
Abstract
Soybean rust caused by Phakopsora pachyrhizi Sydow is a devastating foliar disease that has spread to most soybean growing regions throughout the world, including the USA. Four independent rust resistance genes, Rpp1–Rpp4, have been identified in soybean that recognize specific isolates of P. pachyrhizi. A suppressive subtraction hybridization (SSH) complementary DNA (cDNA) library was constructed from the soybean accession PI200492, which contains Rpp1, after inoculation with two different isolates of P. pachyrhizi that result in susceptible or immune reactions. Both forward and reverse SSH were performed using cDNA from messenger RNA pooled from 1, 6, 12, 24, and 48 h post-inoculation. A total of 1,728 SSH clones were sequenced and compared to sequences in GenBank for similarity. Microarray analyses were conducted on a custom 7883 soybean-cDNA clone array encompassing all of the soybean-rust SSH clones and expressed sequence tags from four other soybean cDNA libraries. Results of the microarray revealed 558 cDNA clones differentially expressed in the immune reaction. The majority of the upregulated cDNA clones fell into the functional category of defense. In particular, cDNA clones with similarity to peroxidases and lipoxygenases were prevalent. Downregulated cDNA clones included those with similarity to cell-wall-associated protein, such as extensins, proline-rich proteins, and xyloglucan endotransglycosylases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Soybean rust caused by Phakopsora pachyrhizi Sydow is an aggressive foliar fungal pathogen that was first described in Japan in 1902 (Hennings 1903). The pathogen has since spread to most soybean-growing countries in Asia, Africa, and South America (Miles et al. 2003a), and in 2004, it was discovered for the first time in the continental USA (Schneider et al. 2005). Yield losses ranging from 40% to 80% have been reported in countries where this disease has become endemic (Bromfield 1984; Patil and Basavaraja 1997). Fungicides are effective in managing soybean rust; however, the application of fungicides increases production costs (Miles et al. 2003b). In addition, there are concerns about fungicide resistance developing in the pathogen population and the potential negative impact to the environment if fungicides are applied to large acreage (Sconyers et al. 2006).
Several germplasm screens have been conducted to identify soybean accessions with resistance to P. pachyrhizi (Hartman 1996; McLean and Byth 1976; Miles et al. 2006; Mo et al. 1994; Patil and Basavaraja 1997; Singh et al. 1974; Tan et al. 1996; Tschanz et al. 1986). Four single dominant genes, Rpp1, Rpp2, Rpp3, and Rpp4, have been described that confer resistance to specific isolates of P. pachyrhizi (Bromfield 1984; Bromfield and Hartwig 1980; Hartwig 1986; Hartwig and Bromfield 1983; McLean and Byth 1980). Three infection types have been described on soybean accessions after inoculation with P. pachyrhizi: (1) Tan, a susceptible reaction characterized by tan lesions with many uredinia and prolific sporulation, (2) RB, a resistant reaction typified by reddish-brown lesions with few uredinia and little to moderate sporulation, and (3) immune, a resistant reaction with no visible lesions or uredinia (Bromfield 1984; Bromfield and Hartwig 1980). Soybean accession PI 200492, the Japanese cultivar Komata, contains Rpp1 and produces an immune reaction after inoculation with the P. pachyrhizi isolates Australia 79-1, India 72-1, and Hawaii 94-1, while other isolates of P. pachryhizi, including Taiwan 72-1, result in a fully susceptible Tan reaction (Bonde et al. 2006; Bromfield and Hartwig 1980; Hartwig and Bromfield 1983). The location of Rpp1 has been mapped between Sct187 and Sat064 on linkage group (LG) G (Hyten et al. 2007). Recently, the Japanese cultivar Hyuuga was found to produce an RB reaction when inoculated with P. pachyrhizi urediniospores collected from field-grown soybean plants and kudzu in Georgia in 2005 (Monteros et al. 2007). The resistance gene in Hyuuga maps between Satt460 and Sat307 on LG-C2 (Monteros et al. 2007). It is not known if the resistance gene in Hyuuga is a new Rpp gene or whether it is Rpp2, Rpp3, or Rpp4.
Microscopic observations of susceptible and immune reactions in the cv. Komata (Rpp1) did not show any difference in the initial penetration of the fungus through the epidermis (Keogh and Deverall 1980; McLean and Byth 1981). Spore germination percentage, germ tube length, appressoria formation, and cell penetration were not different in the susceptible and immune reactions. However, unlike the susceptible reaction, secondary haustoria did not develop in the immune reaction (Keogh and Deverall 1980). Differences in the rate of lignification and the presence of a papillar structure in a resistant soybean rust interaction have also been reported (Fei et al. 1996).
Although Rpp1 has been mapped to LG-G, the function of the gene(s) involved is still unknown. Suppression subtractive hybridization (SSH; Diatchenko et al. 1996) has been used successfully in several plant–pathogen interactions to identify changes in host gene expression after infection (Hu et al. 2006; Kurkcuoglu et al. 2007; Lin et al. 2007; Ray et al. 2003; Li and Asiegbu 2004; Verica et al. 2004; Zhulidov et al. 2004). Similarly, microarrays are an ideal method for surveys of gene expression across thousands of genes both known and unknown, giving a broad sample to mine for new resistance candidates (Alkharouf et al. 2006; Golkari et al. 2007; Schenk et al. 2000; Zabala et al. 2006). In this study, we constructed SSH complementary DNA (cDNA) libraries from messenger RNA (mRNA) extracted from Rpp1-susceptible and immune reactions and used cDNA microarrays to identify transcripts that are differentially expressed in the immune reaction at 6, 12, 24, and 48 h post-inoculation (hpi).
Materials and methods
Biological materials
The soybean cvs. Komata (USDA germplasm accession PI200492) and Williams were grown two per 10.2-cm-diameter clay pot filled with Sunshine LC1 mix (Sun Grown Horticulture Products, Belleview, WA) in a greenhouse under natural light. Urediniospores of the two P. pachyrhizi isolates Taiwan 72-1 (TW72-1) and Hawaii 94-1 (HW94-1) were harvested approximately 14 days after inoculation onto the soybean cv. Williams using a mechanical harvester (Cherry and Peet 1966) and stored in liquid nitrogen at the USDA-ARS Foreign Disease-Weed Science Research Unit Biological Safety Level-3 Plant Pathogen Containment Facility at Fort Detrick, MD, USA (Melching et al. 1983) under the appropriate USDA Animal Plant Health Inspection Service permit. Before inoculating soybean plants, urediniospores were removed from liquid nitrogen, heat shocked at 40°C for 5 min, and hydrated at 100% relative humidity at room temperature for 16 h. Spores were suspended in sterile distilled water containing 0.01% (v/v) Tween 20 and adjusted to a concentration of 2 × 104 spores ml−1 with a hemacytometer. Twenty-two-day-old plants of the soybean cv. Komata were inoculated at the V2 growth stage (Fehr and Caviness 1977) with 2 ml per plant of either P. pachyrhizi isolate HW94-1 or isolate TW72-1 using an atomizer attached to an air compressor at 20 psi. Mock inoculations were conducted by inoculating soybean cvs. Komata and Williams plants with distilled water/Tween 20 as described above. In addition, control inoculations were performed with the two P. pachyrhizi isolates onto the susceptible soybean cv. Williams to verify infection and virulence of both isolates. Plants were incubated in a dew chamber at 20°C overnight (approximately 16 h) and placed in a greenhouse at 20°C to 25°C under a 16-h photoperiod. Supplemental lighting was provided using 1,000-W Metalarc lights (Sylvania, Danvers, MA, USA). For the soybean-rust SSH library, trifoliates were pooled from two plants at 1, 6, 12, 24, and 48 hpi, immediately frozen in liquid N2, and stored at −80°C. For the microarray hybridization probes, leaflets were pooled from four to six plants at each time point in each of three independent inoculations. The 1 hpi sample was omitted from the microarray study to keep the experiment to a manageable size.
RNA extraction
Total RNA was extracted from soybean leaflets using a Trizol/guanidinium isothiocyanate protocol (Chomczynski and Sacchi 1987) with an additional lithium chloride purification step. For the SSH library construction, 20 μg of RNA from each of the time points were pooled (100 μg total), incubated with 16 units of DNaseI (Ambion, Austin, TX, USA) for 30 min at 37°C and purified using the RNeasy mini protocol (Qiagen, Valencia, CA, USA) according to manufacturers’ directions. The mRNA was isolated using an Oligotex mRNA kit (Qiagen) following the manufacturer’s protocols. All purified RNA was quantified using a spectrophotometer (BioRad Laboratories, Hercules, CA, USA) and separated on a 1.2% (w/v) agarose-formaldehyde gel to verify integrity.
Soybean-rust SSH library
SSH libraries were constructed using the polymerase chain reaction (PCR)-select cDNA subtraction kit (BD Biosciences Clonetech, Palo Alto, CA, USA) according to the manufacturer’s protocol with minor modifications. The first-strand cDNA synthesis was performed using 3 μg of mRNA. The forward library’s tester was made using mRNA from cv. Komata inoculated with P. pachyrhizi isolate HW94-1 (immune reaction), and the driver was prepared using mRNA extracted from cv. Komata inoculated with P. pachyrhizi isolate TW72-1 (susceptible reaction). The reverse subtraction library was constructed as above except that the mRNA used for the tester and driver was reversed. A control subtraction was performed using skeletal muscle cDNA provided in the kit. The cDNA fragments were ligated into the vector pT7Blue using a Perfectly Blunt cloning kit (Novagen, San Diego, CA, USA) and transformed into Novablue Singles Competent Cells (Novagen) following the manufacturer’s protocol.
DNA sequencing and data analysis of soybean-rust SSH library
Before sequencing, all white colonies were checked for the presence of an insert by colony-PCR using the SP6 and T7 promoter primers. PCR products were separated by electrophoresis using 1.5% agarose gels. Clones from the forward subtraction and reverse subtraction libraries were sent to the USDA Agricultural Research Service (ARS), Eastern Regional Research Center, Nucleic Acids Facility in Wyndmoor, PA, USA for plasmid preparation and sequencing. DNA was prepared for sequencing reactions using a Qiagen BioRobot 9600 and a Beckman Biomek 2000. Single-pass sequencing was performed from the 5′ end using the plasmid DNA as template and either the T7 promoter primer or the M13 reverse primer using an Applied Biosystems (ABI; Foster City, CA, USA) PRISM big dye terminator kit and an ABI 3700 DNA analyzer. Nucleotide sequences were manually edited using Chromas v2.23 (Technelysium Pty Ltd., Helensvale, Australia). An in-house script “Scanseq” was used to identify redundant expressed sequence tags (ESTs) based on nucleotide overlap >50 nt, with e-value ≤10−5, reducing the number to 979 and 555 low-redundant ESTs (ESTlr) from the forward and reverse subtraction libraries, respectively. ESTs were identified by comparing their predicted amino acid sequences to the Entrez Protein database (http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=search&db=protein) using the BLAST X search algorithm (Altschul et al. 1997) as part of the Genetics Computer Group computer software package (version 10.3, Accelrys, San Diego, CA, USA) at the Advanced Biomedical Computing Center of the National Cancer Institute, Frederick, MD, USA. ESTs that achieved e-values ≤10−3 were retained for further analysis, and ESTs without similarity to any proteins in the database at the e ≤ 10−3 cutoff were annotated as “unknown.” Corresponding Uniprot identifiers were queried using PIR-PSD (Wu et al. 2004, http://pir.georgetown.edu/) to determine the Gene Ontology biological process identifier (Ashburner et al. 2000). The ESTs were classified into functional categories based on their Uniprot and PFam annotations following the rationale detailed in Table 1, which was adapted from Zabala et al. (2006).
cDNA microarray clones and microarray fabrication
Inserts from the SSH libraries were amplified by PCR using the primers U19 and T7 in a Gene Amp PCR System 9700 thermocycler (Applied Biosystems, Foster City, CA, USA) in a 50-μl reaction containing approximately 5 ng of the purified plasmids, 1.5 mM MgCl2, 0.2 μM each primers, 0.02 U Taq in a 1× concentration reaction buffer (Promega, Madison, WI, USA). Cycling conditions were 94°C denaturation for 2 min, 35 cycles at 94°C for 45 s, 55°C for 45 s, 72°C for 2 min, followed by an extension at 72°C for 7 min. PCR products were precipitated with isopropanol, centrifuged and resuspended in 6 μl 50% (v/v) dimethyl sulfoxide (DMSO)/Tris-EDTA (TE) to a final concentration of 80–200 ng/μl. PCR products were checked by gel electrophoresis to identify clones that produced no or multiple amplicons. Only those with single PCR products were randomly re-racked into 384-well plates and stored at 4°C until printing.
Each of the 1,728 clones from the forward and reverse SSH libraries along with 6,155 ESTs from other soybean EST libraries were printed on each microarray slide (7,883 total). We realized that the 1,728 SSH clones produced for this study would not fill the microarray, and so we were able to include additional EST libraries that had been produced for other projects and were available in the laboratory. The clones from additional cDNA libraries used in assembling the microarray were as described by Alkharouf et al. (2006). Briefly, one library was constructed from cvs. Peking and PI437654, soybean genotypes resistant to soybean cyst nematode (Hetrodera glycines) population NL1-RHp. A cv. Peking Lambda ZAP II cDNA library was made from the roots and shoots of cv. Peking plants, 48 h after nematode infection as described (Alkharouf et al. 2004). Two suppressive subtraction libraries prepared from roots of PI437654, 24 h after nematode inoculation followed by a 10-h infection period, were obtained from Clemson University Genomics Institute and were constructed as described in (Tomkins et al. 1999). cDNA clones from soybean roots obtained from E.I. Dupont de Nemours, Newark, DE, USA were also used in the experiment and were described in Alkharouf et al. (2006).
Clones in the cv. Peking cDNA library duplicating those in the SSH library were identified by pairwise comparison of each clone against the EST database housed at the USDA-ARS Soybean Germplasm Improvement Laboratory, Beltsville, MD, USA (http://psi081.ba.ars.usda.gov/SGMD/Default.htm) and then removed, reducing the total number of cDNAs in the experiment. The insert from each clone was amplified by PCR using a PTC 225 thermocycler (MJ Research, Waltham, MA, USA) with the T3 and T7 universal primers in a 100-μl reaction similar to the protocol of Hegde et al. (2000). The amplified product from each clone was checked on a 1% agarose/1× Tris–borate–EDTA gel for the presence of a single band and brought to a final volume of 5 ml in 50% DMSO/50% TE solution. The clones from all libraries were randomized and re-racked before printing. A total of 7,883 PCR products were printed simultaneously in triplicate onto CMT-GAPS coated slides (Corning, NY, USA) using a Cartesian robot model PixSys 5500 PA workstation with a telechem printing head and stealth quill pins. A total of 30 slides were printed and UV crosslinked at 50 mJ/cm2 using a UV Stratalinker 2400 (Stratagene, Cedar Creek, TX, USA).
Fluorescent probe preparation
Trifoliates were harvested from at least four plants per treatment (mock inoculation, immune reaction, and susceptible reaction) per time point (6, 12, 24, and 48 hpi). Total RNA (20 μg) from each sample was reverse-transcribed using SuperScript Indirect cDNA Labeling system (Invitrogen, Carlsbud, CA, USA) to incorporate the amino-modified nucleotides as recommended by the manufacturer. After purification, the cDNA was split equally into two tubes and labeled with either Alexa Fluor Fluorescent dye AF555 or AF647 (Molecular Probes, Eugene, OR, USA). Probes were labeled from all three biological replicates, and the absorption at 550 and 650 nm was measured using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, USA). The percentage of fluorescent dye incorporation and amount of labeled probe was calculated using the NanoDrop ND-1000 software. The labeled probes were stored at −20°C until use.
Microarray hybridization
The microarray slides were pre-washed in 0.1% (w/v) sodium dodecyl sulfate (SDS) in Coplin jars on a shaker for 1 h at room temperature (RT), rinsed in sterile water, washed in isopropanol, and dried by centrifugation at 600 rpm. Equal amounts of AF555- and AF647-labeled probes were denatured at 95°C for 3 min and added to BM hybridization buffer [5× saline-sodium citrate (SSC), 0.5% BDH Casein Hammarsten, 0.1% N-Lauroyl Sarcosine, 0.02% SDS, 1.0 mM dithiothreitol (DTT)], with 0.6 μg poly(A), kept at 60°C, and applied to the array under a 22 × 60 mm lifter coverslip (Erie Scientific, Portsmouth, NH, USA). Slides were placed in sealed hybridization chambers containing 50 μl water and incubated at 60°C for 48 h. After hybridization, the slides were washed in 1× SSC/0.2% SDS for 10 min at 42°C, 0.1× SSC/0.2% SDS for 4 min at RT, 0.5× SSC for 10 s at RT, 0.1× SSC for 3 min at RT and dried by centrifugation at 600 rpm. A total of 30 slides were hybridized, ten for each biological replicate (inoculation experiment). For each biological replicate, there were five time points (mock, 6, 12, 24, and 48 hpi), and the corresponding dye swaps with AF555 or AF647.
Scanning and microarray data analysis
The microarrays were scanned for either AF555 or AF647 at 10-μm resolution using a ScanArray 4000 (GSI Lumonics, Meriden, CT, USA). Fluorescence intensities were extracted from the scanned images using the image processing software package SPOT (http://www.hca-vision.com/product_spot.html). Background subtraction was performed before calculating ratios. Only spots with an intensity of at least 2.0 times above the local background in both channels were used for subsequent analysis. This filtering process usually removed 1% to 10% of the elements. In addition, chimeric and redundant clones identified by sequencing were filtered out and not used in the microarray expression analysis. The extracted data from each slide was then log2-transformed and normalized using the Lowess print-tip group normalization method (Yang et al. 2002).
Potential artifacts and false positives were eliminated by selecting only for those clones that exhibited similar expression patterns between the original hybridization and the corresponding dye swap (Yang et al. 2002). These clones were determined by one-way analysis of variance (1-ANOVA) tests, which were used to detect similar expression levels across replicated slides and across the three biological samples. Only clones with an F value high enough to produce a probability of 0.1 or higher were selected as being similar across biological replicates. Structured query language (SQL) procedural scripts were written to perform t tests and 1-ANOVA tests for every clone post-filtration and normalization.
The Student’s t test was used to calculate t values on log2 expression ratios to identify genes with statistically significant expression ratios. Self–self hybridized slides with equal amounts of AF555 or AF647-labeled RNA from uninfected cv. Komata (K-/K-) were used as control groups for the t test. The clones printed in K-/K- showed a ratio of −1.20 to 1.20 after Lowess print-tip normalization.
The t tests were used to analyze the expression of each clone in immune/susceptible (K:HW94–1/K:TW72-1) hybridizations using background corrected and normalized expression ratios. The results of the t tests were used to determine statistical significance (p ≤ 0.05) of gene expression. A gene was considered differentially expressed if the t test p value was <0.05 and the average fold change for both replicates was >2.0. If the gene expression passed the t test, then a cutoff value of twofold was applied for extra stringency. The high stringency of selection kept the possibility of false positives to a minimum but at the same time increased the likelihood of false negatives. This statistical method takes into account the variability within slides and between replicated slides as well as biological samples to distinguish gene expression changes caused by treatments from gene expression changes attributable to biological and measurement variability.
SQL and online analytical processing (OLAP; Codd et al. 1993; Alkharouf et al. 2005) were used to produce lists of differentially expressed genes in each of the time points. In addition, the clustering function within Analysis Services (Microsoft, Redmond WA, USA) was used to find distinct expression profiles in the differentially expressed genes. A web-based user interface was developed to query the data and post the raw and normalized data sets. The data can be accessed at http://bioinformatics.towson.edu/SGMD/MicroarrayExps/soyrust.htm.
Quantitative real-time RT-PCR analysis
Quantitative real-time reverse transcriptase PCR (RT-PCR) was carried out on total RNA from cv. Komata leaves harvested at 6, 12, 24, and 48 hpi with P. pachyrhizi isolates TW72-1 or HW94-1 using a Smart Cycler (Cepheid, Santa Clara, CA, USA). Four genes analyzed by RT-PCR were chosen to be representative of the differentially expressed genes. The selected genes include two defense-related genes, starvation-associated message 22 (SAM-22) and anthocyanidin synthase (ANS); a housekeeping gene, nitrate transporter 1 (NRT1–5); and an unknown gene. Ten nanograms of the same RNA that was used to create probes for the microarray analysis was used as the template for one-step RT-PCR using the Quantitect SYBR Green One Step RT-PCR kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Gene-specific primers were designed using Primer3 software (Rosen and Skaletsky 2000; http://primer3.sourceforge.net/; Table 2). Each sample was tested in triplicate for all primers. Melting curve analysis was performed on all samples to ensure amplification of a single product with the expected melting temperature and the absence of primer-dimers. The products of each primer set were tested by agarose gel electrophoresis to verify that a single product of the expected size was produced. Relative RNA quantities were determined using the ΔΔCt method (Pfaffl 2001; Pfaffl et al. 2002) by comparing the data for each gene of interest with the data for mock-inoculated control samples at each time point. The data was normalized by comparison to mock-inoculated samples.
Results
Sequencing and characterization of SSH library clones
A total of 1,056 and 672 clones from the forward and reverse SSH libraries, respectively, were sequenced. The size of the cDNA inserts ranged from 52 to slightly >600 bp. No full-length transcripts were obtained. Multiple inserts were identified in 14.7% of the clones in the forward SSH library and 12.1% of the clones in the reverse SSH library. The in-house program Scanseq was used to detect redundant sequences within the forward and reverse SSH libraries, and low-redundancy subsets (ESTlr) of 979 ESTs from the forward and 555 from the reverse libraries were identified. The two most abundant ESTs observed in the forward SSH library shared similarity with the “putative senescence associated protein” from pea [dbj|BAB33421.1] and “hypothetical protein” [dbj|BAF01000.1] from Arabidopsis, having 28 and 14 copies, respectively. The most abundant ESTs from the reverse library shared similarity with “putative senescence associated protein” from pea [dbj|BAB33421.1|] with eight copies and “NADH-dependent hydroxypyruvate reductase” [gb|AAO73867.1|] from soybean with five copies. The sequences of the EST clones were submitted to NCBI as dbEST IDs 34850316–34851929 and GenBank accession nos. DW246150–DW247763. Clones containing multiple inserts were assigned individual accession numbers for each insert.
When queried against the NCBI EST database, 94% of the clones from the forward SSH library ESTlr with e-value ≤10−6 shared identity with Glycine max, 5% shared identity with other species in the Fabaceae (legume) family (Glycine soja, Glycine clandestine, Medicago, Phaseolus, and Lotus), and 1% shared identity with other organisms. Of the ESTlr with e-value ≤10−6 in the reverse SSH library, 90% of the clones shared identity with G. max, 8% shared identity with other Fabaceae (G. soja, G. clandestine, Medicago, Phaseolus, Lotus, Cajanus spp.), and 2% shared identity with other organisms.
Differential gene expression
The SSH library was made by pooling RNA from five time points (1, 6, 12, 24, and 48 hpi) before subtraction. We focused on the earliest stages of the host–pathogen interaction, believing this time frame best suited for identifying genes important for the immune response. We were expecting a small number of ESTs after subtraction but instead recovered 1,728 clones. This prompted us to make a custom cDNA microarray from the ESTs to both confirm changes in gene expression observed in the SSH library and to determine when the changes occurred.
The custom cDNA microarray was used to assess the expression of the ESTs in immune and susceptible reactions at 6, 12, 24, and 48 hpi. In addition to the SSH ESTs, cDNAs from four other soybean libraries (Khan et al. 2004; Alkharouf et al. 2006) were included on the array for a total of 7,883 cDNA inserts. Boxplots were used to assess variation across three biological replicates of the microarray analysis (Fig. 1). Replicate 2 showed abnormally high levels of background fluorescence, as indicated by the wider range of values observed in the boxplot. This resulted in excluding almost all differentially induced genes when all three replicates were used in the analysis. The reason for this is not due to biological variation but rather due to experimental variation resulting from operator error. Replicate 2 was subsequently removed from the study to provide greater sensitivity to the analysis. Removing one biological replicate results in a reduction in statistical power, but as the ESTs are represented in triplicate on each microarray, it was acceptable to lose one replication of the arrays to ensure the highest quality of data. Therefore, all of our analysis reported in this study is from two biological samples in which there was at least a twofold change in the gene expression ratio of immune vs. susceptible reactions.

Box plots showing variation in log2 expression ratios of immune vs. susceptible soybean inoculated with P. pachyrhizi and sampled at four time points after inoculation. Aggregate data of 6, 12, 24, and 48 h post-inoculation is shown for three biological replicates of the inoculation
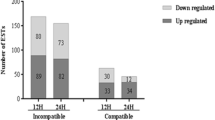
A total of 558 ESTs were differentially expressed over all the time points at p ≤ 0.05. Of these, 300 ESTs were upregulated and 258 were downregulated in the immune reaction. At 6 hpi, 56 ESTs were upregulated in the immune reaction, whereas approximately 80 ESTs were upregulated at each of the other time points. Similarly, 40 ESTs were downregulated in the immune reaction at 6 hpi, and approximately twice as many were downregulated at 12 and 24 hpi (Table 3).
The other functional categories appeared in both up- and downregulated gene list and usually at more than one time point. Oxidation-related genes were upregulated at 6 hpi (lipoxygenase) and 24 hpi (superoxide dismutase, peroxidase ATP8a, and lipoxygenase) and also downregulated at 6 hpi (thyroid-stimulating hormone beta subunit, bell-like homeodomain protein 2) and 48 hpi [Zinc finger C3HC4-type RING, Homeobox protein knotted-1 like 3 (KNAT3), and WD-40 repeat family protein]. Genes related to transcription, including transcription factors and DNA binding proteins, were upregulated at 12 hpi (NtWRKY4) and 24 hpi (Myb-related transcription activator) and downregulated at 24 hpi (putative WRKY-like protein; Table 4). Genes involved in cell growth and maintenance were downregulated at 6 hpi (ribulose-1,5-bisphosphate carboxylase, small nuclear ribonucleoproetin E homolg, and nam-like protein), 24 hpi (cyclin family protein), and 48 hpi (ribosomal protein L12 family protein and ribulose bisphosphate carboxylase small chain 1; Table 4).
Clustered mean expression profiles of ESTs sharing similar expression ratio variation over time are depicted in Fig. 3, and the corresponding genes from each of these profiles are listed in Table 5. The first profile (profile 1 in Fig. 3, Table 5) consists of ESTs downregulated at 6 and 12 hpi and upregulated at 24 and 48 hpi. This profile features ESTs that gradually increase expression over time. Members of this group have similarity to peroxidases and other proteins involved in oxidoreductase activity, such as putative senescence-associated protein. Cryptochrome 1 and pathogen-related protein, both involved in defense, were represented, as were bZIP and AP2-domain transcription factors. ESTs involved in the regulation of translation included translation initiation factor, ribosomal protein L1, 26S ribosomal RNA, and putative ATP-dependent RNA helicase. There were also 11 ESTs in this profile with unknown/no significant hits to the database.

Functional classification of differentially expressed genes in the immune vs. susceptible reaction of soybean cv. Komata to infection by P. pachyrhizi based upon homology search results
The strength of microarray analysis is in its ability to reveal similarities in expression patterns of groups of genes, rather than in pinpointing specific expression changes for individual genes. Particularly with an array produced from ESTs, the identity and behavior of an individual spot on the array has a high degree of uncertainty, while multiple observations of similar behavior in a group of functionally related genes are far more informative (Dardick 2007). For this reason, the differentially expressed ESTs from all libraries were classified into functional groups based upon similarity to known sequences in the NCBI databases (Fig. 2). A list of selected genes from each of the EST libraries at 6, 12, 24, and 48 hpi is presented in Table 4. Several functional categories appeared at multiple time points and in both the up- and downregulated sets of genes. Defense was the most represented functional category, upregulated at 6 hpi (Bax inhibitor, caffeic acid O-methyltransferase, and phenylalanine ammonia lyase), 12 hpi (putative cinnamoyl-CoA reductase, β-xylosidase, Bax inhibitor, chalcone synthase 2, and heat shock transcription factor 21), 24 hpi (elicitor-inducible gene product EIG-I24 and pectinesterase), and 48 hpi [NAD(P)H dependent 6′-deoxychalcone synthase, naringenin-chalcone synthase, cytochrome P450, and 6a-hydroxymaaciain methyltransferase] (Table 4). Some defense genes were also downregulated at all time points, including xyloglucan endotransglycosylase I at 6 hpi; DnaJ protein-like, LRR receptor protein kinase, and heat shock transcription factor HSF1 at 12 hpi; heat shock transcription factor homolog T32G6.21, putative receptor-like protein kinase, and PR-4 typr protein at 24 hpi; and cytochrome P450, MYB-related protein B, and elicitor response element WRKY3 at 48 hpi (Table 4).

Expression profiles of genes differentially expressed in leaves of the soybean cv. Komata to infection by P. pachyrhizi. The expression ratios of the immune vs. susceptible reactions were calculated from normalized, rescaled ratios of the fluorescent ratios of intensity in the red and green channels. Ratios represent the average from the two biological samples that were used. Error bars show the standard deviation of expression levels at each time point
ESTs in the second profile (profile 2 in Fig. 3, Table 5) are downregulated at 6 hpi and continue to be downregulated over time in the immune reaction. Many of the ESTs in this group are predicted to be involved in the biogenesis of cell wall, for example, extensins, xyloglucan endotransglycosylases, and repetitive proline-rich proteins. Several ESTs encode for proteins involved in transport, such as aquaporin, permeases, and translocators. In addition, an EST was identified with similarity to the oxidoreductase fis1, which has been shown to facilitate biotrophic relationship between rust and flax (Ayliffe et al. 2002; Roberts and Pryor 1995). Other ESTs in the functional category of defense including abscisic stress-ripening protein homolog, endochitinase, catalase, cytochrome P450 monooxygenase, heat shock protein 70, and a putative pathogenesis-related protein from Arabidopsis were found, as well as ESTs involved in signaling such as remorin and ADR12-2 protein, which is known to be downregulated by auxin. Ten ESTs with no significant/null hit to the database were observed in this profile.
The third profile (profile 3 in Fig. 3, Table 5) included ESTs downregulated at 6 and 12 hpi and upregulated in the immune reaction starting at 24 hpi. The majority of annotated ESTs in this group included defense-related genes such as heat shock protein 70, pectinesterase, cytochrome P450, salt-induced AAA-type ATPase, and trypsin inhibitor. Also present were transcription regulators such as IAA7-like protein and isoflavone reductase-like protein, as well as 14-3-3-like protein, which is involved in signaling. Nine members of profile 3 had unknown/no significant hit to the NCBI databases.
The final profile (profile 4 in Fig. 3, Table 5) included ESTs upregulated for the first 12 hpi in the immune reaction and then downregulated after 24 hpi. ESTs in this group had similarity to peroxidases, lipoxygenases, and to oxylase-like proteins involved in oxidoreductase activity and antibiotic synthesis. Defense-related ESTs include Bax inhibitor, NtPRp27, which is a secreted pathogenesis-related protein that is inducible by ethylene and JA (Okushima et al. 2000), peptidylprolyl isomerase ROF1 that is induced by wounding and accelerates the folding of proteins (Vucich and Gasser 1996), and diphosphomevalonate decarboxylase-like protein involved in phenylpropanoid synthesis. ESTs involved in transcription regulation include a MYB-like transcription factor and WD-40 repeat protein. ESTs involved in signaling included somatic embryogenesis receptor-like kinase-like protein and SOS2-like protein kinase. Twenty-one ESTs with no significant/null hit to the database were observed in this profile.
Quantitative real-time RT-PCR
PCR primers were designed to 14 genes representing a range of functional categories. Of the 14 primer pairs, seven amplified single DNA products. Four primer pairs displayed linear amplification across the range of RNA concentrations tested (data not shown) and were chosen for further analysis. Three of the gene encode for known proteins, SAM-22, ANS, and nitrate transporter 1 (NTR1-5), while the fourth gene product is unknown. The fold changes were determined for each of these genes by quantitative real-time RT-PCR and microarrays and are shown Fig. 4. The RT-PCR data for the gene encoding NTR1-5 showed upregulation (a KH/KT expression ratio greater than 1) at both 6 and 12 hpi and downregulation (expression ratio less than 1) at 24 and 48 hpi. The microarray data showed a similar pattern (Fig. 4a). ANS showed upregulation at all four time points by RT-PCR and microarrays (Fig. 4b). The unknown gene was more problematic because, while both the microarrays and the RT-PCR revealed downregulation at 6 and 12 hpi, the data disagreed at 24 hpi, with the RT-PCR analysis indicating downregulation and the microarray indicating upregulation (Fig. 4c). The 48 hpi data is more consistent, with both RT-PCR and microarray analysis showing upregulation (Fig. 4c). SAM-22 showed downregulation at 6 and 24 hpi and upregulation at 12 and 48 hpi (Fig. 4d). Overall, our quantitative real-time RT-PCR data confirm the relative expression trends of the respective genes obtained by our microarray analysis.

Differential expression ratio of selected genes in microarray analysis and quantitative real time RT-PCR in the soybean cv. Komata to infection by P. pachyrhizi: a nitrate transporter 1 (NTR1-5), b anthocyanidin synthase (ANS), c unknown protein, and d starvation associated message 22 (SAM-22)
Discussion
The gene expression of soybean after inoculation with two different isolates of P. pachyrhizi was analyzed using SSH and cDNA microarrays. Specifically, we inoculated the soybean cv. Komata, which contains the Rpp1 rust resistance gene, separately with P. pachyrhizi isolates HW94-1 and TW72-1 and compared the gene expression patterns in the corresponding immune and susceptible reactions. This allowed us to make direct comparisons without being concerned about differences in soybean genotype influencing gene expression patterns. Unlike the RB-resistant reaction that occurs on soybean cvs. containing Rpp2, Rpp3, or Rpp4, there are no visible macroscopic symptoms on cv. Komata in an immune reaction (Bromfield 1984). The initial infection process by P. pachyrhizi is similar in the Rpp1 immune and susceptible reactions. Spore germination, appressorium formation, and penetration of the epidermal cell occur within 24 h (Keogh and Deverall 1980; McLean and Byth 1981). In the susceptible reaction, haustoria are visible by 48 h and hyphal growth proceeds intercellularly. However, in the immune reaction, fungal growth does not continue beyond the formation of the penetration peg (Keogh and Deverall 1980; McLean and Byth 1981). Therefore, we chose to examine gene expression during the first 48 hpi. By analyzing gene expression at 6, 12, 24, and 48 hpi, we were able to construct temporal expression profiles.
We initially created a SSH library enriched for sequences significantly more or less abundant in the immune reaction and sequenced the library to identify novel genes important for soybean rust resistance. We were surprised to find that the most common EST in both the forward and reverse libraries shared similarity to the same protein, a “putative senescence-associated protein” from pea [dbj|BAB33421.1]. This suggests that the SSH enrichment was not particularly successful. It is also plausible that these two ESTs represent distinct members of a gene family. Soybean has been shown to have several gene families whose members are highly similar at the sequence level while exhibiting different expression patterns (Grandbastien et al. 1986; D’ovidio et al. 2006; Nelson and Shoemaker 2006; Webb et al. 2007). The small size of the ESTs, an unavoidable feature of the SSH construction method, makes it problematic to unambiguously identify members of gene families. The large number of clones in the SSH library was unpredicted, and we were unable to identify ESTs playing a key role in soybean rust resistance without additional information. In addition, pooling RNA from multiple time points did not allow for resolution of temporal changes in gene expression. Therefore, we chose to construct a cDNA microarray to confirm differentially expressed clones and ascertain changes in gene expression over time.
Our overall goal in this experiment was to identify soybean genes involved in the early stages of the immune response to P. pachyrhizi. We were successful in this endeavor, measured by the fact that so many of the differentially expressed genes had similarilty to defense-related genes identified in other plant–pathogen interactions. The differentially expressed gene list at each time point contained many defense-related genes, and these genes often had complementary functions. While no single gene was upregulated at every time point, classes of genes encoding oxidizing enzymes, such as lipoxygenases and peroxidases, were upregulated at all time points. In addition, antioxidant genes that help the cell protect itself against the damaging effects of oxidative stress (Camera et al. 2004; Foyer and Noctor 2005; Pinto et al. 2006) were also upregulated. Reactive oxygen species (ROS) play numerous roles in defense, from catalysis of cell wall fortification (Bradley et al. 1992), to signaling cascades (Levine et al. 1994), to toxic free radical production (Brisson et al. 1994). In addition to the protective role of ROS, oxidative damage of the host cell can also lead to necrosis. It has been suggested that a key indicator of the cell’s ability to survive pathogen attack is the oxidative balance in the cell (Foyer and Noctor 2003, 2005; Kawano 2003; Levine et al. 1994; Winkel-Shirley 2001). Soybeans (cv. Williams 82) inoculated with Pseudomonas syringae had an initial weak transient H2O2 burst followed approximately 3 hpi with a second massive burst of O2 production that was sustained for several hours (Levine et al. 1994). H2O2 was shown to function as both a diffusible signal triggering expression of defensive compounds in cells surrounding the infection site and as a programmed cell death (PCD) trigger in the infected cell (Levine et al. 1994). Peroxidases and lipoxygenases are both highly represented in profiles 2 and 4, suggesting that a complex interplay of oxide concentrations/levels may play an important role in the ability of the host cell to resist further infection (Passardi et al. 2005; Shah 2005). Lipoxygenases in profile 4 are induced early, returning to basal levels 12 hpi. Their expression could be responsible for an early oxidative burst resulting in cell-wall modification and induction of defensive compounds such as glutathione S-transferase (GST) at the site of infection and in the neighboring cells. Peroxidases in profile 1 begin with low expression in resistant soybeans but, by 48 hpi, are induced in resistant cells and could provide a second oxidative burst in infected cells that might trigger PCD. Additional enzymes related to the oxidative state of the cell, including senescence-associated protein (an ACC oxidase, profile 1) and isoflavone reductase (high expression at 24 and 48 hpi, profile 3) are also coordinately regulated. The lack of visible lesions in Rpp1 resistant soybeans could indicate that PCD is not occurring in Rpp1 plants, or perhaps only the initially infected cell becomes necrotic, resulting in a lesion so small as to be invisible to the naked eye. Further investigation into the nature of the Rpp1-mediated immune response will surely shed light on the reason for the lack of a visible lesion.
Genes in the phenylpropanoid synthesis pathway are among the most powerful antioxidants in the cell and are significantly upregulated by soybean in response to P. syringae (Zabala et al. 2006; Zou et al. 2005). Phenylpropanoid synthesis also leads to the production of defensive phytoalexins and cell-wall-reinforcing lignin (Winkel-Shirley 2001). Several genes similar to those involved in phenylpropanoid synthesis were differentially expressed in this study, suggesting a role of phenylpropanoids in maintaining the oxidative balance of soybean cells and in rust resistance. Recently, a commercial oligonucleotide microarray was developed containing approximately 36,500 soybean genes (GeneChip Soybean Genome Array, Affymetrix, Santa Clara, CA, USA). The Soybean Genome Array was used to analyze gene expression in Rpp2-mediated rust resistance (van de Mortel et al. 2007). That study also identified the phenylpropanoid synthesis pathway as important in the RB resistance reaction (van de Mortel et al. 2007). It will be interesting to see whether additional studies can identify which consequence of phenylpropanoid synthesis (antioxidant production, phytoalexin production, or lignin accumulation) is most important for the immune reaction.
In addition to the importance of phenylpropanoids in rust resistance, van de Mortel et al. (2007) also found that WRKY transcription factors were regulated in the Rpp2-mediated resistance response of soybean rust. Similarly, MYB and bHLH transcription factors are induced in nitrogen-stressed Arabidopsis (Unni et al. 2007). These families of transcription factors were also differentially regulated in the immune response to soybean rust, with MYB transcription factors upregulated at 12 and 24 hpi and downregulated at 48 hpi. WRKY transcription factors were upregulated at 12 hpi and downregulated at 24 and 48 hpi. Differential expression of multiple transcription factor family members suggests that a complex positive and negative regulation pattern of defense pathways allows the host to prevent rust infection.
A recent study of susceptible soybeans infected with P. pachyrhizi (Panthee et al. 2007) reveals moderate induction of general defense-related genes, including GSTs, heat shock proteins, and salicylic-acid-dependent genes, while these same genes and defense pathways are induced earlier and much stronger in the Rpp2 resistant reaction (van de Mortel et al. 2007). This suggests that rust resistance may be more a matter of timing and degree of induction of innate immunity pathways rather than the result of specific induction of a few genes.
This study identified 538 differentially expressed ESTs using a custom-designed cDNA microarray. By extending our study with the use of the Affymetrix GeneChip Soybean Genome Array, we anticipate the discovery of additional differentially expressed genes that would allow for a more comprehensive understanding of Rpp1-mediated resistance. In addition, a comparison could be made to the differentially expressed genes identified from the Rpp2 resistant reaction. Such a comparison would provide insight as to whether Rpp1 and Rpp2 share similar resistance mechanisms.
References
Alkharouf N, Khan R, Matthews B (2004) Analysis of expressed sequence tags from roots of resistant soybean infected by the soybean cyst nematode. Genome Biol 47:380–388
Alkharouf N, Jamison DC, Matthews BF (2005) Online analytical processing (OLAP): a fast and effective data mining tool for gene expression databases. J Biomed Biotechnol 2:181–188
Alkharouf N, Klink V, Chouikha I, Beard H, Macdonald M, Meyer S, Knap H, Khan R, Matthews B (2006) Timecourse microarray analyses reveal global changes in gene expression of susceptible Glycine max (soybean) roots during infection by Heterodera glycines (soybean cyst nematode). Planta 224:838–852
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29
Ayliffe MA, Roberts JK, Mitchell HJ, Zhang R, Lawrence GJ, Ellis JG, Pryor TJ (2002) A plant gene up-regulated at rust infection sites. Plant Physiol 129:169–180
Bateman A, Coin L, Durbin R, Finn R, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer E, Studholme D, Yeats C, Eddy S (2004) The Pfam protein families database. Nucleic Acids Res 32:D138–D141
Bonde MR, Nester SE, Austin CN, Stone CL, Frederick RD, Hartman GL, Miles MR (2006) Evaluation of virulence of Phakopsora pachyrhizi and P. meibomiae isolates. Plant Dis 90:708–716
Bradley DJ, Kjellbom P, Lamb CJ (1992) Elicitor- and wound-induced oxidative cross-linking of a proline-rich plant cell wall protein: a novel, rapid defense response. Cell 70:21–30
Brisson LF, Tenhaken R, Lamb C (1994) Function of oxidative cross-linking of cell wall structural proteins in plant disease resistance. Plant Cell 6:1703–1712
Bromfield KR (1984) Soybean rust. Monograph No. 11. America Phytopathological Society, St. Paul
Bromfield KR, Hartwig EE (1980) Resistance to soybean rust [Phakopsora pachyrhizi] and mode of inheritance. Crop Sci 20:254–255
Camera SL, Gouzerh G, Dhondt S, Hoffmann L, Fritig B, Legrand M, Heitz T (2004) Metabolic reprogramming in plant innate immunity: the contributions of phenylpropanoid and oxylipin pathways. Immunol Rev 198:267–284
Cherry E, Peet C (1966) An efficient device for the rapid collection of fungal spores from infected plants. Phytopathology 56:1102–1103
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Codd E, Codd S, Salley C (1993) Providing OLAP (on-line analytical processing) to user-analysts: an IT mandate. Technical Report, EF Codd & Associates
Dardick C (2007) Comparative expression profiling of Nicotiana benthamiana leaves systemically infected with three fruit tree viruses. Mol Plant Microb Interact 20:1004–1017
Diatchenko L, Lau Y-FC, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Gurskaya N, Sverdlov E, Siebert PD (1996) Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci U S A 93:6025–6030
D’ovidio R, Roberti S, Di Giovanni M, Capodicasa C, Melaragni M, Sella L, Tosi P, Favaron F (2006) The characterization of the soybean polygalacturonase-inhibiting proteins (Pgip) gene family reveals that a single member is responsible for the activity detected in soybean tissues. Planta 224:663–645
Fehr WR, Caviness CE (1977) Stages of soybean development. Iowa State University, Ames, IA
Fei F, Tan Y, Zhou M (1996) Histopathological study of rust (Phakopsora pachyrhizi Syd.) resistance in soybeans. Oil Crops China 18:48–50
Foyer CH, Noctor G (2003) Redox sensing and signaling associated with reactive oxygen in chloroplasts, peroxisomes and mitochondria. Physiol Plantarum 119:355–364
Foyer CH, Noctor G (2005) Redox homeostasis and antioxidant signaling: a metabolic interface between stress perception and physiological responses. Plant Cell 17:1866–1875
Golkari S, Gilbert J, Prashar S, Procunier JD (2007) Microarray analysis of Fusarium graminearum-induced wheat genes: identification of organ-specific and differentially expressed genes. Plant Biotechnol J 5:38–49
Grandbastien MA, Berry-Lowe S, Shirley BW, Meagher RB (1986) Two soybean ribulose-1,5-bisphosphate carboxylase small subunit genes share extensive homology even in distant flanking sequences. Plant Mol Biol 7:451–465
Hartman GL (1996) Highlights of soybean rust research at the Asian Vegetable Research and Development Center. In: Sinclair JB, Hartman GL (eds) Proceedings of the soybean rust workshop, 9–11 August 1995. College of Agricultural, Consumer, and Environmental Sciences, National Soybean Research Laboratory, Urbana, IL, pp 19–28
Hartwig E (1986) Identification of a fourth major gene conferring resistance to soybean rust. Crop Sci 26:1135–1136
Hartwig E, Bromfield K (1983) Relationships among three genes conferring resistance to soybean rust. Crop Sci 23:237–239
Hegde P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, Hughes JE, Snesrud E, Lee N, Quackenbush J (2000) A concise guide to cDNA microarray analysis. Biotechniques 29:548–556
Hennings P (1903) Some new Japanese Uredinales. Hedwiga 4(suppl.):107–108
Hyten DL, Hartman GL, Nelson RL, Frederick RD, Concibido VC, Narvel JM, Cregan PB (2007) Map location of the Rpp1 locus that confers resistance to soybean rust in soybean. Crop Sci 47:835–838
Hu HY, Zhuang JY, Chai RY, Wu JL, Fan YY, Zheng KL (2006) Isolation and characterization of defense response genes involved in neck blast resistance of rice. Yi Chuan Xue Bao 33:251–261
Khan R, Alkharouf N, Beard HS, MacDonald M, Chouikha I, Meyer S, Grefenstette J, Knap H, Matthews BF (2004) Resistance mechanisms in soybean: gene expression profile at an early stage of soybean cyst nematode invasion. J Nematol 36:241–248
Kawano T (2003) Roles of the reactive oxygen species-generating peroxidase reactions in plant defense and growth induction. Plant Cell Rep 21:829–837
Keogh R, Deverall B (1980) Comparison of histological and physiological responses to Phakopsora pachyrhizi in resistant and susceptible soybean. Trans Brit Mycol Soc 74:329–333
Kurkcuoglu S, Degenhardt J, Lensing J, Al-Masri AN, Gau AE (2007) Identification of differentially expressed genes in Malus domestica after application of the non-pathogenic bacterium Pseudomonas fluorescens Bk3 to the phyllosphere. J Exp Bot 58:733–741
Levine A, Tenhaken R, Dixon R, Lamb CJ (1994) H2O2 from the oxidative burst orchestrates the plant hypersensitive disease resistance response. Cell 79:583–589
Li GS, Asiegbu FO (2004) Use of Scots pine seedling roots as an experimental model to investigate gene expression during interaction with the conifer pathogen Heterobasidion annosum. J Plant Res 117:155–162
Lin H, Doddapaneni H, Takahashi Y, Walker MA (2007) Comparative analysis of ESTs involved in grape responses to Xylella fastidiosa infection. BMC Plant Biol 7:8
Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki C, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH (2005) CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res 33:D192–D196
McLean R, Byth D (1976) Resistance of soybean to rust in Australia. Aust Plant Pathol Soc Newsl 5:34–36
McLean R, Byth D (1980) Inheritance of resistance to rust (Phakopsora pachyrhizi) in soybeans. Aust J Agric Res 31:951–956
McLean R, Byth D (1981) Histological studies of the pre-penetration development and penetration of soybeans by rust. Phakopsora pachyrhizi Syd. Aust J Agric Res 32:435–443
Melching JS, Bromfield KR, Kingsolver CH (1983) The plant pathogen containment facility at Frederick, Maryland. Plant Dis 67:717–722
Miles MR, Hartman GL, Frederick RD (2003a) Soybean rust: Is the U.S. soybean crop at risk? Online. APSnet Feature, American Phytopathological Society, St. Paul, MN
Miles MR, Hartman GL, Levy C, Morel W (2003b) Current status of soybean rust control by fungicides. Pestic Outlook 14:197–200
Miles MR, Frederick, RD, Hartman, GL (2006) Evaluation of soybean germplasm for resistance to Phakopsora pachyrhizi. Online. Plant Health Progress. DOI 10.1094/PHP-2006-0104-01-RS
Mo J, Zhu G, Chen Y, Huang L, Sun H (1994) Preliminary survey and identification of soybean varietal resistance to rust. In: Dept. of Science and Technology, MARPC; Asian Regional Center, AVRDC; and Oil Crops Research Institute, CAAS (eds) Advance of soybean rust research. Proceedings of a meeting held in 1992. Hubei Science and Technology Publishing House, Hubei, People’s Republic of China, pp 116–122
Monteros MJ, Missaoui AM, Phillips DV, Walker DR, Boerma HR (2007) Mapping and confirmation of the “Hyuuga” red-brown lesion resistance gene for Asian soybean rust. Crop Sci 47:829–836
Nelson RT, Shoemaker R (2006) Identification and analysis of gene families from the duplicated genome of soybean using EST sequences. BMC Genomic 7:204
Okushima Y, Koizumi N, Kusano T, Sano H (2000) Secreted proteins of tobacco cultured BY2 cells: identification of a new member of pathogenesis-related proteins. Plant Mol Biol 42:479–488
Panthee DR, Yuan JS, Wright DL, Marois JJ, Mailhot D, Stewart CN Jr (2007) Gene expression analysis in soybean in response to the causal agent of Asian soybean rust (Phakopsora pachyrhizi Sydow) in an early growth stage. Funct Integr Genom 7:291–301(11)
Pfaffl MW (2001) A mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36
Passardi F, Cosio C, Penel C, Dunand C (2005) Peroxidases have more functions than a Swiss army knife. Plant Cell Rep 24:255–265
Patil PV, Basavaraja GT (1997) A prospective source of resistance to soybean rust. Karnataka J Agric Sci 10:1241–1243
Pinto MCD, Paradiso A, Leonetti P, Gara LD (2006) Hydrogen peroxide, nitric oxide and cytosolic ascorbate peroxidase at the crossroad between defense and cell death. Plant J 48:784–795
Ray S, Anderson JM, Urmeev FI, Goodwin SB (2003) Rapid induction of a protein disulfide isomerase and defense-related genes in wheat in response to the hemibiotrophic fungal pathogen Mycosphaerella graminicola. Plant Mol Biol 53:741–754
Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, Miller N, Mueller LA, Mundodi S, Reiser L, Tacklind J, Weems DC, Wu Y, Xu I, Yoo D, Yoon J, Zhang P (2003) The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res 31:224–228
Roberts JK, Pryor A (1995) Isolation of a flax (Linum usitatissimum) gene induced during susceptible infection by flax rust (Melampsora lini). Plant J 8:1–8
Rosen S, Skaletsky H (2000) Primer 3 on the WWW for general users and for biologist programmers. In: Krawtez S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana, Totowa, NJ, pp 365–386
Schneider RW, Hollier CA, Whitham HK, Palm ME, McKemy JM, Hernandez JR, Levy L, DeVries-Patterson R (2005) First report of soybean rust caused by Phakopsora pachyrhizi in the continental United States. Plant Dis 89:774
Schenk PM, Kazan K, Wilson I, Anderson JP, Richmond T, Somerville S, Manners JM (2000) Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc Natl Acad Sci U S A 97:11655–11660
Sconyers LE, Kemerait RC, Brock J, Phillips DV, Jost PH, Sikora EJ, Gutierrez-Estrada A, Mueller JD, Marois JJ, Wright DL, Harmon CL (2006) Asian soybean rust development in 2005: A perspective from the Southeastern United States. Online. APSnet Feature, American Phytopathological Society, St. Paul, MN
Shah J (2005) Lipids, lipases, and lipid-modifying enzymes in plant disease resistance. Ann Rev Phytopathol 43:229–260
Singh BB, Gupta SC, Singh BD (1974) Sources of field resistance to rust [Phakopsora pachyrhizi] and yellow mosaic diseases of soybean. Indian J Genet Plant Breed 34:400–404
Tan YJ, Yu Z, Yang CY (1996) Soybean rust. China Agricultural Press, Beijing, China
Tomkins JP, Mahalingam R, Smith H, Goicoechea JL, Knap HT, Wing RA (1999) A bacterial artificial chromosome library for soybean PI 437654 and identification of clones associated with cyst nematode resistance. Plant Mol Biol 41:25–32
Tschanz AT, Wang TC, Tsai BY (1986) Recent advances in soybean rust research at Asian Vegetable Research and Development Center. In: Shanmugasundaram S, Sulberger EW (eds) Soybeans in tropical and subtropical cropping systems. Asian Vegetable Research and Development Center, Shanhua, Tainan, Taiwan, pp 237–245
Unni SL, Rune S, Pål S, Cathrine L (2007) Nitrogen deficiency enhances expression of specific MYB and bHLH transcription factors and accumulation of end products in the flavonoid pathway. Planta 225:1245–1253
van de Mortel M, Recknor JC, Graham MA, Nettleton D, Dittman JD, Nelson RT, Godoy CV, Abdelnoor RV, Almeida AMR, Baum TJ, Whitham SA (2007) Distinct biphasic mRNA changes in response to Asian soybean rust infection. Mol Plant-Microb Interact 20:887–899
Verica JA, Maximova SN, Strem MD, Carlson JE, Bailey BA, Guiltinan MJ (2004) Isolation of ESTs from cacao (Theobroma cacao L.) leaves treated with inducers of the defense response. Plant Cell Rep 23:404–413
Vucich V, Gasser C (1996) Novel structure of a high molecular weight FK506 binding protein from Arabidopsis thaliana. Mol Gen Genet 252:510–517
Webb CJ, Chan-Weiher C, Johnson DA (2008) Isolation of a novel family of genes related to 2-oxoglutarate-dependent dioxygenases from soybean and analysis of their expression during root nodule senescence. J Plant Physiol (in press). DOI 10.1016/j.jplph.2007.10.004
Winkel-Shirley B (2001) Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol 126:485–493
Wu CH, Huang H, Nikolskaya A, Hu Z, Barker WC (2004) The iProClass integrated database for protein functional analysis. Comput Biol Chem 28:87–96
Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O’Donovan C, Redaschi N, Suzek B (2006) The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res 34:D187–D191
Yang Y, Dudoit S, Luu P, Lin D, Peng V, Ngai J, Speed T (2002) Normalization of cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acid Res 30:e15
Zabala G, Zou J, Tuteja J, Gonzalez DO, Clough SJ, Vodkin LO (2006) Transcriptome changes in the phenylpropanoid pathway of Glycine max in response to Pseudomonas syringae infection. BMC Plant Biol 6:26
Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, Matz MV, Meleshkevitch E, Moroz LL, Lukyanov SA, Shagin DA (2004) Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res 32:e37
Zou J, Rodriguez-Zas S, Aldea M, Li M, Zhu J, Gonzalez DO, Vodkin LO, DeLucia E, Clough SJ (2005) Expression profiling soybean response to Pseudomonas syringae reveals new defense-related genes and rapid HR-specific downregulation of photosynthesis. Mol Plant-Microb Interact 18:1161–1174
Acknowledgments
We gratefully acknowledge Christine Stone and Craig Austin for maintenance and propagation of P. Pachyrhizi isolates and with the inoculations. We also would like to thank Hunter Beard for microarray slide spotting and technical expertise. This project was funded in part by the United Soybean Board as Projects 2229, 3217, and 4217 and supports the goals of the USDA National Strategic Plan for the Coordination and Integration of Soybean Rust Research. The use of trade, firm, or corporation names in this publication is for the information and convenience of the reader. Such use does not constitute an official endorsement or approval by the US Department of Agriculture or the Agricultural Research Service of any product or service to exclusion of others that may be suitable.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Transcripts found to be differentially expressed by custom microarray analysis of immune vs. susceptible soybean leaves inoculated with P. pachryhizi and sampled at four times after inoculationa (DOC 1.05 mb)
Rights and permissions
About this article
Cite this article
Choi, J.J., Alkharouf, N.W., Schneider, K.T. et al. Expression patterns in soybean resistant to Phakopsora pachyrhizi reveal the importance of peroxidases and lipoxygenases. Funct Integr Genomics 8, 341–359 (2008). https://doi.org/10.1007/s10142-008-0080-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-008-0080-0