)
Rights and permissions
About this article
Cite this article
Karri, V., Pulugurtha Bharadwaja, K. Tandem combination of Trigonella foenum-graecum defensin (Tfgd2) and Raphanus sativus antifungal protein (RsAFP2) generates a more potent antifungal protein. Funct Integr Genomics 13, 435–443 (2013). https://doi.org/10.1007/s10142-013-0334-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-013-0334-3