
Abstract
Autophagy has been demonstrated to play an important role in the immunity against intracellular pathogens, but very little is known about its role in the host defense against fungal pathogens such as Candida albicans. Therefore, the role of autophagy for the host defense against C. albicans was assessed by complementary approaches using mice defective in autophagy, as well as immunological and genetic studies in humans. Although C. albicans induced LC3-II formation in macrophages, myeloid cell-specific ATG7−/− mice with defects in autophagy did not display an increased susceptibility to disseminated candidiasis. In in vitro experiments in human blood mononuclear cells, blocking autophagy modulated cytokine production induced by lipopolysaccharide, but not by C. albicans. Furthermore, autophagy modulation in human monocytes did not influence the phagocytosis and killing of C. albicans. Finally, 18 single-nucleotide polymorphisms in 13 autophagy genes were not associated with susceptibility to candidemia or clinical outcome of disease in a large cohort of patients, and there was no correlation between these genetic variants and cytokine production in either candidemia patients or healthy controls. Based on these complementary in vitro and in vivo studies, it can be concluded that autophagy is redundant for the host response against systemic infections with C. albicans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Candida albicans is a commensal fungus that colonizes the gastrointestinal tract, skin, and mucosa of more than 50 % of healthy individuals. Candida colonization does not cause disease in healthy individuals, but in patients in whom the immune system is compromised, Candida can cause both mucosal and systemic disease, the latter with a mortality rate reaching up to 30–40 % [1]. C. albicans is recognized by the innate immune system through pathogen recognition receptors (PRRs) such as C-type lectin receptors or Toll-like receptors (TLRs) that interact with pathogen-associated molecular patterns on the Candida cell wall. Candida mannans are recognized by the macrophage mannose receptor and Dectin-2 [2, 3], Dectin-1 recognizes β-glucan [4], while DC-SIGN (CD209) recognizes fucose and mannose/mannan residues [5]. Furthermore, TLRs such as TLR2 [6] and TLR4 [7] also play an important role in the recognition of C. albicans. These interactions between C. albicans and the immune system lead to phagocytosis of the fungus [8] and the induction of proinflammatory cytokines, further promoting clearance of the infection [9].
In addition to these well-known effects of PRR engagement, recent studies have shown that TLRs can also engage autophagy proteins [10], and this, in turn, modulates the inflammatory reaction against pathogens [11–13]. Autophagy is an essential process for cell survival that allows the cell to efficiently regulate its biomass via the degradation of individual proteins (chaperone-mediated autophagy), cytosolic content, and whole-cell organelles (macroautophagy) [14]. Autophagy is characterized by the formation of a double-membrane vesicle, the autophagosome, which engulfs the cytosolic content to be degraded [15]. Subsequent fusion of the autophagosome with the lysosome and the breakdown of the inner membrane expose the content to hydrolases.
In addition to its role in cell homeostasis, autophagy has also been linked to the host defense against viruses [16] and to the processing of invading pathogens (xenophagy) [17, 18]. Inactivation of autophagy genes increases the replication of intracellular pathogens: Singh et al. demonstrated that, in mice, IRGM induces autophagy to eliminate intracellular Mycobacterium tuberculosis [19]; Zhao et al. demonstrated that ATG5−/− mice have decreased resistance to the intracellular bacterium Listeria monocytogenes and the protozoan Toxoplasma gondii [20]. Interestingly, ATG5 and ATG10 have been implicated in the defense against fungi in plants [21].
Little is known about whether autophagy is also involved in the immune response against fungal infections in mammals. Since several of the PRRs involved in the recognition of C. albicans have been demonstrated to induce autophagy, such as recruitment of the autophagosome marker LC3-II by TLR2 and TLR4 [10, 22], it is rational to hypothesize that autophagy might play an important role in the anti-Candida host immune response. In this study, the role of autophagy in the immune response against C. albicans was investigated using mouse knockout models, as well as human genetic association studies and in vitro experiments. We could not identify a major role for autophagy in anti-Candida host defense.
Materials and methods
Study population
To investigate the correlation between autophagy and candidemia, 338 adult candidemia patients (positive blood culture) and 351 healthy controls were enrolled in a study between January 2003 and January 2009 [23]. The candidemia study was approved by the Institutional Review Boards from the Duke University Hospital (Durham, NC, USA) and the Radboud university medical center (Nijmegen, The Netherlands). Participants were included after giving written informed consent, with the exception of patients who were no longer hospitalized or died before a positive blood culture report was made. To investigate the link between autophagy and cytokine production, 67 healthy individuals donated blood. The age of the patients ranged from 23 to 73 years and 77 % was male. Blood was collected by venipuncture into 10-ml EDTA syringes (Monoject, ’s-Hertogenbosch, The Netherlands). The study with the healthy blood donors was approved by the Ethical Committee of the Radboud university medical center (Nijmegen, The Netherlands). Participants were included after giving written informed consent. The studies were performed in accordance with the Declaration of Helsinki.
Mice
The LysM-Cre+ or LysM-Cre−ATG7flox/flox GFP-LC3+ (the conditional ATG7flox/flox mice is a kind gift from Masaaki Komatsu, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) was described previously (kindly provided by Douglas R. Green, St. Jude Children’s Research Hospital, Memphis, TN, USA). All mice were housed in a pathogen-free facility. The animal study was approved by the Animal Care and Use Committee from the St. Jude Children’s Research Hospital (protocol 482-100097-10/11). The study was performed in accordance with the guidelines set by the National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Microorganism
Candida yeast [American Type Culture Collection (ATCC) MYA-3573 (UC820)], a strain well described elsewhere [24], were grown overnight in Sabouraud broth at 37 °C. Cells were harvested by centrifugation, washed twice, and resuspended in culture medium. C. albicans yeasts or hyphae were heat-killed for 1 h at 100 °C.
Macrophage differentiation, stimulation, and Western blotting
Bone marrow-derived macrophages (BMDMs) were differentiated from the total cells isolated from the femurs of 6–10-week-old mice by using supernatant from L929 cells as the differentiation medium. BMDM cells in 12-well tissue culture plates (5 × 105/well) were infected or treated with various Candida ligands [live Candida yeast form (moi 5), heat-killed Candida yeast form (moi 5), live Candida hyphae form, and heat-killed Candida hyphae form) for 8 h. The cells were lysed in RIPA lysis buffer supplemented with complete protease inhibitor mixture (Roche) and PhosSTOP (Roche). The whole-cell lysates were separated on 15 % SDS-PAGE and transferred to PVDF membranes. Membranes were blocked in 5 % non-fat milk and incubated overnight with primary antibody at 4 °C and for 45 min with secondary HRP-tagged antibody at room temperature. The membranes were developed with SuperSignal West Femto Chemiluminescent Substrate (Pierce).
Candida albicans infection model
The WT (LysM-Cre− ATG7f/f GFP-LC3+) and ATG7−/− (LysM-Cre+ ATG7f/f GFP-LC3+) mice were injected intravenously with the inoculum of C. albicans blastoconidia [1 × 106 colony-forming units (CFU)/mouse] in a 100-μl volume of sterile pyrogen-free phosphate-buffered saline (PBS). Survival was assessed daily for 30 days. For assessing fungal burden, subgroups of 5–10 mice were humanely terminated on days 3 or 7 of infection. To measure the fungal burden, the kidneys of the sacrificed animals were removed aseptically and homogenized in sterile PBS using a tissue grinder. The CFU values of the viable Candida from the kidney homogenates were measured by plating serial dilutions on Sabouraud dextrose agar plates (50 μg/ml of gentamicin), as described previously.
Fluorescence microscopy
HeLa cells were transfected with a plasmid containing GFP-LC3 (kindly provided by Dr. T Yoshimori, Osaka, Japan) using the transfection medium Fugene 6 (Roche), according to the manufacturer’s instructions. GFP-LC3+ HeLa cells were grown and stimulated on coverslips (19-mm diameter) in 12-well plates. Cells were fixed with 2 % paraformaldehyde for 15 min at room temperature and permeabilized for 10 min with cold methanol (100 %). After washing with PBS (three times), the coverslips were mounted onto glass slides with Vectashield + DAPI and analyzed on a fluorescence microscope.
Phagocytosis and killing assays
Phagocytosis and killing was performed as described previously [24, 25]. In short, 5 × 105 peripheral blood mononuclear cells (PBMCs) (in a volume of 100 μl) were put in a flat-bottom well. The plate was incubated at 37 °C for 1 h, to allow the monocytes to adhere to the plastic surface, in the absence or presence of 3MA (10 mM). Thereafter, the supernatant was removed, and the monolayer was rinsed with modified Eagle’s medium (MEM). 200 μl of live C. albicans (5 × 104/ml) in MEM, 2.5 % serum was added, and the plate was incubated for 15 min to allow phagocytosis of the yeast. The supernatant was removed, and the monolayer was rinsed with MEM. 200 μl of MEM/Sabouraud was added to the monolayer, after which the plate was incubated at 37 °C for 2 h and 45 min to allow intracellular killing of the yeast. After this incubation period, the monocytes were lysed. Both the supernatants with non-phagocytosed Candida and the lysed monocyte suspension with non-killed Candida were directly plated in duplicate in two different dilutions on agar plates. These plates were cultured for 24 h, after which the number of colonies was counted. The percentage of phagocytosed and killed Candida was calculated.
PBMCs isolation
The separation and stimulation of PBMCs was performed as described previously [26]. Briefly, the PBMC fraction was obtained by density centrifugation of diluted blood (one part blood to one part pyrogen-free saline) over Ficoll-Paque (Pharmacia Biotech, Uppsala, Sweden). PBMCs were washed twice in saline and suspended in culture medium. The cells were counted in a Coulter counter (Coulter Electronics, Buckinghamshire, England) and their number was adjusted to 5 × 106/ml.
Cell stimulation
A total of 5 × 105 human PBMCs in a 100-ml volume of RPMI was added to round-bottom 96-well plates (Greiner). Cells were stimulated with live C. albicans UC820 (1 × 104/ml) or E. coli-derived lipopolysaccharide (LPS) (E. coli O55:B5 LPS, Sigma Chemical Co.), in the absence or presence of 3MA (Sigma). After 24 h, supernatants were stored at −20 °C. IL-1β, IL-8, and IL-10 was measured in cell culture supernatants using an enzyme-linked immunosorbent assay (ELISA) (R&D Systems, MN, USA and Sanquin, Amsterdam, The Netherlands).
Autophagy gene SNP genotyping
Genomic DNA was isolated from EDTA blood of patients, matched controls, and a cohort of healthy volunteers using standard methods, and 5 ng of DNA was used for genotyping. We selected 18 single-nucleotide polymorphisms (SNPs) from 13 autophagy-related genes (Table 1). Multiplex assays were designed using Mass ARRAY Designer Software (Sequenom) and genotypes were determined using Sequenom matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) according to the manufacturer’s instructions (Sequenom Inc., San Diego, CA, USA). Briefly, the SNP region was amplified by a locus-specific polymerase chain reaction (PCR) assay. After amplification, a single base extension from a primer adjacent to the SNP was performed to introduce mass differences between alleles. This was followed by salt removal and product spotting onto a target chip with 384 patches containing matrix. MALDI-TOF MS was then used to detect mass differences and genotypes were assigned in real-time using Typer 4 software (Sequenom Inc., San Diego, CA, USA). As quality control, 5 % of samples were genotyped in duplicate and each 384-well plate also contained at least eight positive and eight negative controls; no inconsistencies were observed. DNA samples of which SNPs failed were excluded from the analyses. Variants with call rates below 90 % were also excluded from further analyses (n = 0).
Statistics
Data were analyzed using SPSS version 20.0 (SPSS Inc., Chicago, IL, USA) and SAS (version 9.3, SAS Institute, Cary, NC, USA). All statistical analyses were two-sided and p < 0.05 was considered to be statistically significant (*). The survival was analyzed using the log-rank Mantel–Cox test, and the CFUs were analyzed with the Student’s t-test. Differences in cytokine production were analyzed using the Wilcoxon signed-rank test. The Hardy–Weinberg equilibrium (HWE) was checked for each SNP using the program HWE Version 1.10 (Rockefeller University, New York, NY, USA). The associations between autophagy SNPs and candidemia susceptibility and clinical outcome of disease (30-day survival, persistent disease, and disseminated disease) were assessed using the Chi-squared test or Fisher’s exact test, as appropriate. With the application of the Bonferroni correction for multiple testing, p < 0.003 and p < 0.001 were considered to be statistically significant, respectively.
Results
Candida albicans induces LC3-II shift in mouse BMDMs
Two forms of LC3 exist; LC3-I is located in the cytoplasm, while LC3-II is a processed form of LC3, which is associated with the (auto)phagosome membrane. Mouse BMDMs were stimulated with live and heat-killed C. albicans yeasts and hyphae. LC3-I and LC3-II were measured using Western blot. All forms, except heat-killed C. albicans yeasts, induced a strong upregulation of LC3-II expression (Fig. 1).

Candida albicans induces LC3-II in mouse bone marrow-derived macrophages (BMDMs). Mouse BMDMs were stimulated with live and heat-killed (HK) C. albicans yeasts and hyphae. LC3-II was measured using Western blot (UT = unstimulated, +ve control = positive control)
ATG7 is redundant for host defense against systemic Candida albicans infection
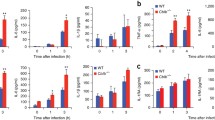
Because C. albicans induced autophagy in murine macrophages, we studied the effects of autophagy deficiency in myeloid cells on the outcome of systemic candidiasis. Although ATG7−/− KO mice appeared to have a slightly lower survival rate compared to WT mice, the difference was small and did not reach statistical significance (Fig. 2a). Furthermore, there were no differences in the fungal burdens in the kidneys, the target organ of disseminated candidiasis, 3 or 7 days after C. albicans infection of wild-type or autophagy-defective mice (Fig. 2b).

No difference in survival between wild-type (WT) and ATG7−/− mice. a 13 WT (open circles) and 14 ATG7−/− (filled circles) mice were injected with live Candida albicans at day 1. Survival was monitored for 30 days. The survival was analyzed using the log-rank Mantel–Cox test. b After 3 and 7 days of infection, colony-forming units (CFUs) were counted in the kidneys (n = 7 mice/group on day 3 and n = 9 mice/group on day 7). The CFUs were analyzed with the Student’s t-test
Candida albicans induces LC3-II shift in HeLa cells
Although autophagy does not seem to have a major impact on the outcome of murine candidiasis, different effects may be seen in humans. To investigate whether autophagy is induced upon Candida stimulation in human cells, we transfected HeLa cells with GFP-LC3 and stimulated them with heat-killed C. albicans yeast. LC3-II was analyzed using immunofluorescence microscopy. Heat-killed C. albicans yeasts induced a strong upregulation of LC3-II expression, demonstrating the induction of autophagy (Fig. 3a). The induction of LC3-II was reverted in the presence of the autophagy inhibitors 3MA and wortmannin (Fig. 3b, c).

Candida-induced LC3-II in GFP-LC3 transfected HeLa cells. a Fluorescence microscopy image (40×) showing LC3-II induction upon heat-killed C. albicans stimulation in GFP-LC3 transfected HeLa cells. In the presence of the autophagy inhibitors 3MA (b) and wortmannin (c), LC3-II is no longer induced, demonstrating the specificity of the assay
Inhibition of autophagy does not influence the phagocytosis and killing capacity of monocytes, or their cytokine production upon stimulation with C. albicans
In the next set of experiments, we investigated the importance of autophagy for the human anti-Candida host response. Firstly, we investigated whether autophagy is important for the phagocytosis and killing of live C. albicans by human monocytes. Freshly isolated human primary monocytes were stimulated with live C. albicans in the absence or presence of 3MA. Blocking autophagy with 3MA did not affect the capacity of human monocytes to phagocytose and kill C. albicans (Fig. 4a).

Blocking autophagy does not inhibit the phagocytosis and killing capacity of human monocytes, nor the C. albicans-induced cytokine response. a Freshly isolated monocytes were stimulated with live C. albicans. The amount of phagocytosed and killed Candida was determined after 15 min and 3 h, respectively. The bars represent the mean ± standard error of the mean (SEM) of four healthy volunteers. b Human peripheral blood mononuclear cells (PBMCs) were stimulated for 24 h with live C. albicans or Escherichia coli-derived lipopolysaccharide (LPS), in the absence or presence of 3MA. The concentration of IL-1β was measured in cell culture supernatants using enzyme-linked immunosorbent assay (ELISA). The bars represent the mean ± SEM of 19 healthy volunteers. Differences in cytokine production were analyzed using the Wilcoxon signed-rank test (***p < 0.001)
In a subsequent set of experiments, human PBMCs were stimulated with live C. albicans or E. coli-derived LPS, in the absence or presence of the autophagy inhibitor 3MA. While 3MA strongly increased LPS-induced IL-1β production, the increase in IL-1β production in C. albicans-stimulated cells was more modest and did not reach statistical significance (Fig. 4b). Stimulation of other proinflammatory cytokines such as TNF and IL-6 by C. albicans was not influenced by the modulation of autophagy (not shown).
SNPs in autophagy genes are not associated with candidemia
In order to further assess the importance of autophagy in the anti-Candida host defense in humans, we investigated whether polymorphisms in autophagy genes were correlated to susceptibility with systemic candidiasis. Although 18 SNPs in 13 different autophagy genes were studied, chosen based on their likelihood to influence the autophagy process, none of them were significantly associated with susceptibility to disseminated candidiasis (Table 1). Furthermore, none of the 18 SNPs studied were correlated with circulating cytokine concentrations in patients with candidemia or clinical outcome of disease (data not shown).
SNPs in autophagy genes do not correlate with Candida-induced cytokine production
Finally, we investigated whether the same SNPs in autophagy genes influenced Candida-induced cytokine production in PBMCs isolated from healthy volunteers. There were no statistically significant associations between the autophagy genotypes and Candida-induced cytokine production. A small number of these SNPs showed a tendency to influence cytokine production, but the associations were not statistically significant (Fig. 5).

No significant correlation between cytokine production and genotype in healthy volunteers. PBMCs of healthy volunteers were stimulated with heat-killed C. albicans conidia for 24 h. Cytokines were measured in cell culture supernatants using ELISA. Data are presented as the mean ± 95 % confidence interval (CI)
Discussion
In this study, we investigated the role of autophagy in the anti-Candida host immune response. Using complementary immunological and genetic approaches in both mice and humans, we show that autophagy is redundant for the systemic host defense against C. albicans.
C. albicans is recognized by PRRs on the surface of the innate immune cells, and recent studies have shown that the engagement of PRR receptors can induce autophagy. Huang and Brumell demonstrated that LC3-II is recruited to the phagosome upon zymosan stimulation, through a Dectin-1-dependent pathway [27]. Indeed, both C. albicans and the Dectin-1 ligand β-glucan induce LC3 lipidation [28]. Furthermore, Nicola et al. also demonstrated that Cryptococcus neoformans can induce LC3 recruitment to the phagosome, although to a lesser extent than C. albicans [29]. LC3 lipidation does not necessarily implicate autophagy activation, but could also be a sign of LC3-associated phagocytosis (LAP) [30]. Here, we confirm the LC3-inducing activity of C. albicans by demonstrating that Candida stimulation was able to induce LC3-II in mouse BMDMs. Altogether, these data demonstrate that fungi could induce the process of autophagy, and this prompted us to investigate its role in host defense against disseminated candidiasis.
Surprisingly, however, mice with a specific deletion of the autophagy gene ATG7 in their myeloid cells did not display an increased mortality due to disseminated candidiasis. Furthermore, there was no difference in the fungal burden in the kidneys between wild-type and ATG7−/− mice. These data are paralleled by those of Nicola and colleagues, who also failed to find any difference in the survival between wild-type and conditional ATG5−/− mice infected with C. neoformans [29]. Although the authors reported that ATG5−/− mice die slightly sooner compared to wild-type mice upon infection with C. albicans, this difference was small (2 days), and they did not replicate this finding. Furthermore, the effect on Candida burdens in the kidneys was not reported [29]. In short, although both Nicola et al. and our group show that LC3-II can be induced upon fungal stimulation, the absence of autophagy has no major effect in in vivo infection models.
Because the immune system of mice can differ substantially from that of humans [31], we next investigated the potential role of autophagy in the immune response against C. albicans in humans. Similarly to what we have seen in mouse cells, we observed that LC3-II activation was induced upon Candida stimulation in the human HeLa cell line. Due to the fact that autophagy modulates inflammation induced by TLR ligands in human cells [32–34], we also tested whether anti-Candida immune responses are modulated by autophagy. Firstly, blocking autophagy with pharmacological inhibitors did not affect important aspects of the Candida-induced immune response, such as phagocytosis and killing of the fungus, or Candida-induced cytokine production. Of note, the inhibitors we used here are not completely specific. 3MA has been demonstrated to also be able to actually induce autophagy in some specific situations, and to influence cell survival through AKT1 [35]. Fortunately, similar results were obtained with 3MA and wortmannin. More importantly, Ma et al. previously demonstrated that the phagocytosis and killing of C. albicans is unaffected in LC3β-deficient mouse bone marrow-derived cells (BMDCs) [28]. The same is true for Salmonella enterica, which can be recognized by autophagy machinery in the absence of LC3 recruitment [36].
Secondly, the (mostly) non-synonymous SNPs in autophagy genes (ATG10, ATG16L1, ATG16L2, ATG2A, ATG2B, ATG5, ATG9B, EREG, IRGM, LAMP1, LAMP3, P2RX7, and WIPI1) did not influence susceptibility to candidemia, nor did they influence serum cytokine levels in the patients or the clinical outcome of disease. However, several of these SNPs have been demonstrated to be associated with immune function. For example, rs2241880 in ATG16L1 influences IL-1β and IL-6 production upon NOD2 stimulation [34], and rs72553867 in IRGM has been associated with inflammatory bowel disease [37]. The lack of association here could be explained by the fact that systemic candidiasis is a relatively rare disease, with a population frequency of 6:100,000 [38]. However, the genetic association study presented here has been performed in the largest cohort available to date. With the current sample size, we should be able to detect differences in proportions from 9 % and higher with a power of 80 % [39]. So, although we cannot fully exclude that these polymorphisms do influence the susceptibility to candidiasis, at least we can conclude that these effects, if existent, are very small. While several PRR and cytokine polymorphisms have been shown to be associated with susceptibility to candidemia [40, 41], the fact that none of the autophagy SNPs is associated with an increased susceptibility or severity of candidemia is another argument for a redundant role of autophagy for the systemic anti-Candida host defense in humans. Thirdly, we have identified no correlation between genetic variants in autophagy genes and ex vivo cytokine production by PBMCs of healthy controls. In line with this, Ma et al. demonstrated that cytokine production was normal in LC3β-deficient cells that completely lack functional autophagy [28].
Despite these complementary data demonstrating that autophagy does not play a central role for the systemic host defense against Candida spp., we cannot exclude a role of autophagy in other anti-Candida host defense mechanisms, e.g., mucosal antifungal defense. In order to prevent lysosomal degradation, Candida actively stimulates the recycling of LAMP-1 from the phagosome [42], an important protein involved in chaperone-mediated autophagy [43]. The C-type lectin receptor Dectin-1 is crucial for the recognition of β-glucans from Candida [44, 45], and defects in Dectin-1 have been previously shown to be associated with mucosal and skin Candida infections, but not systemic candidiasis [23, 46, 47]. While Dectin-1-dependent mechanisms induce autophagy [27], interestingly, it has also been demonstrated that the autophagy protein Rubicon can bind CARD9, dampening the signaling downstream of Dectin-1 [48]. Furthermore, Ma et al. showed that the recruitment of MHCII to the phagosome was reduced in LC3β-deficient cells, demonstrating that autophagy-related proteins may play a role in enhancing antigen presentation and adaptive immune responses [28]. Indeed, adaptive Th17 and Th1 responses are known to play an important role, especially for mucosal antifungal infections, as demonstrated in STAT1 mutations and STAT3 deficiency syndromes characterized by defective Th17 responses and chronic mucocutaneous candidiasis [49–51]. Taking into consideration this entire body of information, autophagy induction by Candida through Dectin-1-dependent mechanisms may play a role in the modulation of adaptive Th17 responses and mucosal antifungal defense, but this hypothesis remains to be demonstrated.
In conclusion, although C. albicans can induce LC3-II in both mice and human cells, the consequences at the level of phagocytosis, killing, and cytokine induction are limited, and autophagy is redundant for the host defense against systemic candidiasis. However, this does not exclude that autophagy could play a role in the mucosal anti-Candida immune response through antigen presentation and/or T-helper cell activation, and future studies are warranted to assess this possibility.
References
Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB (2004) Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39(3):309–317
van de Veerdonk FL, Marijnissen RJ, Kullberg BJ, Koenen HJ, Cheng SC, Joosten I, van den Berg WB, Williams DL, van der Meer JW, Joosten LA, Netea MG (2009) The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 5(4):329–340
Cheng SC, van de Veerdonk FL, Lenardon M, Stoffels M, Plantinga T, Smeekens S, Rizzetto L, Mukaremera L, Preechasuth K, Cavalieri D, Kanneganti TD, van der Meer JW, Kullberg BJ, Joosten LA, Gow NA, Netea MG (2011) The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J Leukoc Biol 90(2):357–366
Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S (2003) Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 197(9):1119–1124
Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, van Kooyk Y (2003) Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol 170(4):1635–1639
Villamón E, Gozalbo D, Roig P, O’Connor JE, Fradelizi D, Gil ML (2004) Toll-like receptor-2 is essential in murine defenses against Candida albicans infections. Microbes Infect 6(1):1–7
Tada H, Nemoto E, Shimauchi H, Watanabe T, Mikami T, Matsumoto T, Ohno N, Tamura H, Shibata K, Akashi S, Miyake K, Sugawara S, Takada H (2002) Saccharomyces cerevisiae- and Candida albicans-derived mannan induced production of tumor necrosis factor alpha by human monocytes in a CD14- and Toll-like receptor 4-dependent manner. Microbiol Immunol 46:503–512
Heinsbroek SE, Taylor PR, Martinez FO, Martinez-Pomares L, Brown GD, Gordon S (2008) Stage-specific sampling by pattern recognition receptors during Candida albicans phagocytosis. PLoS Pathog 4(11):e1000218
Netea MG, Brown GD, Kullberg BJ, Gow NA (2008) An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 6(1):67–78
Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR (2007) Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450(7173):1253–1257
Deretic V (2005) Autophagy in innate and adaptive immunity. Trends Immunol 26(10):523–528
Deretic V (2009) Multiple regulatory and effector roles of autophagy in immunity. Curr Opin Immunol 21(1):53–62
Virgin HW, Levine B (2009) Autophagy genes in immunity. Nat Immunol 10(5):461–470
Ashford TP, Porter KR (1962) Cytoplasmic components in hepatic cell lysosomes. J Cell Biol 12:198–202
Deretic V, Levine B (2009) Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5(6):527–549
McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM (2011) Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J Virol 85(9):4212–4221
Fabri M, Realegeno SE, Jo EK, Modlin RL (2011) Role of autophagy in the host response to microbial infection and potential for therapy. Curr Opin Immunol 23(1):65–70
Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T (2004) Autophagy defends cells against invading group A Streptococcus. Science 306(5698):1037–1040
Singh SB, Davis AS, Taylor GA, Deretic V (2006) Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313(5792):1438–1441
Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW (2008) Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 4(5):458–469
Lenz HD, Haller E, Melzer E, Kober K, Wurster K, Stahl M, Bassham DC, Vierstra RD, Parker JE, Bautor J, Molina A, Escudero V, Shindo T, van der Hoorn RA, Gust AA, Nürnberger T (2011) Autophagy differentially controls plant basal immunity to biotrophic and necrotrophic pathogens. Plant J 66(5):818–830
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27(1):135–144
Rosentul DC, Plantinga TS, Oosting M, Scott WK, Velez Edwards DR, Smith PB, Alexander BD, Yang JC, Laird GM, Joosten LA, van der Meer JW, Perfect JR, Kullberg BJ, Netea MG, Johnson MD (2011) Genetic variation in the dectin-1/CARD9 recognition pathway and susceptibility to candidemia. J Infect Dis 204(7):1138–1145
Vonk AG, Wieland CW, Netea MG, Kullberg BJ (2002) Phagocytosis and intracellular killing of Candida albicans blastoconidia by neutrophils and macrophages: a comparison of different microbiological test systems. J Microbiol Methods 49(1):55–62
Kullberg BJ, van ’t Wout JW, Hoogstraten C, van Furth R (1993) Recombinant interferon-γ enhances resistance to acute disseminated Candida albicans infection in mice. J Infect Dis 168(2):436–443
Netea MG, Gow NA, Munro CA, Bates S, Collins C, Ferwerda G, Hobson RP, Bertram G, Hughes HB, Jansen T, Jacobs L, Buurman ET, Gijzen K, Williams DL, Torensma R, McKinnon A, MacCallum DM, Odds FC, Van der Meer JW, Brown AJ, Kullberg BJ (2006) Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest 116(6):1642–1650
Huang J, Brumell JH (2009) NADPH oxidases contribute to autophagy regulation. Autophagy 5(6):887–889
Ma J, Becker C, Lowell CA, Underhill DM (2012) Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem 287(41):34149–34156
Nicola AM, Albuquerque P, Martinez LR, Dal-Rosso RA, Saylor C, De Jesus M, Nosanchuk JD, Casadevall A (2012) Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect Immun 80(9):3065–3076
Lai S-C, Devenish RJ (2012) LC3-associated phagocytosis (LAP): connections with host autophagy. Cells 1(3):396–408
Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172(5):2731–2738
Crişan TO, Plantinga TS, van de Veerdonk FL, Farcaş MF, Stoffels M, Kullberg BJ, van der Meer JW, Joosten LA, Netea MG (2011) Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One 6(4):e18666
Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC (2011) Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286(11):9587–9597
Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, van der Meer JW, Girardin SE, Joosten LA, Netea MG (2011) Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 60(9):1229–1235
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Bánhegyi G, Bartholomew CR, Bassham DC, Bast RC Jr, Batoko H, Bay BH, Beau I, Béchet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahová M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen X, Chen X, Chen YG, Chen Y, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronjé MJ, Cuervo AM, Cullen JJ, Czaja MJ, D’Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CA, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht AM, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen EL, Espert L, Espina V, Fan H, Fan J, Fan QW, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farré JC, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fésüs L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao FB, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, González-Estévez C, González-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan JL, Guan KL, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S, Hazelhurst LA, He C, He YW, Hébert MJ, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Höhfeld J, Holyoake TL, Hong MH, Hood DA, Hotamisligil GS, Houwerzijl EJ, Høyer-Hansen M, Hu B, Hu CA, Hu HM, Hua Y, Huang C, Huang J, Huang S, Huang WP, Huber TB, Huh WK, Hung TH, Hupp TR, Hur GM, Hurley JB, Hussain SN, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidoro C, Isobe K, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jäättelä M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon JH, Jeung EB, Jiang H, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiang X, Jiang X, Jiménez A, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GV, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhász G, Juillerat-Jeanneret L, Jung CH, Jung YK, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang YA, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke PY, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JA, Kiger AA, Kihara A, Kim DR, Kim DH, Kim DH, Kim EK, Kim HR, Kim JS, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim YS, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BC, Koch JC, Koga H, Koh JY, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong WJ, Korolchuk VI, Kotake Y, Koukourakis MI, Kouri Flores JB, Kovács AL, Kraft C, Krainc D, Krämer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Krüger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, László L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJ, Lee BW, Lee GM, Lee J, Lee JH, Lee M, Lee MS, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei HY, Lei QY, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li JL, Li L, Li S, Li W, Li XJ, Li YB, Li YP, Liang C, Liang Q, Liao YF, Liberski PP, Lieberman A, Lim HJ, Lim KL, Lim K, Lin CF, Lin FC, Lin J, Lin JD, Lin K, Lin WW, Lin WC, Lin YL, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu CF, Liu K, Liu L, Liu QA, Liu W, Liu YC, Liu Y, Lockshin RA, Lok CN, Lonial S, Loos B, Lopez-Berestein G, López-Otín C, Lossi L, Lotze MT, Lőw P, Lu B, Lu B, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KF, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MC, Malorni W, Man N, Mandelkow EM, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Mariño G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, McInerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Meléndez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RF, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao CY, Miao JY, Michels PA, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE, Müller S, Muller S, Münger K, Münz C, Murphy LO, Murphy ME, Musarò A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakatogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nürnberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou JH, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Pöggeler S, Poirot M, Poletti A, Poüs C, Pozuelo-Rubio M, Prætorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian SB, Qin L, Qin ZH, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Régnier-Vigouroux A, Reichert AS, Reiners JJ Jr, Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, Rodríguez de Córdoba S, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KM, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EB 3rd, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AM, Sánchez-Alcázar JA, Sánchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AH, Scharl M, Schätzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schüller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen HM, Shen WC, Sheng ZH, Shi Y, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin EC, Simmons A, Simon AK, Simon HU, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Strålfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun SY, Sun ZJ, Sung JJ, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy LK, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takács-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KS, Tanaka K, Tanaka K, Tang D, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Tönges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai TF, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doorn W, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan XB, Wang A, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H, Wang HG, Wang HD, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang X, Wang YJ, Wang Y, Wang Z, Wang ZC, Wang Z, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen LP, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu DC, Wu WK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xie Z, Xu DZ, Xu J, Xu L, Xu X, Yamamoto A, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang DS, Yang E, Yang JM, Yang SY, Yang W, Yang WY, Yang Z, Yao MC, Yao TP, Yeganeh B, Yen WL, Yin JJ, Yin XM, Yoo OJ, Yoon G, Yoon SY, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu L, Yu SW, Yu WH, Yuan ZM, Yue Z, Yun CH, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJ 3rd, Zeitlin SO, Zhang H, Zhang HL, Zhang J, Zhang JP, Zhang L, Zhang L, Zhang MY, Zhang XD, Zhao M, Zhao YF, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou CZ, Zhu C, Zhu WG, Zhu XF, Zhu X, Zhu Y, Zoladek T, Zong WX, Zorzano A, Zschocke J, Zuckerbraun B (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8(4):445–544
Noda T, Kageyama S, Fujita N, Yoshimori T (2012) Three-axis model for Atg recruitment in autophagy against Salmonella. Int J Cell Biol 2012:389562
Moon CM, Shin DJ, Kim SW, Son NH, Park A, Park B, Jung ES, Kim ES, Hong SP, Kim TI, Kim WH, Cheon JH (2013) Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm Bowel Dis 19(1):106–114
Diekema DJ, Messer SA, Brueggemann AB, Coffman SL, Doern GV, Herwaldt LA, Pfaller MA (2002) Epidemiology of candidemia: 3-year results from the emerging infections and the epidemiology of Iowa organisms study. J Clin Microbiol 40(4):1298–1302
Wang H, Chow S-C (2007) Sample size calculation for comparing proportions. John Wiley & Sons, Inc., Published Online: 14 Dec 2007. doi:10.1002/9780471462422.eoct005
Plantinga TS, Johnson MD, Scott WK, van de Vosse E, Velez Edwards DR, Smith PB, Alexander BD, Yang JC, Kremer D, Laird GM, Oosting M, Joosten LA, van der Meer JW, van Dissel JT, Walsh TJ, Perfect JR, Kullberg BJ, Netea MG (2012) Toll-like receptor 1 polymorphisms increase susceptibility to candidemia. J Infect Dis 205(6):934–943
Johnson MD, Plantinga TS, van de Vosse E, Velez Edwards DR, Smith PB, Alexander BD, Yang JC, Kremer D, Laird GM, Oosting M, Joosten LA, van der Meer JW, van Dissel JT, Walsh TJ, Perfect JR, Kullberg BJ, Scott WK, Netea MG (2012) Cytokine gene polymorphisms and the outcome of invasive candidiasis: a prospective cohort study. Clin Infect Dis 54(4):502–510
Fernández-Arenas E, Bleck CK, Nombela C, Gil C, Griffiths G, Diez-Orejas R (2009) Candida albicans actively modulates intracellular membrane trafficking in mouse macrophage phagosomes. Cell Microbiol 11(4):560–589
Eskelinen EL (2006) Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 27(5–6):495–502
Brown GD, Gordon S (2001) Immune recognition. A new receptor for beta-glucans. Nature 413(6851):36–37
Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SY, Gordon S (2002) Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med 196(3):407–412
Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, Elbers CC, Johnson MD, Cambi A, Huysamen C, Jacobs L, Jansen T, Verheijen K, Masthoff L, Morré SA, Vriend G, Williams DL, Perfect JR, Joosten LA, Wijmenga C, van der Meer JW, Adema GJ, Kullberg BJ, Brown GD, Netea MG (2009) Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 361(18):1760–1767
Plantinga TS, van der Velden WJ, Ferwerda B, van Spriel AB, Adema G, Feuth T, Donnelly JP, Brown GD, Kullberg BJ, Blijlevens NM, Netea MG (2009) Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin Infect Dis 49(5):724–732
Yang CS, Rodgers M, Min CK, Lee JS, Kingeter L, Lee JY, Jong A, Kramnik I, Lin X, Jung JU (2012) The autophagy regulator Rubicon is a feedback inhibitor of CARD9-mediated host innate immunity. Cell Host Microbe 11(3):277–289
van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG (2011) STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 365(1):54–61
Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulácsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bué M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Maródi L, Boisson-Dupuis S, Puel A, Casanova JL (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208(8):1635–1648
Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452(7188):773–776
Acknowledgments
The authors would like to thank Mark van Boxtel for helping with the in vitro experiments and Dennis Kremer for the help with designing the Sequenom assays.
M.G.N. was supported by an ERC Consolidator Grant [310372]. T.S.P. was supported by a VENI grant from the Dutch Organization for Scientific Research. F.L.vdV. was supported by a VENI grant from the Dutch Organization for Scientific Research.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Smeekens, S.P., Malireddi, R.K., Plantinga, T.S. et al. Autophagy is redundant for the host defense against systemic Candida albicans infections. Eur J Clin Microbiol Infect Dis 33, 711–722 (2014). https://doi.org/10.1007/s10096-013-2002-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-013-2002-x