
Abstract
The gene encoding Trichosanthes kirilowii defensin (TDEF1) was cloned by reverse transcriptase-polymerase chain reaction (RT-PCR). The newly discovered TDEF1 cDNA contains 231 bp (Genbank accession number DQ526373) and encodes a 76-amino acid protein, which consists of a 29-amino acid signal peptide and a 47-amino acid mature peptide. The partial cDNA, corresponding to the mature peptide coding region of TDEF1, was inserted into bacterial expression vector pET32a(+). Subsequent expression showed that TDEF1 was produced as a 26-kDa fusion protein in the form of inclusion body in Escherichia coli BL21 (DE3). After protein refolding and purification, the fusion TDEF1 displayed an inhibitive activity against the fungal pathogen, Fusarium oxysporum, with EC50 of 247 μg/ml by means of fungal growth inhibition method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Defensins are a cysteine-rich family of peptides with antimicrobial activity. These proteins, found in animals, plants, insects, and mammals, exhibit structural similarities: low molecular weight peptides with 45 to 54 amino acids and Cys-rich with two to six disulfide bridges to stabilize the structure (Thomma et al. 2002; Garcia-Olmedo et al. 1999). Plant defensins, however, display a specific eight-cysteine arrangement that is related with stabilization of their particular globular structure (Meyer et al. 1996). Moreover, plant defensins consist of a signal peptide domain to target the protein for extracellular secretion and a mature peptide domain to exhibit biological activity (Broekaert et al. 1997). Plant defensins were initially identified in seeds, but in the last years, they have been reported in other plant organs and tissues such as leaves (Terras et al. 1995), flowers (Urdangarin et al. 2000), fruits (Meyer et al. 1996), pods (Chiang and Hadwiger 1991), and tubers (Stiekema et al. 1988). Some of the genes encoding these antimicrobial proteins are specifically expressed in response to pathogen infection (Chiang and Hadwiger 1991; Do et al. 2004); others are constitutively expressed (Urdangarin et al. 2000).
As most plant defensins are involved in plant defense response to fungal attack and exhibit antifungal activity against various plant pathogens (Broekaert et al. 1995), they have been applied to control the fungal pathogens in agriculture. Expression of the defensin from Medicago sativa in transgenic potato plants provides robust resistance to the pathogen, Verticillium dahliae (Gao et al. 2000). The transgenic tobacco plants with radish defensin showed a sevenfold reduction in the lesion of the untransformed plants upon infection with foliar fungal pathogen, Alternaria longipes (Terras et al. 1995).
In addition to the transgenic researches, defensins can also be expressed in a functional form in yeast. rDFN1, a defensin in bark and fruit tissues of peach, has been produced as a recombinant defensin in Pichia pastoris, and activity assay showed that it could inhibit germination of the fungal pathogens, Penicillium expansum and Botrytis cinerea (Wisniewski et al. 2003). In the following sections, we report the cloning of a cDNA encoding a novel defensin (TDEF1) from Trichosanthes kirilowii leaves. The defensin cDNA isolated in this work has GenBank accession number DQ526373. To screen its antifungal activity in vitro, bacterial expression of the defensin was subsequently investigated.
Materials and methods
Plant material, bacterial strains, vectors, and fungal strain
T. kirilowii was used to isolate cDNA sequence of TDEF1. Its leaves were collected 14 days after sprouting. Escherichia coli DH5α and vector pMD18-T (TaKaRa) were used for DNA manipulation. E. coli BL21 (DE3) and vector pET32a(+) (Novagen) were used for expression of TDEF1 gene. Transformants of E. coli were grown at 37°C in Luria–Bertani (LB) medium containing 50 μg/ml of ampicillin. Fusarium oxysporum f. sp. vasnifectum Ag117 was obtained from the Institute of Plant Protection, Chinese Academy of Agricultural Sciences.
Cloning of TDEF1 cDNA
Total RNA was isolated from leaves of T kirilowii, by using a Flash UNIQ-10 total RNA extraction reagent Kit (Shanghai Biotechnology Company, China). Upon the nucleotide sequence of the radish defensin (GenBank accession number U18556), a sense primer with BamHI site, called SP1 [5′-CGA GGATCC ATG GCT AA(A/G) TT(T/C) G-3′], and an antisense primer with HindIII site, SP2 [5′-GC AAGCTT TTA (A/G)CA AGG (A/G)AA (A/G)T-3′], were deduced from the open reading frame of radish defensin. Reverse transcriptase-polymerase chain reaction (RT-PCR) was conducted based on the protocol of RNA LA PCR™ Kit (TaKaRa, Japan). Primer SP2 was used in reverse transcription to generate the double stranded cDNA in a 25-μl reaction mixture containing 10 μg total RNA, and the mixture was incubated at 45°C for 20 min. Primer SP1 and SP2 were used in PCR amplification with 1 μl double-stranded cDNA reaction mixture obtained above as template. PCR program included pre-denaturation for 4 min at 94°C, then 35 cycles of 30 s at 94°C, 45 s at 55°C, 30 s at 72°C, and finally, 10 min at 72°C. The PCR system without total RNA template was used as negative control. The resulting product was subjected to agar electrophoresis, then the appropriate band was removed and purified with PCR Fragment Recovery Kit (TaKaRa). The purified fragment was further cloned into pMD18-T vector. The sequence of the clone was confirmed by DNA sequencing (TaKaRa).
Construction of the vector pET32a(+)-TDEF1
To obtain clones merely with the mature peptide coding region of TDEF1, a primer, called MP1 with BamHI site (5′-CGA GGATCC AGA ACA TGT CAG-3′), was designed based on the mature peptide coding sequence of the target clone obtained above. PCR reaction (with MP1 and SP2 as primers) were as follows: 94°C for 30 s, 55°C for 45 s, 72°C for 30 s, and finally, 72°C for a 10-min extension. The amplified product was cloned into pMD18-T and sequenced. The recombinant vector pMD18-T-TDEF1 was digested with BamHI and HindIII, and the resulting fragment of TDEF1 was cloned into the BamHI and HindIII sites of the expression vector pET32a(+), which was designated pET32a(+)-TDEF1. It was introduced into E. coli BL21 (DE3) cells. The empty vector was also introduced into BL21 (DE3) as a control.
Fusion protein production in E. coli
E. coli BL21 (DE3) cells containing a plasmid of interest were grown at 37°C for 14 h in 5 ml LB medium containing appropriate antibiotics (50 μg/ml of ampicillin). The culture was diluted 1:100 (v/v) into 300 ml of the same medium, followed by shaking at 37°C until an OD600 of 0.5 was reached. Isopropyl β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1.0 mM, and cells were grown at 37°C for 3 h.
Refolding of fusion protein
Recombinant bacteria induced by IPTG were collected by centrifugation and suspended in 50 mM Tris–HCl buffer (pH 8.0). Then, the bacteria cell lysate was prepared by sonication at 4°C. The supernatant was removed by centrifugation at 12,000×g for 15 min, and the pellet (inclusion bodies) was used for purification. The inclusion bodies were dissolved in 8 M urea (containing 50 mM Tris–HCl buffer, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 10 mM dithiothreitol (DTT), pH 8.0), and centrifuged at 12,000×g for 15 min to collect the supernatant for 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) electrophoresis. The protein refolding was processed by dialysis against the buffer systems (50 mM Tris–HCl buffer, 3 mM glutathione reduced, 1 mM glutathione oxidized, pH 8.0), containing 6, 4, 2, 1, and 0.5 M urea, respectively, to decrease, gradually, the concentration of urea in the protein solution to 0.5 M, then kept it overnight at 4°C, and finally, the residual urea was eliminated by dialysis against the same buffer but containing no urea.
Purification of fusion protein
Purification of the fusion protein, tagged with thioredoxin (TRX) and hexa-His at the N-terminus, was carried out following the ProBond™ Purification System Kit for proteins tagged with histidines (Invitrongen). The solution of the fusion protein (500 mM NaCl supplied) was mixed with 10 ml Ni-NTA agarose and loaded onto a ProBond Resin column. Unbound proteins were washed away with 100 mM phosphate buffer (pH 7.5, containing 500 mM NaCl). Bound proteins were fractionally eluted out with 100 mM phosphate buffer (pH 7.5, containing 500 mM NaCl) supplemented with 100, 150, and 250 mM imidazole, respectively. The fusion proteins eluted by imidazole-containing buffer were pooled and dialyzed against 100 mM phosphate buffer (pH 7.5), then stored at −20°C until used for antifungal activity assay. SDS-PAGE was performed using a 15% gel, and the gel was stained with Coomassie Brilliant Blue. The concentration of the fusion protein was determined by the Lowry method with bovine serum albumin (BSA) as standard.
Antifungal activity assay
The assay was manipulated by means of fungal growth inhibition method (Fang et al. 2005) with certain revision. The procedure was as follows: (1) F. oxysporum was cultured on potato–dextrose–agar (PDA) medium plates at 28°C for 2 weeks; then, its hyphae discs with 5 mm in diameter were cut off from petri dishes; (2) solution of the fusion TDEF1 was added to 20 ml PDA medium to produce the PDA plates with final concentration of 450, 400, 350, 300, 250, 200, 150, and 100 μg/ml, respectively; solution of the purified tags of TRX-His from bacterial cell with empty pET32a(+) vector was similarly treated as negative control, and the purified TDEF1 from T. kirilowii leaves, as positive control; (3) each of the hyphae disc above was inoculated on the PDA plates with different concentrations of the fusion protein; (4) inhibition of the fungal growth was determined by the diameter of hyphal growth at 28°C for 3 days after inoculation. Inhibition ratio was calculated using the following formulation: inhibition ratio (%) = [(diameter of negative control − diameter of experimental hyphae)/diameter of negative control] × 100.
Results
Cloning of TDEF1 gene
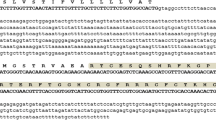
With the designed primers, a cDNA fragment with the size of approximately 231 bp was obtained by RT-PCR from T. kirilowii leaves. The sequencing result shows that the cDNA contains 231 bp, which encodes a deduced protein of 76 amino acids, TDEF1 (Fig. 1). It consists of a predicted signal peptide of 29 amino acids at the N-terminus domain (underlined in Fig. 1b) by sequence homology with other defensins in Genbank and a mature peptide of 47 amino acids with a calculated molecular mass of 5.6 kDa.

Alignment of the nucleotide sequence of TDEF1 with the sequences of other genes encoding defensins (a) and deduced amino acid sequence of TDEF1 (b). a RSU18556 and RSU18557 from Raphanus sativus (GenBank accession number U18556 and U18557), alfAFP from alfalfa (Medicago sativa, GenBank accession number AF319468). The predicted signal sequence cleavage site is indicated by an arrow. The conserved sites for cysteines are boxed. b The signal peptide is underlined and eight cysteine residues are in bold
Bacterial expression of fusion TDEF1
Upon PCR amplification, a 144-bp fragment was obtained. This partial cDNA, corresponding to the mature peptide coding region of TDEF1 gene (Fig. 1a), was further cloned into pMD18-T, then constructed into expression vector pET32a(+). A BamHI and HindIII digestion was used to screen the recombinant pET32a(+)-TDEF1, which was subsequently transformed into E. coli BL21 (DE3).
Induced by IPTG, the bacteria BL21 transformed with pET32a(+)-TDEF1 could express fusion TDEF1 protein (Fig. 2, lane 3). The fusion protein consisted of 20.4 kDa TRX-His tags (Fig. 2, lane 2) and a 5.6-kDa mature peptide of TDEF1. Thus, the total size of the predicted fusion protein is 26 kDa. This roughly corresponds to the molecular size of the expressed TRX-TDEF1 on SDS-PAGE (Fig. 2). The amount of the fusion protein could reach up to about 11% of total BL21 proteins. In this work, the bacteria cell lysate was centrifuged to collect the supernatant and the pellet for SDS-PAGE analysis, respectively. The SDS-PAGE result showed that the fusion TDEF1 protein has been expressed in the form of inclusion body (Fig. 2, lane 4 and lane 7).

SDS-PAGE analysis of the expressed fusion protein. 1 Uninduced BL21/pET32a(+)-TDEF1, 2 induced BL21/pET32a(+), 3 total protein expression of BL21/pET32a(+)-TDEF1, by IPTG induction, 4 the inclusion bodies containing fusion TDEF1, after the induced bacteria BL21/pET32a(+)-TDEF1 were sonicated, 5 the purified fusion TDEF1, 6 low molecular mass marker proteins, 7 the supernatant containing soluble fraction released from the induced bacteria BL21/pET32a(+)-TDEF1 after sonication
Antifungal activity of fusion TDEF1
Via purification through the resin with affinity to the proteins tagged with His, the fusion protein with more than 98% purity was obtained (Fig. 2, lane 5). The yield of the purified fusion protein was about 23 μg/ml of original bacterial culture. The expressed TDEF1 showed a strong activity against spores germination and hyphal growth of the fungal pathogen, F. oxysporum (Fig. 3). Incubated with the control protein (TRX-His) for 3 days, the fungal spores were germinated continuously and the fungal hyphae were grown rapidly, leading to a precipitous increase in diameter of the fungal disc (left one in Fig. 3a). At the same time, in the presence of the fusion TDEF1 (250 μg/ml) and the purified TDEF1 (20 μg/ml) from T. kirilowii leaves, their germination and hyphal elongation were inhibited, resulting in small fungal discs (central and right ones in Fig. 3a). When the time of incubation was prolonged for a week, the fusion TDEF1 still inhibited the hyphal growth with the inhibition ratio of 62.3% and the purified TDEF1 was fully inhibitory at 20 μg/ml (Fig. 3b). Compared with the negative control protein, the fusion TDEF1 could inhibit the fungal growth with EC50 (median effective concentration) value of 247 μg/ml (Fig. 3c).

Antifungal activity of TDEF1 on F. oxysporum. The inhibition ratio was monitored by incubating the fungi on PDA plates containing different concentration of the fusion TDEF1: filled diamonds (♦). F. oxysporum was incubated on PDA plate with 250 μg/ml fusion TDEF1 (central petri dish), negative control plate (left petri dish), and 20 μg/ml purified TDEF1 from T. kirilowii leaves as positive control plate (right petri dish) for 3 days (a) and a week (b), respectively. Bars = 10 mm. c Inhibition curve at third day after inoculation. The data shown are means of three experiments. Regressive equation: \(Y = 13.4X + 16.925\), correlation coefficient: 0.9423, EC50: 247 μg/ml
Discussion
In the present study, we have described cloning and bacterial expression of the defensin (TDEF1) originating from T.kirilowii leaves. We have demonstrated here that this defensin could effectively be produced as the fusion protein of TRX-TDEF1 with an inhibitive activity on the fungal pathogen, F. oxysporum.
TDEF1 gene exhibited high nucleotide sequence homology to the defensin genes from Raphanus sativus (GenBank accession number U18556 and U18557) with identity 65.45 and 62.96%, respectively. While compared with another antifungal peptide from M. sativa (GenBank accession number AF319468), they showed no significant homology (with identity 40.95%). By sequence alignment, however, the conserved sequence encoding eight cysteine residues to form four disulfide bridges in TDEF1 gene (Fig. 1) is consistent with homologues in other plant defensins. Therefore, TDEF1 could be sorted as a novel member of defensins family.
We chose to express the defensin using prokaryotic system for a number of reasons. Compared with other expression systems, although yields of correctly folded and functional protein are frequently low due to protein aggregation, E. coli, as a host for recombinant protein, provides an economical and fast way to produce the molecules in relatively large amounts. The expressed TDEF1 in the form of inclusion body could reach up to about 11% of the total BL21 proteins, suggesting that the expression system is highly efficient. During the protein refolding, we have introduced the strategy by which the concentration of urea was gradually decreased by dialysis before the process of protein purification, to avoid the protein aggregation when the denaturant was suddenly extracted away from the buffer system (Hevehan and Clark 1997). Moreover, in pET32a(+) system used in this study, the TRX fusion tag expressed from the vector could enhance the solubility of the target protein isolated from inclusion bodies (Sachdev and Chirgwin 1998).
The presence of a typical secretion signal peptide is one of the characteristics of plant defensins (Broekaert et al. 1997). According to the report by Xu and Reddy, bacteria failed to produce preprotein of a PR5-like protein from Arabidopsis because its N-terminus affected E. coli growth (Xu and Reddy 1997). Our findings are consistent with their result. In our previous work, we found that if TDEF1 gene with its signal peptide coding region were inserted into pET32a(+), no fusion protein could be induced under the same conditions (data not shown).The work by Wisniewski et al. showed that rDNF1 (a defensin from peach), with or without its signal peptide, could be expressed in the yeast, P. pastoris (Wisniewski et al. 2003). These results may indicate that the E. coli system could not recognize the endogenous signal peptide of the defensin. Therefore, the partial TDEF1 cDNA, corresponding to a mature peptide, was inserted into the expression vector, and TDEF1 was produced as the fusion protein in E. coli.
While the fusion TDEF1 inhibited the growth of F. oxysporum with an EC50 of 247 μg/ml, no appreciable effect was discovered at 450 μg/ml on V. dahliae (data not shown). Native alfAFP from M. sativa could inhibit the hyphal elongation of V. dahliae by 50% at 5 μg/ml (Gao et al. 2000). Theses results support that the antifungal activity of plant defenses is dependent on the target fungus. Full growth inhibition at 20 μg/ml against F. oxysporum for the purified TDEF1 from T. kirilowii leaves was relatively low, compared with the concentration required for inhibiting the fungal growth by the fusion TDEF1. It is possibly due to the fact that the N-terminal in TRX-TDEF1 fusion protein might have decreased the TDEF1’s activity.
F. oxysporum may cause wilt disease of certain plants such as cotton and potato, resulting in their poor yield and quality for usage. Chemical pesticides play a main role in controlling the fungal pathogen in agricultural crops. Although they do have effects somewhat, their residues may result in serious ecologic and social problems, whereas, defensins have not been found to affect the viability of human and plant cells (Broekaert et al. 1995). Thus, the antifungal activity of the defensin and its availability with genetic engineering should make it possible to be applied for certain fungal pathogen control.
References
Broekaert WF, Cammue BPA, De Bolle MFC, Thevissen K, De Samblanx GW, Osborn RW (1997) Antimicrobial peptides from plants. Crit Rev Plant Sci 16:297–323
Broekaert WF, Terras FRG, Cammue BPA, Osborn RW (1995) Plant defensins: novel antimicrobial peptides as components of the host defence system. Plant Physiol 108:1353–1358
Chiang CC, Hadwiger LA (1991) The Fusarium solani-induced expression of a pea gene family encoding high cysteine content proteins. Mol Plant Microbe Interact 4:324–331
Do HM, Lee SC, Jung HW, Sohn KH, Hwang BK (2004) Differential expression and in situ localization of a pepper defensin (CADEF1) gene in response to pathogen infection, abiotic elicitors and environmental stress in Capsicum annuum. Plant Sci 166:1297–1305
Fang Y, Li X, Yang W (2005) Preliminary evaluations of four actinomyces strains on inhibition against plant pathogenic fungi. Journal of Yunnan Agricultural University 20:343–345
Gao A, Hakimi SM, Mittanck CA, Wu Y, Woerner BM, Stark DM, Shah DM, Liang J, Rommens CMT (2000) Fungal pathogen protection in potato by expression of a plant defensin peptide. Nat Biotechnol 18:1307–1310
Garcia-Olmedo F, Molina A, Alamillo JM, Rodriguez-Palenzuela P (1999) Plant defense peptides. Biopolymers 47:479–491
Hevehan DL, Clark FDB (1997) Oxidative renaturation of lysozyme at high concentration. Biotechnol Bioeng 54:221–230
Meyer B, Houlne G, Pozueta-Romero J, Shantz ML, Shantz R (1996) Fruit-specific expression of a defensin-type gene family in bell pepper. Plant Physiol 112:615–622
Sachdev D, Chirgwin JM (1998) Solubility of proteins isolated from inclusion bodies is enhanced by fusion to maltose-binding protein or thioredoxin. Protein Expr Purif 12:122–132
Stiekema WJ, Heidekamp F, Dirkse WG, Van Beckum J, De Haan P, Louwerse J D (1988) Molecular cloning and analysis of four potato tuber mRNAs. Plant Mol Biol 11:255–269
Terras SB, Eggermont K, Kovaleva V, Raikhel NV, Osborn RW, Kester A, Ress SB, Torrekens S, Van Leuven F, Vanderleyden J (1995) Small cysteine-rich antifungal proteins from radish: their role in host defense. Plant Cell 7:573–588
Thomma BPHJ, Cammue BPA, Thevissen K (2002) Plant defensins. Planta 216:193–202
Urdangarin MC, Norero NS, Broekaert WF, de la Canal L (2000) A defensin gene expressed in sunflower inflorescence. Plant Physiol Biochem 38:253–258
Wisniewski ME, Bassett CL, Artlip TS, Webb RP, Janisiewicz WJ, Norelli JL, Goldway M, Droby S (2003) Characterization of a defensin in bark and fruit tissues of peach and antimicrobial activity of a recombinant defensin in the yeast, Pichia pastoris. Physiol Plant 119:563–572
Xu H, Reddy ASN (1997) Cloning and expression of a PR5-like protein from Arabidopsis: inhibition of fungal growth by bacterially expressed protein. Plant Mol Biol 34:949–959
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Da-Hui, L., Gui-Liang, J., Ying-Tao, Z. et al. Bacterial expression of a Trichosanthes kirilowii defensin (TDEF1) and its antifungal activity on Fusarium oxysporum . Appl Microbiol Biotechnol 74, 146–151 (2007). https://doi.org/10.1007/s00253-006-0631-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0631-z