
Abstract
The Dynactin 1 (DCTN1) encodes the p150 subunit of dynactin, which engages retrograde axonal transport. Missense mutations in DCTN1 have been linked to a series of neurodegenerative diseases, including distal hereditary motor neuropathies (dHMN) and Perry syndrome. A few pathogenic DCTN1 mutations related with Perry syndrome have been described within, or adjacent to, the highly conserved N-terminal cytoskeleton-associated protein, glycine-rich (CAP-Gly) domain. But to our best knowledge, only the pathogenic G59S mutation in DCTN1 has been reported in dHMN7B families. Herein, we provided a novel heterozygous mutation in DCTN1 which caused both dHMN7B and Perry syndrome from a Chinese family. Whole exome sequencing (WES) was performed to identify the disease-associated genes. Single nucleotide variants (SNVs) and small insertions/deletions (INDELs) were further predicted with Mutation Taster, Polymorphism Phenotyping v2 (PolyPhen-2), and Sorting Intolerant From Tolerant (SIFT) and compared to the Single Nucleotide Polymorphism Database(dbSNP), Exome Aggregation Consortium (ExAC), and the 1000 Genomes Project. Furthermore, a novel missense mutation c.279G>C (Q93H) in DCTN1 was identified as the candidate loci. The mutation was confirmed with Sanger sequencing in the family members and cosegregated with various phenotypes. In silico analysis and molecular structural modeling, the mutation not only caused the loss of a hydrogen bond within the p150 protein but also affected the formation of hydrogen bonds between p150 and EB. Therefore, the new Q93H mutation in DCTN1 caused both familial dHMN7B and Perry syndrome. Our findings could expand the clinical and pathogenic spectrum and strengthen the clinical diagnostic role of the DCTN1 gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The DCTN1 gene encodes the p150 subunit of the transporter protein dynactin, a microtubule-associated protein, which plays a crucial role in axon maintenance, and regulating vesicle and organelle transport [1]. The p150 subunit is required for retrograde axonal transport of vesicles and organelles along microtubules, the functions of which rely on its CAP-Gly domain (aa 48–90) [2]. The CAP-Gly domain interacts with microtubules and EB, the binding to which is impaired in mutant constructs [2, 3]. To date, mutations in DCTN1 have been identified to result in several kinds of neurodegenerative diseases, mainly including dHMN, Perry syndrome, and amyotrophic lateral sclerosis (ALS) [4].
The dHMN are genetically heterogeneous motor neuron diseases which share the cardinal feature of a length-dependent predominantly motor neuropathy [5]. dHMN are classified into various types based on the mode of inheritance and phenotype (Table 1). dHMN7, including type 7A and 7B, is characterized by adult onset with vocal-cord paralysis and autosomal dominant inheritance pattern [5]. Of the two, dHMN7B is caused by mutations in the DCTN1 gene located on 2p13 [6]. Patients with dHMN7B exhibited dyspnea due to bilateral vocal cord paralysis, progressive facial weakness, and weakness and muscle atrophy in the hands [6, 7]. In 2003, Puls et al. firstly reported a family with dHMN7B caused by a DCTN1 mutation (p.G59S) in the CAP-Gly domain [6]. DCTN1 p.G59S mutation was later identified in two unrelated families with dHMN from Korea [7]. Mutations lying without the CAP-Gly domain could also result in dHMN, including p.L210Afs*90 and p.E340G [8, 9]. The significance of other mutations in DCTN1 gene, including p.V685G [10], p.Y670F [11], and p.D424H [12], has remained unclear. So far, a few mutations in DCTN1 have been associated with dHMN7B, the phenotypes of which vary among patients with different pathogenic mutations. The interesting thing was that DCTN1 mutations could also lead to Perry syndrome, a quite different neurodegenerative disease from dHMN7B. Perry syndrome, a rare neurodegenerative disorder characterized by autosomal dominant inheritance, psychiatric symptoms, parkinsonism, weight loss, and central hypoventilation, was previously linked to other p150 mutations, such as p.Gly71Arg/Ala/Glu, p.Thr72Pro, and p.Gln74Pro [13]. In families with Perry syndrome, mean onset is 48 years (range, 35–61), with a 5-year (range, 2–10) duration to death attributed to respiratory failure or suicide [14]. Brain autopsy reveals a pallidonigral TDP-43 proteinopathy affecting the ventrolateral medulla respiratory center but sparing the cortex, hippocampus, and motor neurons [13, 15, 16].
Here we identified a Chinese family with dHMN and Perry syndrome simultaneously due to a novel DCTN1 mutation (p.Q93H). The main clinical manifestations were adult-onset widespread weakness and muscle atrophy among dHMN patients. On the contrary, one patient developed depression, parkinsonism, and hypoventilation and was diagnosed with Perry syndrome. On the basis of thorough clinical, pathological, and genetic analysis, we aimed to explore the phenotype–genotype relationship in the complicated family.
Methods
Cases and clinical assessment
Detailed medical history taking and standard neurological examinations were conducted for family members. Electromyography (EMG) after obtaining informed consent was performed for the proband and his affected nephew. Mucsle biopsy (left gastrocnemius muscle) was also conducted on the proband. For histopathological analysis, serial frozen sections (12 μm) were stained with hematoxylin and eosin (H&E), modified Gomori trichrome (MGT), oil red O (ORO), and periodic acid-Schiff (PAS). Histochemical (cytochrome c oxidase (COX), succinate dehydrogenase (SDH), NADH, and ATPase (pH 4.20, 9.60) reactions) analyses were performed as well. Peripheral blood samples from 8 family members (II-1, II-3, II-5, II-7, II-9, III-11, III-13, III-15) were collected for DNA extraction. DNA was isolated from peripheral blood with CWE9600 Automated Nucleic Acid Extraction System using CWE2100 Blood DNA Kit V2 (CWBiotech, China) according to the manufacturer’s instructions.
WES and bioinformatics analysis
Whole exome sequencing was performed to identify the disease-associated variants in two affected and one unaffected individuals. DNA libraries were prepared with KAPA Library Preparation Kit (Kapa Biosystems, KR0453). Exome sequence capture was performed using the IDT xGen Lockdown Probes (Integrated DNA Technologies, USA). DNA libraries were sequenced using paired-end 150 bp reads on an Illumina Novaseq 6000 platform (Illumina, USA) according to the standard manual. The raw data were subjected to quality control using Illumina Sequence Control Software (SCS) and then aligned to the human reference genome (hg19) using the BWA Aligner. The single-nucleotide polymorphisms (SNPs) and small insertions/deletions (INDELs) were analyzed using the Genome Analysis ToolKit (GATK). Variants were annotated using ANNOVAR. Variants with a low quality score (depth < 10 or genotype quality < 20) were filtered out. Variants were also excluded with the minor allele frequency (MAF) > 0.5% in the dbSNP, genomeAD, and EXAC. The pathogenicity of variants was predicted with SIFT, Polyphen-2, and Mutation Taster programs. To estimate the evolutionary conservation of the mutated site, protein sequences from different species were aligned using Clustal X1.8.
Causative mutation identification
An autosomal dominant inheritance pattern should be responsible for the different clinical features in this family due to the diseases affecting both male and female individuals in three generations. To identify causative mutations, we looked for deleterious autosomal dominant mutations shared by two patients and absent in one unaffected individual.
PCR and sanger sequencing
The candidate causal gene discovered by WES was then confirmed by Sanger sequencing and cosegregation analyses were also conducted. We also screened the mutations in 200 unrelated Chinese individuals. The primers were designed by Primer Premier 5.0 (Premier Biosoft, USA) and PCR was carried out to amplify the fragments covering the mutated sites on LifeECO Thermal Cycler TC-96/G/H(b)C (Bioer Technology, China). The PCR products were further purified with agarose gel electrophoresis and then sequenced by ABI 3730XL DNA Sequencer (Applied Biosystems, USA). Sanger sequencing results were analyzed by Chromas Lite v2.01 (Technelysium, Australia).
Molecular modeling of p150 protein
The mutant protein structure for p150 was constructed by SWISS-MODEL (http://www.swissmodel.expasy.org), using a crystal structure of the CAP-Gly domain of human p150 (PDB: 2HKN) as a structural template. The three-dimensional (3D) protein modeling for wild-type and mutant p150 was obtained using the Swiss-PdbViewer 4.1 molecular visualization software.
Results
Clinical manifestation
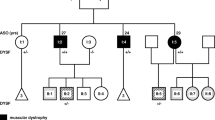
A three-generation family from Shanxi Province of China was recruited (Fig. 1). The proband (II-9), a 49-year-old male worker, noticed weakness and amyotrophy in his hands 10 years ago. Muscle weakness and atrophy in four limbs and trunk developed gradually. He can walk independently on his heels or toes. However, walking fast could induce dyspnea. The patient did not complain sensory symptoms. Physical examination revealed bilateral facial weakness, dysarthria, and widespread muscle atrophy (Fig. 2a and b). The Medical Research Council scale values were grade 2 for bilateral hand intrinsic muscles and grade 4 for muscles at four limbs. The deep-tendon reflexes were diminished, and the Babinski signs were negative. Sensory examination was normal. Tongue fasciculation and atrophy, dysphagia, and abnormal gag reflex were not noticed. Mental psychological and cognitive tests were normal. An elevated serum creatine kinase (1106 U/L) was noted. Arterial blood gas analysis indicated hypercapnia and hypoxemia (PH 7.34; PCO2 49 mmHg; PO2 70 mmHg). The electrophysiological studies showed giant motor unit action potentials and spontaneous activity, indicating extensive acute and chronic neurogenic lesions. The motor conduction studies were normal except for prolonged distal latencies and reduced velocities and CMAP amplitudes in bilateral median nerves. The sensory conduction studies were normal (Table 2). The biopsy of left gastrocnemius showed neurogenic pathological changes (Fig. 3). Nerve ultrasound at bilateral upper limbs was normal. The cross-sectional areas of the left median nerve, right median nerve, left ulnar nerve, and right ulnar nerve were respectively 0.24 cm2, 0.25 cm2, 0.4 cm2, and 0.5 cm2. Indirect laryngoscopy did not show vocal-fold paralysis. The pulmonary ventilation function was in the normal range, and the diffuse function was moderately decreased. There were no significant abnormalities in MRI of the brain and the cervical spine, chest CT, and abdominal ultrasound.

Pedigree of the family with DCTN1 mutation. Generations are shown as I–III. Males are indicated by a square, females by circles, affected members indicated by a shaded black square or circles, and the proband (II-9) indicated by an arrow. A slash indicates deceased family members. Plus sign in parentheses represents carrying the mutation site, and minus sign in parentheses represents not

Clinical manifestations of the proband and patient II-1. a Scapular winging and widespread muscle atrophy of the proband. b Atrophy of hand intrinsic muscles of the proband. c, d Atrophy of hand intrinsic muscles of patient II-1

Muscle biopsy from the proband. a Hematoxylin and eosin staining muscular fibers are moderate variable in size and the shapes of small fibers are mostly angular and elongated. Degeneration muscle fibers can be seen. b Modified Gomori trichrome staining shows no ragged red fibers (RRFs). c NADH staining shows that the reticulate structure of some muscle fibers is slightly disordered. NADH dehydrogenase is partly reduced, and target fibers can be seen. d ATPase 4.20 staining shows that there are mainly type I muscle fibers and grouping can be observed
The proband’s mother (I-2) got ill in her 40s and died in her 50s. According to the statements of family members, patient I-2 did have a history of hand muscle weakness and atrophy. Two of the proband’s affected sisters II-1 (Fig. 2c and d) and II-7 also had similar symptoms, but what different from the proband was that both of them had postural tremor (Supplementary video). In addition, tongue fasciculation was noticed only in patient II-1.
(MP4 626 kb)
On the other hand, some affected family members presented clinical symptoms consistent with Perry syndrome. Patient II-11, another affected sister of the proband, committed suicide from depression at the age of 53. Based on her son’s recollections, postural tremor, as well as hand muscle weakness and atrophy were observed in patient II-11 for more than a decade. Patient III-1 was diagnosed with depression at the age of 43. Then she developed static tremor 3 years later and was considered for Parkinson’s disease with masklike face, bradykinesia, and abnormal posture and pace. Unfortunately, she died soon after of respiratory failure at the age of 49. Patient III-15 was relatively young (age 32 at examination) and had the mildest clinical symptoms and only presented with postural tremor of the hands. The electrophysiological studies of patient III-15 were normal. The proband’s father (I-1) and the other two sisters (II-3 and II-5) did not have similar symptoms. An autosomal dominant inheritance pattern was evident in this family’s pedigree. The clinical manifestations of the proband and other family members are shown in Table 3.
WES identification of novel missense mutation in DCTN1
To localize the disease gene accurately, sequencing was performed in the proband (II-9), one of his affected sisters (II-7), and one of his unaffected sisters (II-5). The mean depths of the targeted region were 143.16×, 137.71×, and 132.26× for II-5, II-7, and II-9 respectively. Targeted regions with depths greater than 20×reads showed coverage more than 99.70%. Given the autosomal dominant inheritance pattern of this family, 30 heterozygous variants shared by subjects II-7 and II-9 and absent in II-5 were taken for further analysis (Table 4). After filtering out irrelevant mutations (Supplementary table), a novel heterozygous mutation c.279G>C (NM_004082; NP_004073) in DCTN1 gene was identified (Fig. 4a). Mutation c.279G>C converts amino acid 93 from glutamine to histidine (p.Q93H), which is predicted to be deleterious by SIFT (SIFT score: 0.004), Polyphen-2 (probability score: 0.971, sensitivity: 0.77, specificity: 0.96), and Mutation Taster (disease causing, probability score: 0.999). The variant was absent from the Human Gene Mutation Database (HGMD), dbSNP, 1000 Genomes project, ClinVar database, Exome Aggregation Consortium (ExAC) database, and the Genome Aggregation Database (gnomAD). Protein conservation analysis by an alignment of p150 protein sequences revealed that the position is highly conserved among diverse species (Fig. 4b). We assayed the mutation-carrying state in the proband, all his alive siblings, two nephews, and one niece (Fig. 1). The mutation c.279G>C was co-segregated with the clinical phenotypes. In 200 unrelated Chinese controls, the mutation was not found. According to the American College of Medical Genetics and Genomics guidelines, the Q93H mutation in DCTN1 could be classified as a “pathogenic variant” [17].

Mutation analysis of the family. a Sanger sequencing to confirm the mutation in the family members. b Sequence conservation analysis of p150 protein
Molecular modeling of p150 protein
A 3D structural model of p150 was predicted with SWISS-MODEL to determine whether the novel p.Q93H missense mutation affected the protein structure based on a published crystal structure of the CAP-Gly domain of human p150 (PDB: 2HKN). Using Swiss-Pdb Viewer 4.1, the mutation could result in the loss of an H-bond between Gln93 and Arg90 due to the substitution of glutamine to histidine at position 93, possibly perturbing the amino acid side chain (Fig. 5). Furthermore, Gln93 and Arg90 in p150 provide additional specific binding sites for EEY/F-COO2 sequence motifs, which interact with the CAP-Gly domains (aa 48–90) and represent characteristic elements of EB and a-tubulin proteins [3]. There are favorable hydrogen bonding interactions between the two glutamate side chains of the EEY-COO2 tripeptide segment and the side chains of Gln93 and Arg90. The p.Q93H missense mutation may disturb the interaction between p150 and a-tubulin or EB.

Molecular modeling comparison of wild-type and missense mutant (p.Q93H, Gln93-to-His) in the p150 protein. a The wild-type protein has two H-bonds between Gln 93 and Arg90. b The mutant protein His 93 is predicted to loss an H-bond between Gln93 and Arg90 due to the substitution of histidine to glutamine at position 93, possibly perturbing the amino acid side chain. Comparison sites are highlighted by white frame lines and locally zoomed
Discussion
A certain mutation in DCTN1 has been identified to result in dHMN, Perry syndrome, or ALS [9]. In this study, we report a novel heterozygous DCTN1 mutation p.Q93H in a Chinese family with different clinical phenotypes. Four (I-2, II-1, II-7, and II-9) of the seven affected family members presented with adult-onset muscle weakness and atrophy beginning at bilateral hands, which coincided with dHMN. One (III-1) presented with depression and parkinsonism diagnosed with Perry syndrome. One (II-11) had overlapped clinical symptoms of dHMN and Perry syndrome. The other one only had postural tremor, which is also noticed in II-1, II-7, and II-11.
The Q93H mutation in DCTN1 was first reported as a pathogenic mutation, because this mutation (i) was not found in 200 unrelated Chinese controls and cosegregated with the phenotypes in the family (PS4 and PP1); (ii) located in a critical and well-established functional domain (PM1); (iii) was absent from controls in HGMD, dbSNP, 1000 Genomes project, ClinVar database, ExAC database, and gnomAD (PM2); (iv) was predicted to be pathogenic by at least three in silico prediction tools (PP3); and (iv) affected highly conserved amino acid [17].
The dynactin protein complex, a microtubule-associated protein with a molecular mass of 1.2 MDa and 20 subunits, plays a crucial role in axon maintenance, and regulating vesicle and organelle transport via direct binding to microtubules, the molecular motor dynein, and various cargoes [1]. DCTN1 gene on chromosome 2p13 encodes the p150 protein (OMIM 601143, also called Dynactin 1), which is the largest subunit of the dynactin complex with a size of 150KD. The p150 protein, containing the N-terminal CAP-Gly domain and followed by two coiled-coil domains (CC1 and CC2, Fig. 6), is essential in enriching dynactin at neurite tips through the EB, which are both microtubule-binding molecules [18, 19]. The CAP-Gly domain interacts with a-tubulin protein and EB, and CC1 domain interacts with dynein [2].

Domain structure of p150 subunit of dynactin
A link between DCTN1 gene and dHMN was first proposed in 2003 by Puls et al., and this type of dHMN was termed 7B [3]. They identified that a G59S missense mutation in the CAP-Gly domain of DCTN1 gene was pathogenic in a family with slowly progressive, autosomal dominant form of lower motor neuron disease without sensory symptoms. The disease presented in adulthood with breathing difficulty due to vocal-cord paralysis, progressive facial and hand weakness, with leg weakness and dysphagia developing later. However, clinical heterogeneity was observed in different families carrying the G59S mutation. Hwang et al. reported that affected members from two families did not exhibit facial muscle weakness, dysphagia, and dysarthria, and in one family, hand muscle weakness was the first major symptom [7]. The clinical features of dHMN7B were more heterogeneous among patients carrying different pathogenic mutations in DCTN1. Nam et al. reported one patient presented in childhood with distal dominant weakness in upper and lower limbs and respiratory distress while talking caused by a p.E340G mutation [8]. Tian et al. described a patient with a de novo frameshift heterozygous variant c.626dupC (p.L210Afs*90) in DCTN1 exhibited lifelong weakness of distal limbs, abnormal gait, and congenital foot malformation [9].
Patients with mutation p.Q93H and p.G59S in DCTN1 shared similar clinical features, including adult onset and hand muscular atrophy presenting as the initial manifestation, which might be due to the overlap of their function. EEY/F-COO2 sequence motifs of EB and a-tubulin proteins interact with the CAP-Gly domains (aa 48–90) [3]. In vitro evidence demonstrated that the p.G59S substitution resulted in reduced microtubule binding [6]. Gln93 and Arg90 of p150 protein provide additional specific binding sites for EEY/F-COO2 sequence motifs [3]. Patients suffered from dHMN in the present study had their own characteristics. Postural tremor and widespread muscular atrophy including muscles in proximal limbs and trunk had not been reported in the literature. In addition, our patients did not exhibit apparent vocal-cord paralysis. Dyspnea or stridor induced by vocal-cord paralysis often presented at onset in patients carrying p.G59S mutation [20].
Up to date, there has been no literature of one mutation in DCTN1 causing dHMN or Perry syndrome. However, in the pedigree we reported, one patient (III-1) presented with depression and parkinsonism, which were in accord with Perry syndrome. The mechanism was not known. We speculated it might be due to that the mutant p150 proteins in dHMN and in Perry syndrome had similar functional deficits. All the mutations associated with Perry syndrome map to CAP-Gly domain. Most of the dHMN7B-related mutations tend to locate on the N-ter half. On the contrary, the majority of the ALS-related mutations tend to locate on C-ter half of p150 protein (Fig. 7) [9]. Functional analyses of the mutant p150 proteins associated with Perry syndrome (p.P52L, p.G71R, p.Q74P, and p.Y78C) and dHMN7B (p.G59S) showed reduced microtubule binding [4, 6, 13, 21, 22]. However, the ALS-associated mutant p150 (p.M571T, p.A785W, p.R1101K, and p.T1249I) did not show abnormal microtubule binding abilities [23, 24]. The mutant of dHMN7B (p.G59S) was more prone to inducing aggregation of p150 than Perry syndrome mutants, which were highly toxic [25]. The mutation p.G59S affects a residue important for maintaining the structure of the CAP-Gly domain which promotes misfolding and aggregation, and the aggregation decreases the stability of the dynactin complex, preventing effective association between dynein and dynactin and ultimately disrupts axonal transport [25]. In contrast, mutations associated with Perry syndrome are more specifically disrupting protein-protein interactions. The primary pathogenic mechanism in Perry syndrome may be a loss of CAP-Gly function [25]. Gln93 and Arg90 are located on the surface of p150 protein and provide additional specific binding sites for EEY/F-COO2 sequence motifs of EB and a-tubulin proteins [3]. The p.Q93H missense mutation may disturb the interaction between p150 and a-tubulin or EB and reduce microtubule binding capacity. Further functional studies are needed to verify whether p.Q93H mutation promotes misfolding and aggregation of p150.

Variants in DCTN1 reported in dHMN7B, Perry syndrome, and ALS
It is interesting why specific cell types, dopaminergic neurons or motor neurons, are damaged preferentially in each disease. Patients with Perry syndrome had lower glutamate and GABA content in the frontal cortex and substantia nigra [26]. The neocortex, hippocampus, and upper and lower motor neurons, which were typically affected in ALS, were spared. In one autopsied case of a dHMN7B patient, neuronal loss was seen in the hypoglossal nucleus and the ventral horn of the spinal cord, but no pathology in the substantia nigra was described [4]. There were also DCTN1 mutations reported in ALS patients, whereas their role in motor neuron degeneration was unclear [27]. DCTN1 was found to be downregulated in both cortexes and the spinal cord in sporadic ALS patients, resulting in a lower protein expression, suggesting altered expression of this protein could be involved in the pathophysiological process [28, 29]. Recently, it was proposed that Dynactin1a (a zebrafish ortholog for DCTN1) depletion represented an early event in NMJ degeneration and that ALS-related mutations in this gene were likely not causative but indeed had a place in the oligogenic etiology of ALS pathogenesis [30].
In conclusion, we identified a Chinese family with dHMN and Perry syndrome due to a novel DCTN1 mutation (p.Q93H). Up to now, very few mutations in DCTN1 are associated with dHMN7B and there has been no literature of one DCTN1 mutation causing dHMN or Perry syndrome at the same time. As the mutational spectrum in DCTN1 is expanding, clinical heterogeneity of DCTN1-related diseases will be more significant. These results have enriched the clinical and mutational spectrum of dHMN7B and Perry syndrome, and will provide assistance for the molecular diagnosis of DCTN1 in the future.
References
Urnavicius L et al (2015) The structure of the dynactin complex and its interaction with dynein. Science 347(6229):1441–1446
Cronin MA, Schwarz TL (2012) The CAP-Gly of p150: One domain, two diseases, and a function at the end. Neuron 74(2):211–213
Honnappa S et al (2006) Key interaction modes of dynamic +TIP networks. Mol Cell 23(5):663–671
Konno T et al (2017) DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat Disord 41:14–24
Rossor AM et al (2012) The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 83(1):6–14
Puls I et al (2003) Mutant dynactin in motor neuron disease. Nat Genet 33(4):455–456
Hwang SH et al (2016) Distal hereditary motor neuropathy type 7B with Dynactin 1 mutation. Mol Med Rep 14(4):3362–3368
Nam SH et al (2016) Identification of genetic causes of inherited peripheral neuropathies by targeted gene panel sequencing. Mol Cell 39(5):382–388
Tian WT et al (2020) New phenotype of DCTN1-related spectrum: early-onset dHMN plus congenital foot deformity. Ann Clin Transl Neurol 7(2):200–209
Chae JH et al (2015) Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J Med Genet 52(3):208–216
Schabhüttl M et al (2014) Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J Neurol 261(5):970–982
Klein CJ et al (2014) Application of whole exome sequencing in undiagnosed inherited polyneuropathies. Journal of Neurology. Neurosurg Psychiatry 85(11):1265–1272
Farrer MJ et al (2009) DCTN1 mutations in Perry syndrome. Nat Genet 41(2):163–165
Wider C et al (2010) Elucidating the genetics and pathology of Perry syndrome. J Neurol Sci 289(1–2):149–154
Tsuboi Y et al (2008) Neurodegeneration involving putative respiratory neurons in Perry syndrome. Acta Neuropathol 115(2):263–268
Wider C et al (2009) Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord 15(4):281–286
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Schroer TA (2004) Dynactin. Annu Rev Cell Dev Biol 20(1):759–779
Waterman-Storer CM, Karki S, Holzbaur EL (1995) The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc Natl Acad Sci U S A 92(5):1634–1638
Li Y, Garrett G, Zealear D (2017) Current treatment options for bilateral vocal fold paralysis: a state-of-the-art review. Clin Exp Otorhinolaryngol 10(3):203–212
Araki E et al (2014) A novel DCTN1 mutation with late-onset parkinsonism and frontotemporal atrophy. Mov Disord 29(9):1201–1204
Tacik P et al (2014) Three families with Perry syndrome from distinct parts of the world. Parkinsonism Relat Disord 20(8):884–888
Dixit R et al (2008) Regulation of dynactin through the differential expression of p150GluedIsoforms. J Biol Chem 283(48):33611–33619
Stockmann M et al (2013) The dynactin p150 subunit: cell biology studies of sequence changes found in ALS/MND and Parkinsonian syndromes. J Neural Transm (Vienna) 120(5):785–798
Moughamian AJ, Holzbaur ELF (2012) Dynactin is required for transport initiation from the distal axon. Neuron 74(2):331–343
Perry TL et al (1990) Dominantly inherited apathy, central hypoventilation, and Parkinson’s syndrome: clinical, biochemical, and neuropathologic studies of 2 new cases. Neurology 40(12):1882–1887
Cooper-Knock J et al (2017) Targeted genetic screen in amyotrophic lateral sclerosis reveals novel genetic variants with synergistic effect on clinical phenotype. Front Mol Neurosci 10:370
Kuźma-Kozakiewicz M et al (2013) Dynactin deficiency in the CNS of humans with sporadic ALS and mice with genetically determined motor neuron degeneration. Neurochem Res 38:2463–2473
Tanaka F et al (2012) Neuropathology and omics in motor neuron diseases. Neuropathology 32(4):458–462
Bercier V et al (2019) Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol Neurodegener 14(1):27
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical publication statement
We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this study is consistent with those guidelines.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures were approved by the Ethics committee of First Hospital of Shanxi Medical University (Taiyuan, China) and carried out in accordance with the principles of Helsinki Declaration.
Informed consent
Informed consent was preliminarily obtained from all patients and controls before their inclusion in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 2
(XLS 29 kb)
Rights and permissions
About this article

Cite this article
Zhang, J., Wang, H., Liu, W. et al. A novel Q93H missense mutation in DCTN1 caused distal hereditary motor neuropathy type 7B and Perry syndrome from a Chinese family. Neurol Sci 42, 3695–3705 (2021). https://doi.org/10.1007/s10072-020-04962-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-020-04962-w