
Abstract
During antimicrobial photodynamic inactivation (APDI) in the treatment of an infection, it is likely that microorganisms would be exposed to sub-lethal doses of APDI (sAPDI). Although sAPDI cannot kill microorganisms, it can significantly affect microbial virulence. In this study, we evaluated the effect of sAPDI using methylene blue (MB) on the expression of genes belonging to two quorum sensing (QS) operons (rhl and las systems) and two genes necessary for biofilm formation (pelF and pslA) under QS control in Pseudomonas aeruginosa. Biofilm formation ability of P. aeruginosa ATCC 27853 exposed to sAPDI (MB at 0.012 mM and light dose of 23 J/cm2) was evaluated using triphenyl tetrazolium chloride (TTC) assay and scanning electron microscopy (SEM). The effect of sAPDI on expression of rhlI, rhlR, lasI, lasR, pelF, and pslA were also evaluated by quantitative real-time polymerase chain reaction. Quantitative assay (TTC) results and morphological observations (SEM) indicated that a single sAPDI treatment resulted in a significant decrease in biofilm formation ability of P. aeruginosa ATCC 27853 compared to their non-treated controls (P = 0.012). These results were consistent with the expression of genes belonging to rhl and las systems and pelF and pslA genes. The results suggested that the transcriptional decreases caused by MB-sAPDI did lead to phenotypic changes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Universal increase in drug-resistant bacteria and simultaneous decline towards the discovery of new antibacterial agents now presents a serious threat to the treatment of life-threatening infections. Therefore, it is necessary to search for new antimicrobial strategies that act more efficiently than the current antibiotics, and to which bacteria will not easily develop resistance [1].
One promising approach is antimicrobial photodynamic inactivation (APDI). APDI makes use of the photo-oxidative stress which is elicited by exogenously administered photosensitizers (PSs) that absorb visible light and cause the production of reactive oxygen species (ROS) by energy transfer or electron flow to oxygen [2]. When energy is transferred to oxygen, singlet oxygen (1O2) arises, however, when electrons are transferred to oxygen, radicals such as superoxide anions (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (·OH) arise, and these may trigger the development of inorganic or organic radicals depending on the microenvironment [3]. When the PS passes through the cell wall, ROS are produced in the cytoplasm and cause macromolecule degradation and bacterial killing. If ROS are generated near the outer side of the bacterial envelope, cellular integrity is damaged [4].
Oxidative stress in microbial life is an imbalance between the production of ROS and the ability of a cell to neutralize their harmful effects. Bacteria can respond to oxidative stress in several ways. Some molecules are constitutively present and help to maintain an intracellular reducing environment or to scavenge chemically ROS, including non-enzymatic antioxidants (nicotinamide adenine dinucleotide phosphate (NADPH) and nicotinamide adenine dinucleotide (NADH) pools, β-carotene, ascorbic acid, α-tocopherol, and glutathione). In addition, specific enzymes decrease the constant levels of ROS including catalase and superoxide dismutase [5]. Moreover, after photo-oxidative stress induction, bacteria are able to express heat shock proteins (HSPs) to maintain bacterial integrity by eliminating misfolded proteins [6]. On the other hand, the ability of bacteria to produce biofilm and pigments may contribute to survival to oxidative stress [7]. Some mechanisms by which bacteria may respond to APDI-mediated oxidative stress are protective pigments [8, 9], capsular polysaccharide [10], efflux pumps [11], anti-oxidant enzymes [12], and HSPs [13].
Pseudomonas aeruginosa is a well-known opportunistic human pathogen that has the ability to produce many virulence factors and biofilm [14]. Biofilms are known as organized communities of microbial cells attached onto a living or inert surface and encased in a matrix of extracellular polymeric substances (EPS) [15]. EPS as “house of the biofilm cells” represents 85% of total biofilm biomass [16]. Biofilm is an important factor for P. aeruginosa pathogenesis, plays a role in its infections and avoidance of immune defense mechanisms, and has the ability to protect the bacteria from antibiotics [17].
Extracellular polysaccharides are key components of the biofilm matrix. It seems that P. aeruginosa has the ability to produce multiple types of polysaccharides (alginate, Pel, and psl) [18]. Alginate consists of mannuronic acid and guluronic acid monomers and plays a role in formation of bacterial microcolonies in vivo [19]. The composition of Pel polysaccharide is still unclarified [20], while psl consists of a repeating penta-saccharide consisting of d-mannose, l-rhamnose, and d-glucose [21]. Both Pel and psl polysaccharides are involved at early stages of biofilm formation [18].
It has been shown that a bacterial cell-cell communication mechanism, widely known as quorum sensing (QS), plays a key role in modulating the expression of virulence genes in P. aeruginosa [22]. This microorganism has two acyl homoserine-lactone (AHL) based-QS systems, las and rhl. The las system consisted of the transcriptional regulator LasR and its cognate AHL signal, N-(3-oxododecanoyl)-l-homoserine lactone (3-oxo-C12-HSL), synthesized by the AHL synthase LasI [23]. Similarly, the rhl system consisted of RhlR together with its cognate AHL, N-butyryl-l-homoserine lactone (C4-HSL), synthesized by the RhlI AHL synthase [24]. Reports showed that the QS regulator LasR can bind to the promoter region of the psl operon, suggesting that QS can regulate psl expression [25]. Also, it has been reported that the rhl system moderates P. aeruginosa biofilm formation by increasing Pel polysaccharide biosynthesis [26].
If APDI was used in clinical setting and the PS would extend to the target site at only sub-lethal concentrations, and was activated by light producing sub-lethal of ROS, any bacteria viable at the site of infection would be exposed to doses of APDI that would not cause total cell death, i.e., sub-lethal doses of APDI (sAPDI), exposing survivors to ROS stress [27]. Such stress leads to increased mutational events, which could lead to selection for survival [28].
The effect of sAPDI using different PSs on biofilm formation ability of microorganisms has been reported in several studies [29]. Also, there is a recent study investigating the involvement of QS in response to APDI [30]. However, to our knowledge, there is no report on the effect of sAPDI on expression of genes involved in P. aeruginosa biofilm formation. For the first time, in this study, we aimed to evaluate the effect of sAPDI using methylene blue (MB) on the expression of genes belonging to two QS operons (rhl and las systems) and two genes necessary for biofilm formation (pelF and pslA) under QS control in P. aeruginosa.
Material and methods
Growth conditions
P. aeruginosa ATCC 27853 was grown aerobically on Tryptic soy agar (Merck, Germany) at 37 °C for 18–24 h and then, bacterial suspension was prepared in sterile 0.9% saline to reach the turbidity of 0.5 McFarland. Tryptic Soya Broth (TSB, Merck, Germany) was used as the liquid medium for biofilm culture.
Photosensitizer and light source
Methylene blue (MB, Sigma-Aldrich) was used as the photosensitizing agent. MB stock solution (3.2 mM) was prepared in 0.9% saline, filter sterilized, and stored at 4 °C in the dark no more than 2 weeks prior to use. Stock solution was further diluted to obtain the desired concentrations (0.012–0.8 mM).
The light source used in this study was a diode laser (AZOR, Russia) with an emission at 650 nm. The total output power provided by the device was 30 mW.
Effect of APDI on viability and biofilm formation ability of P. aeruginosa
Viable counts, shown as colony forming units (CFU ml−1) in P. aeruginosa ATCC 27853 cell suspension, were estimated after APDI. To this end, 21 sterile 2-ml microtubes were prepared and filled with 500 μl of cell suspensions (1–2 × 108 CFU/ml) and 500 μl of each concentration of MB (at final concentrations of 0.006–0.4 mM) and incubated for 10 min in the dark. Then, samples were irradiated with a diode red laser light for 10 min (23 J/cm2), followed by washing excess MB. One milliliter TSB was added to each microtube; serially decimal dilutions were prepared in 0.9% saline and streaked on nutrient agar plates. Plates were incubated for 18–24 h at 37 °C in the dark to allow colony formation. The accepted range for countable colonies on a plate was between 30 and 300.
To determine biofilm formation ability, aliquots of 200 μl of each APDI-treated suspension in TSB medium were transferred to wells of 96-well flat-bottom, sterile microtiter plates (SPL, Korea). Plates were incubated for 24 h at 37 °C. Thereafter, each well was aspirated, plates were washed five times with sterile saline and biofilm formation ability was further evaluated by triphenyl tetrazolium chloride (TTC) assay [31]. In this assay, metabolically active cells can convent TTC to a colored formazan derivative, so that the colorimetric measurement of biofilm biomass can be simply quantified.
Experiments were repeated three times in triplicate. Controls included bacterial suspensions incubated with 0.9% saline in the dark (untreated), bacterial suspensions incubated with MB in the dark (dark toxicity), and bacterial suspensions subjected to illumination in the absence of MB (light alone).
Visualization of P. aeruginosa biofilms by scanning electron microscopy
Scanning electron microscopy (SEM) images were taken to evaluate the effect of sAPDI on P. aeruginosa biofilms. To this end, sterile glasses with 1 cm × 1 cm dimensions were placed in the wells of a 12-well sterile microtiter plate, followed by adding 4 ml of bacterial suspension and TSB medium in first well (growth control). Other wells were filled by sAPDI-treated bacterial suspensions in TSB. Plates were covered and incubated at 37 °C for 24 h. After that, glasses were washed by sterile saline and placed in glutaraldehyde 4% for 2 h. Dehydration was done by placing each glass 15 min in concentrations of 40%, 60%, 80%, 90%, and 100% of absolute ethanol (Merck, Germany), respectively. Finally, specimens were observed by scanning electron microscope (Vega3 lmu TESCAN, Czech Republic) [32].
Quantitative polymerase chain reaction experiments
Microorganisms were grown on Luria Bertani (LB) agar overnight at 37 °C. Total RNA extraction of sAPDI-treated and control groups were performed using RNX-Plus solution (SinaClone, Iran) guidelines. Concentration and purity of RNA samples were assayed on a ND-1000 Nanodrop, and absence of degradation was confirmed on 1% agarose gel. cDNAs were then synthesized through random hexamer primed reactions using a Thermo Scientific kit, according to the manufacturer’s protocol.
Quantitative polymerase chain reaction (qPCR) experiments were performed on a Rotor-Gene 6000 thermocycler (Corbett, Qiagen Inc., Toronto, ON, Canada), using Green master mix with fluorescent dye (Genaxxon kit, Germany) according to the manufacturer’s protocol. The reference gene was 16s rRNA. Primers (as shown in Table 1) were used for amplification under the following conditions: 95 °C for 15 min, amplification for 40 cycles with denaturation at 95 °C for 15 s, annealing for 20 s at 61 °C, and extension at 72 °C for 30 min. The specificity of the primers was evaluated using melt curves. Real-time PCR data was analyzed by ∆∆Ct method, and fold change in gene expression levels was determined by 2−∆∆Ct [33]. Relative changes of pslA, pelF, lasI, lasR, rhlI, and rhlR gene expression in samples were evaluated with respect to 16s rRNA as internal control.
Statistical analysis
Values were expressed as means ± standard error of mean (SEM). Comparisons between means of groups were analyzed using one-way ANOVA and post hoc Tukey tests. P < 0.05 was considered statistically significant.
Results
APDI effect on viability and biofilm formation ability of P. aeruginosa
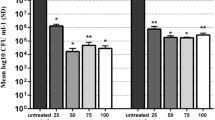
Figure 1 shows the effect of APDI mediated by different concentrations of MB (0.006–0.4 mM) and 23 J/cm2 light irradiation on viability of bacteria (log10 CFU/ml). The results showed that with increasing MB concentration from 0.006 to 0.4 mM, the killing effect of APDI increased and the viability (CFU/ml) of bacteria reduced. For example, > 3 log killing was obtained for P. aeruginosa ATCC 27853 treated with 0.1, 0.2, and 0.4 mM MB at 23 J/cm2. According to the USA Food and Drug Administration’s Tentative Final Monograph for Healthcare Antiseptic Drug Products, a reduction of at least 3 log steps must be achieved so that one can state a bactericide effect of a specific treatment [34]. On the basis of our results, APDI mediated by 0.1, 0.2, and 0.4 mM MB at 23 J/cm2 did have bactericide effects on the studied microorganism. So we used “sub-lethal” term for APDI mediated by MB at concentrations < 0.1 mM.

Antimicrobial photodynamic inactivation mediated by methylene blue (MB) in P. aeruginosa ATCC 27853; 1–2 × 108 CFU/ml were sensitized with different concentrations of MB and exposed to 23 J/cm2 light dose. (Control: untreated group)
There was no cytotoxic effect of light dose or sensitizer alone (data not shown).
Biofilm formation ability of APDI-treated microorganisms at different concentrations of MB (0.006–0.4 mM) is shown in Fig. 2. TTC assay results showed that there was no significant difference between biofilm formation ability of APDI-treated P. aeruginosa ATCC 27853 at different MB concentrations compared to untreated controls except for MB concentration at 0.012 mM. MB at 0.012 mM and light dose of 23 J/cm2 resulted in significant decrease in biofilm formation ability of P. aeruginosa ATCC 27853 compared to their untreated controls (P = 0.012).

Biofilm formation ability of APDI-treated P. aeruginosa ATCC 27853 detected by TTC assay. The amount of biofilm formation after 24 h of incubation was estimated. Absorbance was measured at 492 nm following incubation with TTC (0.5%). Positive control: biofilm formation ability of untreated bacteria
As significant decreasing in biofilm formation ability was observed with 0.012 mM MB and light dose of 23 J/cm2, we used these “sub-lethal” APDI parameters for other experiments.
Scanning electron micrographs
Micrographs (Fig. 3) clearly confirmed TTC assay results. Visualization of 24-h P. aeruginosa ATCC 27853 biofilm by SEM revealed reduced extracellular matrix following sAPDI treatment and was effective at disrupting biofilm because very few P. aeruginosa ATCC 27853 cells remained, and those that were present showed a scattered distribution (Fig. 3a). However, non-treated P. aeruginosa ATCC 27853 showed cell-cell adhesion and proliferation (Fig. 3b).

Scanning electron micrographs of P. aeruginosa ATCC 27853 biofilm. Micrographs a sAPDI-treated group (a scattered distribution), micrographs b control group (cell-cell adhesion and proliferation)
sAPDI changes pelF and pslA expression in P. aeruginosa
Quantitative PCR analysis was carried out to assess the changes in transcriptional levels of QS genes (rhl and las systems) and pelF and pslA in sAPDI-treated samples compared to their controls. Figure 4 shows fold changes in gene expression of the studied microorganism after sAPDI (0.012 mM MB, 23 J/cm2) compared to their untreated control. SAPDI led to the downregulation of the expression of QS-controlled biofilm formation genes (pslA and pelF) and QS genes (lasI, lasR, rhlI, and rhlR) in P. aeruginosa ATCC 27853. This is consistent with the phenotypic changes. These results indicated that the transcriptional decreases caused by sAPDI did lead to phenotypic changes.

Gene expression fold changes in P. aeruginosa ATCC 27853 after sAPDI (MB at 0.012 mM and light dose of 23 J/cm2)
Discussion
Antimicrobial photodynamic inactivation is a promising approach to current antibiotics. However, in human hosts receiving this therapy, pathogens may encounter sub-lethal doses of APDI. As biofilm formation has a key role in the pathogenesis of P. aeruginosa, it is therefore important to understand how sAPDI may affect this ability.
In the present study, APDI regimens mediated by MB (at concentrations < 0.1 mM) at 23 J/cm2 were defined as “sub-lethal” doses. We surprisingly observed that biofilm formation ability of sAPDI-treated organisms (at MB concentrations < 0.1 mM) was not different from the ability of untreated controls except for MB concentration at 0.012 mM. So, 0.012 mM MB and light dose of 23 J/cm2 were used as “sub-lethal” APDI parameters for other experiments (visualization of P. aeruginosa biofilms by scanning electron microscopy and qPCR).
The four studied QS genes, i.e., lasI, lasR, rhlI, and rhlR, were downregulated by a single sAPDI treatment with MB at 0.012 mM and light dose of 23 J/cm2 in P. aeruginosa ATCC 27853. A dramatic reduction of gene expression was also observed with the pslA and pelF genes. pslA and pelF are necessary enzymes in biochemical pathways of biosynthesis of psl and Pel polysaccharides [35]. Morphological observations (SEM) also indicated that sAPDI elicited a significant dispersal effect on the biofilm biomass compared with a control, probably due to the interfering with cell signaling, and repressing QS in the majority of cells.
In bacteria, biofilm formation, which is associated with the activation of quorum-sensing signals, can be induced by conditions that are potentially toxic for the bacterial cell (i.e., exposure to ROS). The link between biofilm formation and oxidative stress has been shown in a number of bacteria, including Escherichia coli [36], Streptococcus mutans [37], and Staphylococcus aureus [29]. However, we did not observed any significant increase in biofilm formation ability of P. aeruginosa ATCC 27853. Dissimilar results observed in this study may be due to the induction of other adaptive responses that protect the organism. According to Palma et al. [38] study, the early response of P. aeruginosa to H2O2 consists of an upregulation of protective mechanisms and a downregulation of primary metabolism. P. aeruginosa antioxidant and protective mechanisms for defense against ROS include two SODs (cofactored by either iron (Fe-SOD) or manganese (Mn-SOD) [38], three catalases (KatA, KatB, and KatC) [39], four alkyl hydroperoxide reductases (AhpA, AhpB, AhpCF, and Ohr) [40], the mucoid phenotype [41], and pigments [9]. Other protective mechanisms such as increasing pigment production which may protect P. aeruginosa against oxidants are currently under investigation in our laboratory.
Although APDI should be generally used at lethal doses to kill bacteria, but it is likely that bacteria will be exposed to sub-lethal doses. Here, if oxidative stress is very high, it can lead to cell death. However, exposure to low levels of stress activates protective mechanisms through a complex pathway involving various regulators so that cell death is avoided [42]. For example, Escherichia coli MazE/MazF system in response to low levels of stress stimulates the activation of protective pathways including the Cpx envelope protein stress system for the refolding or degradation of misfolded proteins in the periplasm, the inhibition of katG mRNA degradation, and MazF-mediated ·OH accumulation [43, 44]. In the case of extreme stress, the same proteins used to trigger ROS scavenging systems contribute to a cascade of ROS, and activate a programmed cell death pathway, essential to reduce the risk of hyper-mutation and loss of genetic integrity [45]. Consequently, bacteria start to produce several combating strategies to resist in such stressful environments as well as changes in their physiology and pathogenesis factors.
Conclusion
In this study, we declared that sAPDI with MB at 0.012 mM and light fluency of 23 J/cm2 reduced biofilm formation ability of P. aeruginosa; however, there is no guarantee that other PSs, light sources, or other bacteria react like this. There is an essential need to investigate more for the effect of sAPDI on other pathogens especially in molecular levels and in vivo situation. It confirms why we must be cautious by using sAPDI.
References
Taylor PW, Stapleton PD, Paul Luzio J (2002) New ways to treat bacterial infections. Drug Discov Today 7(21):1086–1091
Wainwright M (1998) Photodynamic antimicrobial chemotherapy (PACT). J Antimicrob Chemother 42:13–28
Huang L, Xuan Y, Koide Y, Zhiyentayev T, Tanaka M, Hamblin MR (2012) Type I and type II mechanisms of antimicrobial photodynamic therapy: an in vitro study on gram-negative and gram-positive bacteria. Lasers Surg Med 44:490–499
Caminos DA, Spesia MB, Pons P, Durantini EN (2008) Mechanisms of Escherichia coli photodynamic inactivation by an amphiphilic tricationic porphyrin and 5,10,15,20-tetra(4-N,N,Ntrimethylammoniumphenyl) porphyrin. Photochem Photobiol Sci 7:1071–1078
Niederhoffer EC, Naranjo CM, Bradley KL (1990) Control of Escherichia coli superoxide dismutase (sodA and sodB) genes by the ferric uptake regulation (fur) locus. J Bacteriol 172(4):1930–1938
Ziegelhoffer EC, Donohue TJ (2009) Bacterial responses to photo-oxidative stress. Nat Rev Microbiol 7(12):856–863
Boles BR, Singh PK (2008) Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc Natl Acad Sci U S A 105(34):12503–12508
Anderson SM, Krinsky NI (1973) Protective action of carotenoid pigments against photodynamic damage to liposomes. Photochem Photobiol 18(5):403–408
Orlandi VT, Bolognese F, Chiodaroli L, Tolker-Nielsen T, Barbieri P (2015) Pigments influence the tolerance of Pseudomonas aeruginosa PAO1 to photodynamically induced oxidative stress. Microbiology 161(12):2298–2309
Gad F, Zahra T, Hasan T, Hamblin MR (2004) Effects of growth phase and extracellular slime on photodynamic inactivation of gram-positive pathogenic bacteria. Antimicrob Agents Chemother 48(6):2173–2178
Tegos GP, Hamblin MR (2006) Phenothiazinium antimicrobial photosensitizers are substrates of bacterial multidrug resistance pumps. Antimicrob Agents Chemother 50(1):196–203
Nakonieczna J, Michta E, Rybicka M, Grinholc M, Gwizdek-Wiśniewska A, Bielawski K (2010) Superoxide dismutase is upregulated in Staphylococcus aureus following protoporphyrin-mediated photodynamic inactivation and does not directly influence the response to photodynamic treatment. BMC Microbiol 10(1):323
St Denis TG, Huang L, Dai T, Hamblin MR (2011) Analysis of the bacterial heat shock response to photodynamic therapy-mediated oxi- dative stress. Photochem Photobiol 87:707–713
Jayaseelan S, Ramaswamy D, Dharmaraj S (2014) Pyocyanin: production, applications, challenges and new insights. World J Microbiol Biotechnol 30(4):1159–1168
Donlan RM (2002) Biofilms: microbial life on surfaces. Emerg Infect Dis 8(9):881
Watnick P, Kolter R (2000) Biofilm, city of microbes. J Bacteriol 182(10):2675–2679
Al-Wrafy F, Brzozowska E, Górska S, Gamian A (2017) Pathogenic factors of Pseudomonas aeruginosa-the role of biofilm in pathogenicity and as a target for phage therapy. Adv Hyg Exp Med Hig Med Dosw 71:78–91
Colvin KM, Irie Y, Tart CS, Urbano R, Whitney JC, Ryder C et al (2012) The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ Microbiol 14(8):1913–1928
Lam J, Chan R, Lam K, Costerton JW (1980) Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect Immun 28:546–556
Friedman L, Kolter R (2004) Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol 51(3):675–690
Swartjes JJTM, Das T, Sharifi S et al (2013) A functional DNasei coating to prevent adhesion of bacteria and the formation of biofilm. Adv Funct Mater 23(22):2843–2849
Smith RS, Iglewski BH (2003) P aeruginosa quorum-sensing systems and virulence. Curr Opin Microbiol 6:56–60
Passador L, Passador L, Cook JM, Gambello MJ, Rust L, Iglewski BH (1993) Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 260:1127–1130
Latifi A, Winson MK, Foglino M, Bycroft BW, Stewart GS, Lazdunski A, Williams P (1995) Multiple homologues of LuxR and LuxI control expression of virulence determinants and secondary metabolites through quorum sensing in Pseudomonas aeruginosa PAO1. Mol Microbiol 17:333–343
Wei Q, Ma LZ (2013) Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int J Mol Sci 14(10):20983–21005
Sakuragi YKR (2007) Quorum-sensing regulation of the biofilm matrix genes (pel) of Pseudomonas aeruginosa. J Bacteriol 189(14):5383–5386
Kashef N, Hamblin MR (2017) Can microbial cells develop resistance to oxidative stress in antimicrobial photodynamic inactivation? Drug Resist Updat 31:31–42
Moody CS, Hassan HM (1982) Mutagenicity of oxygen free radicals. Proc Natl Acad Sci 79(9):2855–2859
Kashef N, Akbarizare M, Kamrava SK (2013) Effect of sub-lethal photodynamic inactivation on the antibiotic susceptibility and biofilm formation of clinical Staphylococcus aureus isolates. Photodiagn Photodyn Ther 10(4):368–373
Orlandi VT, Bolognese F, Martegani E, Cantaluppi V, Medana C, Barbieri P (2017) Response to photo-oxidative stress of Pseudomonas aeruginosa PAO1 mutants impaired in different functions. Microbiology 163(11):1557–1567
Sabaeifard P, Abdi-Ali A, Soudi MR, Dinarvand R (2014) Optimization of tetrazolium salt assay for Pseudomonas aeruginosa biofilm using microtiter plate method. J Microbiol Methods 105:134–140
Hendiani S, Abdi-Ali A, Mohammadi P, Kharrazi SH (2015) Synthesis of silver nanoparticles and its synergistic effects in combination with imipenem and two biocides against biofilm producing Acinetobacter baumannii. Nanomed J 2:291–298
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3(6):1101–1108
Boyce JM, Pittet D (2002) Guideline for hand hygiene in health-caresettings. Recommendations of the healthcare infection controlpractices advisory committee and the HIPAC/SHEA/APIC/IDSAhand hygiene task force. Am J Infect Control 30:1–46
Franklin MJ, Nivens DE, Weadge JT, Howello PL (2011) Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front Microbiol 2:167
Schembri MA, Hjerrild L, Gjermansen M, Klemm P (2003) Differential expression of the Escherichia coli autoaggregation factor antigen 43. J Bacteriol 185(7):2236–2242
Wen ZT, Suntharaligham P, Cvitkovitch DG, Burne RA (2005) Trigger factor in Streptococcus mutans is involved in stress tolerance, competence development, and biofilm formation. Infect Immun 73(1):219–225
Palma M, DeLuca D, Worgall S, Quadri LEN (2004) Transcriptome analysis of the response of Pseudomonas aeruginosa to hydrogen peroxide. J Bacteriol 186(1):248–252
Brown SM, Howell ML, Vasil ML, Anderson AJ, Hassett DJ (1995) Cloning and characterization of the katB gene of Pseudomonas aeruginosa encoding a hydrogen peroxide-inducible catalase: purification of KatB, cellular localization, and demonstration that it is essential for optimal resistance to hydrogen peroxide. J Bacteriol 177(22):6536–6544
Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ (2000) Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, andahpC-ahpF. J Bacteriol 182(16):4533–4544
Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell J, I A, Jensen P et al (1999) Mucoid conversion of Pseudomonas aeruginos by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145(6):1349–1357
Gambino M, Cappitelli F (2016) Mini-review: biofilm responses to oxidative stress. Biofouling 32(2):167–178
Pogliano J, Lynch AS, Belin D, Lin EC, Beckwith J (1997) Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev 11(9):1169–1182
Zhao X, Drlica K (2014) Reactive oxygen species and the bacterial response to lethal stress. Curr Opin Microbiol 21:1–6
Dorsey-Oresto A, Lu T, Mosel M, Wang X, Salz T, Drlica K, Zhao X (2013) YihE kinase is a central regulator of programmed cell death in bacteria. Cell Rep 3(2):528–537
Acknowledgments
This study was supported by the College of Science, University of Tehran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hendiani, S., Pornour, M. & Kashef, N. Sub-lethal antimicrobial photodynamic inactivation: an in vitro study on quorum sensing-controlled gene expression of Pseudomonas aeruginosa biofilm formation. Lasers Med Sci 34, 1159–1165 (2019). https://doi.org/10.1007/s10103-018-02707-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-018-02707-y