
Abstract
Background
Charcot–Marie–Tooth (CMT) disease is a prevalent and heterogeneous peripheral neuropathy. Most patients affected with the axonal form of CMT (CMT2) do not harbor mutations in the approximately 90 known CMT-associated genes. We aimed to identify causative genes in two CMT2 pedigrees.
Methods
Neurologic examination, laboratory tests and brain MRIs were performed. Genetic analysis included exome sequencing of four patients from the two pedigrees. The predicted effect of a deep intronic mutation on splicing was tested by regular and real-time PCR and sequencing.
Results
Clinical data were consistent with CMT2 diagnosis. Inheritance patterns were autosomal recessive. Exome data of CMT2-101 did not include mutations in known CMT-associated genes. Sequence data, segregation analysis, bioinformatics analysis, evolutionary conservation, and information in the literature strongly implicated HADHA as the causative gene. An intronic variation positioned 23 nucleotides away from following intron/exon border in GDAP1 was ultimately identified as cause of CMT in CMT2-102. It was shown to affect splicing.
Conclusion
The finding of a HADHA mutation as a cause of CMT is of interest because its encoded protein is a subunit of the mitochondrial trifunctional protein (MTP) complex, a mitochondrial enzyme involved in long chain fatty acid oxidation. Long chain fatty acid oxidation is an important source of energy for skeletal muscles. The mutation found in CMT2-102 is only the second intronic mutation reported in GDAP1. The mutation in the CMT2-102 pedigree was outside the canonical splice site sequences, emphasizing the importance of careful examination of available intronic sequences in exome sequence data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Charcot–Marie–Tooth (CMT) disease, also known as hereditary motor sensory neuropathy (HMSN), constitutes a clinically and genetically heterogeneous group of inherited peripheral neuropathies with an estimated prevalence of one in a few thousand in most populations [1,2,3,4,5]. It is the most prevalent category of inherited neuropathies [2]. The clinical features of CMT usually include symmetric slowly progressive distal muscle weakness, atrophy and deformity that first affect the lower limbs, some distal sensory impairment, and depressed tendon reflexes. Onset can be during childhood or adulthood, but is most often during the juvenile or early adulthood years. Genetic heterogeneity of CMT is reflected in its various inheritance patterns, including autosomal dominant, autosomal recessive, and X-linked. The dominant pattern is most frequent. Some sporadic cases caused by de novo mutations have been described [6]. Approximately, 90 genes have been implicated to cause or contribute to CMT pathology [7] (https://neuromuscular.wustl.edu/). They function in processes that include RNA processing, protein synthesis and posttranslational processing, intracellular trafficking, ion channel dysfunction, and mitochondrial dysfunction [8, 9]. An effective pharmacologic treatment for CMT is not presently available.
CMT is traditionally classified on the basis of electrophysiological measurements of median motor nerve conduction velocities (MNCV). These measurements reflect the relative amount of myelin and axonal pathology. The major classifications are CMT type 1 (CMT1; demyelinating, MNCV < 38 m/s), CMT type 2 (CMT2; axonal, MNCV > 38 m/s), and the intermediate form (ICMT; MNCV: 25–45 m/s) [3, 10]. Although CMT2 presents with nearly normal motor NCV, neurographic studies show decreased amplitude of nerve action potential, which suggests damage to the axons [11]. Some recent classifications of CMT include the mode of inheritance and the name of the causative gene. A recent review, while acknowledging caveats in available epidemiological data, reported that CMT1 is the most prevalent CMT subtype in most countries and that the frequency of CMT2 varied from 12 to 35.9% in different populations [12]. Most known CMT genes are associated with CMT1, and more than 80% of CMT1 affected individuals have mutations in the known causative genes [9]. PMP22 that encodes peripheral myelin protein 22 KD is by far the most common CMT1 causative gene; mutations in PMP22 account for disease in up to 70% of CMT1 patients [3, 13, 14]. Approximately, 30 genes have been reported to potentially contribute to CMT2 [11]. MFN2 that encodes mitofusin 2 is the major CMT2 causative gene; MFN2 mutations are present in up to 33% of CMT2 patients [15]. Mitofusin 2 has an important role in mitochondrial function. Mutations in MPZ (myelin protein zero) and HSPB1 (Heat Shock Protein Family B (Small) Member 1) are also found in a few percent of CMT2 patients. Contrary to CMT1, up to 75% of CMT2 affected patients do not have mutations in any of the known causative genes [9]. This suggests that some CMT2 causative genes remain to be identified.
Here, we report clinical data on CMT2 affected individuals of two families and results of genetic analysis that culminated in identification of causative mutations in HADHA and GDAP1.
Methods
This research was performed in accordance with the Declaration of Helsinki and with the approval of the ethics board of the University of Tehran.
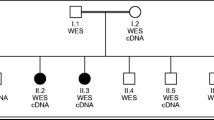
Two CMT2 diagnosed patients, CMT2-101-II1 and CMT2-102-III4, were referred for genetic analysis. CMT2-101-II1 had an affected sibling (CMT2-101-II3), while CMT2-102-III4 had an affected sibling (CMT2-102-III6) and also an affected maternal aunt (CMT2-102-II7) (Fig. 1). CMT2 diagnosis was based on standard criteria (Table 1). The parents and some of the patients were interviewed to obtain family history. Thorough clinical neurologic examinations on the five patients and electrodiagnostic (EDX) testing that included nerve conduction studies (NCS) and electromyography (EMG) in the upper and lower extremities, truncal regions, and cranial regions were performed according to standard procedures (Dantec, Keypoint G4, Natus, CA, USA). Brain magnetic resonance imaging (MRI) was performed on one affected individual of each family. MRI was done using a 1.5-T system (MAGNETOM Avanto 1.5 T, Siemens, Germany). T1- and T2-weighted spin echo protocols were used. Plasma organic acid and acylcarnitine profiles of patients of CMT2-101 were obtained by tandem mass spectrometry.

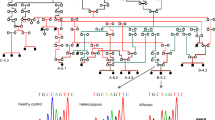
CMT2-101 and CMT2-102 pedigrees. Filled circles and squares: CMT2 affected; probands are identified with arrow. Genotypes of respective putative causative gene in individuals of each pedigree who underwent genetic analysis are shown. MM homozygous mutant genotype, MN heterozygous genotype, NN normal genotype
Genetic analysis was initiated by whole exome sequencing of the DNA of the proband of each family to determine the presence or absence of mutations in any of the many known CMT-associated genes. Exome sequencing was done using the Sure Select V6-POST kit and an Illumina HiSeq 4000 system (Illumina, CA, USA). Sequence alignment was performed against human reference genome GRCh37/hg19, and variant callings were done using ENSEMBL Variant Effect Predictor (https://www.ensembl.org/Tools/VEP) and wANNOVAR (https://wannovar.wglab.org/). Subsequently, filtering was performed by removing SNPs with a minor allele frequency (MAF) of > 0.01 in the dbSNP database (https://www.ncbi.nlm.nih.gov/), the Trans-Omics for Precision Medicine program (https://www.nhlbiwgs.org/), the 1000 Genomes database (www.1000genomes.org), the NHLBI Exome Sequencing Project (https://evs.gs.washington.edu/EVS/), the Exome Aggregation Consortium database (https://exac.broadinstitute.org/), the Genome Aggregation Database (https://genomad.broadinstitute.org/), the Greater Middle East Variome Project (https://igm.ucsd.edu/gme/), ENSEMBL (https://www.ensembl.org/index.html), the Healthy Exomes database (https://www.alzforum.org/exomes/hex), the Sequencing Initiative Suomi database (https://www.sisuproject.fi/), the VarCards database (https://varcards.biols.ac.cn/), or the Iranome database (https://iranome.com/). SNPs observed in in-house exome data belonging to approximately 100 unrelated Iranians affected with non-neurological diseases were also not considered. Among the variations that remained, those that did not affect amino acid change or canonical splicing sites were then removed. A file of homozygous variations and a file of compound heterozygous variations were prepared, and the files were searched to identify variations within any of 74 known CMT-associated genes (Table S1). Subsequently, exome sequencing was performed on the DNA of one additional affected individual of each family and the same analysis protocol was applied to the newly obtained sequence data. For each family, homozygous or compound heterozygous mutations that were identified in both exomed patients were considered as candidate disease-causing variations. These were screened for segregation with disease status among pedigree members by direct sequencing. Novel mutations were also screened in 300 Iranian control individuals by an allele-specific PCR protocol and/or sought in the Iranome database that contains exome data on 800 healthy Iranians.
For assessment of effect of a potential splice site mutation, RNA was isolated from leukocytes of CMT2-102-III4, CMT2-102-III6 and two control individuals, and cDNA was synthesized by standard protocols. The cDNAs were used as template in PCR experiments. The primers used in these PCR experiments were designed to specifically amplify regions that were expected to be present only within the cDNA of patients. All primer sequences are available upon request. Initially, regular PCR followed by gel electrophoresis was done. Subsequently, real-time PCR using a Corbett 65H0 real-time PCR machine (Corbett Research, Sidney, Australia) and the RealQ Plus Master Mix Green (Ampliqon A/S, DK) was performed. β2M (beta-2 microglobulin) and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used as control genes. The experiments pertaining to each individual were done in triplicate. Statistical analysis was done using the Relative Expression Software Tool (REST).
Results
Clinical data on the patients are presented in Table 1. The data, including childhood onset, prominent distal leg weakness, foot deformities, sensory signs, decreased tendon reflexes, electrodiagnostic results, and normal MRI, are consistent with a diagnosis of CMT2 for the five patients of the two families. Commencement of independent walking at 18 months suggests delayed motor development in the two siblings of CMT2-101.
Representative data on exome sequencing results that evidence high-quality sequencing is presented in Table S2. Exome sequencing of the proband of CMT2-101 did not identify homozygous or compound heterozygous sequence variations in known CMT-associated genes (Table S1). The combined sequencing data of the proband and her brother (CMT2-101-II3) proved to be informative. After the described filterings were conducted, nine homozygous variations distributed in nine genes and seven compound heterozygous variations in three genes that were present in both patients were retained (Table 2). Direct sequencing of these 16 variations in 23 members of the proband’s immediate and extended family showed that four homozygous variations in DCST2, HADHA, NAPRT, and ARHGAP39 segregated with disease status (Fig. 2). These genes, respectively, encode DC-stamp domain containing 2, hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha, nicotinate phosphoribosyltransferase, and Rho GTPase activating protein 39. The observed nucleotide variations in the four genes have been previously reported in various databases at low frequencies (Table 2). The affected amino acid in DCST2 is not well conserved during evolution, and five different amino acids with various biochemical properties are observed at corresponding positions in orthologous proteins. The amino acid affected by the variation in HADHA, which is positioned in the “fatty acid oxidation complex, alpha subunit, mitochondrial” domain of the encoded protein, is completely conserved in mammals through fish (Table 3) [16]. Consistent with these observations, all 12 bioinformatics prediction softwares used assessed the DCST2 variation of family CMT2-101 as non-deleterious and 11 assessed the HADHA variation to be deleterious. The affected amino acids in NAPRT and ARHGAP39 are both relatively well conserved. Eleven of 12 bioinformatics tools predicted that the ARHGAP39 variation is neutral, and 6 predicted that the NAPRT variation is neutral. The comparisons reported can cautiously be interpreted to suggest that axonal CMT in family CMT2-101 is caused by the mutation in HADHA. Ultimately, this proposal is supported by descriptions in the literature of clinical features of patients with mutations in HADHA and the closely related gene HADHB (see “Discussion”) [16,17,18,19]. Long-term clinical follow-up (> 10 years), normal creatine phosphokinase levels (measured four times in each patient), and EMG results were not suggestive of myopathy. There were no indications of cardiac problems. The acylcarnitine profiles of the two CMT2-101 patients were normal. Elevated plasma aspartic acid levels in both patients (114 and 120 μM; reference level < 84 μM) is of unknown significance.

Sequence chromatograms of CMT2 causative mutations observed in DNA of CMT2-101 and CMT2-102 patients. Chromatograms of HADHA and GDAP1 mutated and wild-type genotypes are shown
Homozygous or compound heterozygous sequence variations in known CMT-associated genes were also not found in the exome sequencing data of the proband of family CMT2-102 (Table S1). The combined sequencing data of this patient (CMT2-102-III4) and her aunt identified four homozygous or compound heterozygous mutations distributed in three genes (Table 2). Surprisingly, none of these segregated with disease status in the family. The exome sequencing data of the proband and aunt were reanalyzed so as to filter out only variations with a frequency of > 0.01 in databases and variations not shared by both patients. Intronic variations were thus not filtered out. In addition to the aforementioned mutations, a homozygous intronic variation (c.311-23A > G) in GDAP1 that is a well-known CMT-causative gene was thus identified (Fig. 2) [20, 21]. GDAP1 encodes ganglioside-induced differentiation-associated protein 1. The variation, which has not previously been reported, segregated with disease status in the family. It is positioned in the second intron of the gene, upstream of its third exon. It was predicted by NNSPLICE 0.9 (https://www.fruitfly.org/seq_tools/splice.html) and Human Splicing Finder (HSF) version 3.1 (HSF 3.1) (https://www.umd.be/HSF/HSF.shtml) softwares to create a novel acceptor splice site. The new splice site (NNSPLICE score 0.86 out of 1; HSF score 91.99 out of 100) was predicted to be stronger than the splice site at the junction of intron 2 and exon 3 (NNSPLICE score: 0.79; HSF score: 77.16). The use of this acceptor site in lieu of the acceptor site at the border of intron 2 and exon 3 was expected to introduce 22 additional nucleotides to the mature transcript which upon translation would introduce 15 altered amino acids after p.103Asp, followed by two consecutive premature stop codons. Most of the 358 amino acids of the wild-type mutated protein would be absent.
Results of PCR experiments showed the presence of the putative mutated RNA in the blood of the patients. A fragment of expected size was amplified using patient cDNA as template and primers that would function effectively only in the presence of the 22 introduced nucleotides. Sanger sequencing confirmed presence of the 22 nucleotides in the PCR product (Fig. 3). To our initial surprise, the same product that contained the additional 22 nucleotides was also obtained in reactions that used cDNA of unrelated control individuals as template (Fig. 3). Attention to the sequence surrounding position c.311-23A in the wild-type gene suggested that the wild-type sequence may act as a non-canonical acceptor site [22]. Various bioinformatics tools did not recognize the wild-type sequence as a potential splice site, and this suggested it would at best function poorly. Consistent with this, quantification of the mutated cDNA in the blood of two control individuals and patient CMT2-102-III6 by real-time PCR showed that the level of the mutated cDNA was more than tenfold higher in the patient’s blood (Fig. 4). It was therefore concluded that the c.311-23A > G mutation in GDAP1 is the likely cause of CMT2 in family CMT2-102.

Sequence chromatograms of PCR amplicons that evidence insertion of 22 intron 2 nucleotides into GDAP1 mRNAs. Complementary DNAs used as template in the PCR reactions were synthesized using RNA from two patients of pedigree CMT2-102 with the intronic c.311-23A > G mutation and from an unrelated control individual without the mutation. The chromatograms evidence that mRNAs that include 22 nucleotides of intron 2 of GDAP1 are present in the cells of patients with the splice site mutation, and also in the cells of control individuals without the mutation

Increased ratio of GDAP1 mRNAs with the 22 nucleotide insertion from intron 2/ GDAP1 mRNAs without the insertion in CMT2 patient with the c.311-23A > G mutation as compared to control individual without the mutation. The approximately 14-fold increase shown was calculated using real-time PCR data that pertain to patient CMT2-102-III6 as compared to two different control individuals (Control 1 and Control 2). GAPDH was used as the control gene in these experiments. Thirteen-fold to 14-fold increases in the ratio were also evidenced in experiments in which β2M was used as the control gene (not shown)
Discussion
We have described the clinical features of five patients from two families affected by CMT2. Their presentations are within the spectrum of features usually attributed to axonal CMT. The genetic findings are more notable, as the causative gene in one family has not been previously reported as a CMT causative gene and the causative mutation in the other is an intronic mutation in a recognized CMT causative gene.
The homozygous c.955G > A (p.G319S) mutation in HADHA was surmised to be the cause of CMT2 in patients of family CMT2-101. Hydroxyacyl-CoA dehydrogenase-α encoded by HADHA (OMIM: 600890) is a subunit of the mitochondrial trifunctional protein (MTP). MTP is a multimeric enzyme composed of four HADHA and four hydroxyacyl-CoA dehydrogenase-β (HADHB) subunits. This inner mitochondrial membrane bound enzyme catalyzes the three final steps of long chain fatty acid β-oxidation, an important energy source for organs that require large amounts of energy including skeletal muscles [18, 23]. The biochemical profile associated with MTP complex defects reflects accumulation of toxic β-oxidation intermediates, and the associated clinical symptoms mostly involve heart and skeletal muscles. These features present in the framework of two rare related autosomal recessive disorders known as long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD; OMIM 609016) and generalized MTP deficiency (OMIM 609015) that can be early onset or late onset. Early-onset forms are more severe and have high morbidity and mortality rates. Late-onset forms are often associated with myopathy, neuropathy, and/or retinopathy [16, 24].
HADHA and HADHB mutations that cause LCHAD/MTP deficiency have been reported in several studies [19, 25,26,27]. They have also been reported in a patient with recurrent rhabdomyolysis, which is a skeletal muscle disorder [28]. The rhabdomyolysis affected patient showed signs of mild axonal peripheral neuropathy. Most interestingly, in a recent NGS (next generation sequencing) screening of 403 patients with myopathy or neuropathy using a target panel of 1500 genes associated with human diseases, 2 patients each with two mutated HADHA alleles, 1 with two mutated HADHB alleles, and 9 with one mutated HADHA or HADHB allele were identified [16]. One of the mutations in a patient (Patient 2) with compound heterozygous HADHA mutated alleles was the same c.955G > A (p.G319S) mutation found in the CMT2-101 patients in the homozygous state. To the best of our knowledge, this is the only other report of the mutation in the existing literature. The clinical descriptions of Patient 2 (18 years old) in the publication and the patients of CMT2-101 have some similarities, but are not identical. Electrophysiological studies on Patient 2 and the CMT2-101 patients revealed an axonal sensorimotor polyneuropathy, and the acylcarnitine profiles of all three patients were normal. However, it was reported that “the clinical findings [on Patient 2] suggested a metabolic myopathy”. The patient also had weakness of the masticatory muscles. There were no clinical, electrodiagnostic or serologic findings suggestive of myopathy and no evidence of cranial involvement (e.g., extraocular or masticatory muscle weakness) in the patients of family CMT2-101. It is emphasized that the patients of CMT2-101 had a pure CMT presentation. The authors of the earlier publication concluded that late-onset MTP deficiency may mimic hereditary neuropathy. The findings pertaining to CMT2-101 are consistent with this mimicry, but also show that neuropathy may in some cases be the prominent phenotypic feature of disease caused by pathogenic HADHA variants. This consideration is important as therapeutic options for MTP deficiency exist [29,30,31].
In addition to the reports in the literature described above, there are multiple reports of mutations in the related HADHB gene as cause of adult-onset axonal neuropathy or axonal CMT disease [17,18,19, 26, 32]. This also supports the contention that the homozygous c.955G > A (p.G319S) mutation in HADHA is the cause of disease in family CMT2-101. Of course, it is notable that major CMT2 causative genes, including MFN2 and GDAP1, have mitochondrial-related functions [33]. As biochemical confirmatory tests for long-chain fatty acid disorders are challenging, genetic screening of HADHA and HADHB should be considered for early-onset and late-onset CMT2-diagnosed patients.
The intronic c.311-23A > G mutation in GDAP1 that creates an acceptor splice site was considered to be the probable cause of CMT2 in family CMT2-102. Although the acceptor site that includes the mutated nucleotide is predicted by bioinformatics softwares to be better than the acceptor sequence at the junction between intron 2 and exon 3, it is possible that the latter is also used to some extent in the patients’ cells. This could result in production of sub-normal levels of functional protein in the patients. Unfortunately, the position of the mutation precluded design of primers that would exclusively recognize the wild-type product. Based on bioinformatics-based comparison of the acceptor site created by the mutation and the site at the junction of intron 2 and exon 3, it is expected that the majority of the mRNA products would be mutated. Detection of mutated mRNAs in both patients and controls suggests that at least some mutated mRNA molecules escape nonsense-mediated mRNA decay.
GDAP1 has two mature transcripts (NM_018972.4 and NM_001040875.3) that encode proteins with 358 and 290 amino acids (NP_061845.2 and NP_001035808.1). The shorter protein lacks 68 amino acids at the amino terminal of the longer protein. The splice site mutation in CMT2-102 (described with reference to the longer transcript and protein in “Results”) creates early stop codons in both transcripts. Although GDAP1 was first identified as a CMT causative gene almost two decades ago, the manner by which it affects CMT etiology remains unclear [20, 21]. The encoded protein is an integral protein of the mitochondrial outer membrane, expressed mainly in neurons and at lower levels in Schwann cells [34, 35]. Available data suggest that the GDAP1 protein has roles in various important cellular processes, including maintenance of mitochondrial morphology and function [33]. Mutations in GDAP1 have now been repeatedly reported in CMT patients, usually in those with axonal or intermediate forms of the disease [33, 36, 37]. Interestingly, both recessive and dominant inheritance patterns for the causative mutations have been observed. Although symptoms in patients with GDAP1 mutations are quite diverse, CMT presentations are generally more severe in recessively inherited forms [36]. Prior to the mutation being reported here, there has only been one description of a deleterious mutation in GDAP1 outside of amino acid coding sequences or intronic junctions [38]. This earlier report and our findings emphasize the importance of detailed examination of exome sequence data, including all available intronic sequences.
References
Mathis S, Goizet C, Tazir M, Magdelaine C, Lia AS, Magy L, Vallat JM (2015) Charcot–Marie–Tooth diseases: an update and some new proposals for the classification. J Med Genet 52(10):681–690
Morena J, Gupta A, Hoyle JC (2019) Charcot–Marie–Tooth: from molecules to therapy. Int J Mol Sci 20(14):3419
Eggermann K, Gess B, Hausler M, Weis J, Hahn A, Kurth I (2018) Hereditary neuropathies. Dtsch Arztebl Int 115(6):91–97
Gonzaga-Jauregui C, Harel T, Gambin T, Kousi M, Griffin LB, Francescatto L, Ozes B, Karaca E, Jhangiani SN, Bainbridge MN, Lawson KS, Pehlivan D, Okamoto Y, Withers M, Mancias P, Slavotinek A, Reitnauer PJ, Goksungur MT, Shy M, Crawford TO, Koenig M, Willer J, Flores BN, Pediaditrakis I, Us O, Wiszniewski W, Parman Y, Antonellis A, Muzny DM, Baylor-Hopkins Center for Mendelian G, Katsanis N, Battaloglu E, Boerwinkle E, Gibbs RA, Lupski JR (2015) Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep 12(7):1169–1183
Khani M, Taheri H, Shamshiri H, Houlden H, Efthymiou S, Alavi A, Nafissi S, Elahi E (2019) Continuum of phenotypes in hereditary motor and sensory neuropathy with proximal predominance and Charcot–Marie–Tooth patients with TFG mutation. Am J Med Genet A 179(8):1507–1515
Blair IP, Nash J, Gordon MJ, Nicholson GA (1996) Prevalence and origin of de novo duplications in Charcot–Marie–Tooth disease type 1A: first report of a de novo duplication with a maternal origin. Am J Hum Genet 58(3):472–476
Pisciotta C, Shy ME (2018) Neuropathy. Handb Clin Neurol 148:653–665
Jerath NU, Shy ME (2015) Hereditary motor and sensory neuropathies: Understanding molecular pathogenesis could lead to future treatment strategies. Biochim Biophys Acta 1852(4):667–678
Rossor AM, Polke JM, Houlden H, Reilly MM (2013) Clinical implications of genetic advances in Charcot–Marie–Tooth disease. Nat Rev Neurol 9(10):562–571
Laura M, Pipis M, Rossor AM, Reilly MM (2019) Charcot–Marie–Tooth disease and related disorders: an evolving landscape. Curr Opin Neurol 32(5):641–650
Soo Hyun N, Byung-Ok C (2019) Clinical and genetic aspects of Charcot–Marie–Tooth disease subtypes. Prec Fut Med 3(2):43–68
Barreto LC, Oliveira FS, Nunes PS, de Franca Costa IM, Garcez CA, Goes GM, Neves EL, de Souza-Siqueira-Quintans J, de Souza-Araujo AA (2016) Epidemiologic study of Charcot–Marie–Tooth disease: a systematic review. Neuroepidemiology 46(3):157–165
Szigeti K, Garcia CA, Lupski JR (2006) Charcot–Marie–Tooth disease and related hereditary polyneuropathies: molecular diagnostics determine aspects of medical management. Genet Med 8(2):86–92
Nelis E, Van Broeckhoven C, De Jonghe P, Lofgren A, Vandenberghe A, Latour P, Le Guern E, Brice A, Mostacciuolo ML, Schiavon F, Palau F, Bort S, Upadhyaya M, Rocchi M, Archidiacono N, Mandich P, Bellone E, Silander K, Savontaus ML, Navon R, Goldberg-Stern H, Estivill X, Volpini V, Friedl W, Gal A et al (1996) Estimation of the mutation frequencies in Charcot–Marie–Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet 4(1):25–33
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME (2011) Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Ann Neurol 69(1):22–33
Diebold I, Schon U, Horvath R, Schwartz O, Holinski-Feder E, Kolbel H, Abicht A (2019) HADHA and HADHB gene associated phenotypes—Identification of rare variants in a patient cohort by Next Generation Sequencing. Mol Cell Probes 44:14–20
Hong YB, Lee JH, Park JM, Choi YR, Hyun YS, Yoon BR, Yoo JH, Koo H, Jung SC, Chung KW, Choi BO (2013) A compound heterozygous mutation in HADHB gene causes an axonal Charcot–Mari–-tooth disease. BMC Med Genet 14:125
Lu Y, Wu R, Meng L, Lv H, Liu J, Zuo Y, Zhang W, Yuan Y, Wang Z (2018) HADHB mutations cause infantile-onset axonal Charcot–Marie–Tooth disease: a report of two cases. Clin Neuropathol 37(5):232–238
Boutron A, Acquaviva C, Vianey-Saban C, de Lonlay P, de Baulny HO, Guffon N, Dobbelaere D, Feillet F, Labarthe F, Lamireau D, Cano A, de Villemeur TB, Munnich A, Saudubray JM, Rabier D, Rigal O, Brivet M (2011) Comprehensive cDNA study and quantitative analysis of mutant HADHA and HADHB transcripts in a French cohort of 52 patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab 103(4):341–348
Cuesta A, Pedrola L, Sevilla T, Garcia-Planells J, Chumillas MJ, Mayordomo F, LeGuern E, Marin I, Vilchez JJ, Palau F (2002) The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot–Marie–Tooth type 4A disease. Nat Genet 30(1):22–25
Baxter RV, Ben Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, Hentati F, Ben Hamida M, Bel S, Stenger JE, Gilbert JR, Pericak-Vance MA, Vance JM (2002) Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot–Marie–Tooth disease type 4A/8q21. Nat Genet 30(1):21–22
Parada GE, Munita R, Cerda CA, Gysling K (2014) A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res 42(16):10564–10578
Uchida Y, Izai K, Orii T, Hashimoto T (1992) Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J Biol Chem 267(2):1034–1041
Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, Das A, Haase C, Hennermann JB, Karall D, de Klerk H, Knerr I, Koch HG, Plecko B, Roschinger W, Schwab KO, Scheible D, Wijburg FA, Zschocke J, Mayatepek E, Wendel U (2009) Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis 32(4):488–497
Ij L, Ruiter JP, Hoovers JM, Jakobs ME, Wanders RJ (1996) Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest 98(4):1028–1033
Spiekerkoetter U, Sun B, Khuchua Z, Bennett MJ, Strauss AW (2003) Molecular and phenotypic heterogeneity in mitochondrial trifunctional protein deficiency due to beta-subunit mutations. Hum Mutat 21(6):598–607
Brackett JC, Sims HF, Rinaldo P, Shapiro S, Powell CK, Bennett MJ, Strauss AW (1995) Two alpha subunit donor splice site mutations cause human trifunctional protein deficiency. J Clin Invest 95(5):2076–2082
Liewluck T, Mundi MS, Mauermann ML (2013) Mitochondrial trifunctional protein deficiency: a rare cause of adult-onset rhabdomyolysis. Muscle Nerve 48(6):989–991
Gillingham MB, Connor WE, Matern D, Rinaldo P, Burlingame T, Meeuws K, Harding CO (2003) Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab 79(2):114–123
Karall D, Mair G, Albrecht U, Niedermayr K, Karall T, Schobersberger W, Scholl-Burgi S (2014) Sports in LCHAD deficiency: maximal incremental and endurance exercise tests in a 13-year-old patient with long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency (LCHADD) and heptanoate treatment. JIMD Rep 17:7–12
Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, Das A, Haase C, Hennermann JB, Karall D, de Klerk H, Knerr I, Koch HG, Plecko B, Roschinger W, Schwab KO, Scheible D, Wijburg FA, Zschocke J, Mayatepek E, Wendel U (2009) Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis 32(4):498–505
Spiekerkoetter U, Bennett MJ, Ben-Zeev B, Strauss AW, Tein I (2004) Peripheral neuropathy, episodic myoglobinuria, and respiratory failure in deficiency of the mitochondrial trifunctional protein. Muscle Nerve 29(1):66–72
Rzepnikowska W, Kochanski A (2018) A role for the GDAP1 gene in the molecular pathogenesis of Charcot–Marie–Tooth disease. Acta Neurobiol Exp (Wars) 78(1):1–13
Niemann A, Ruegg M, La Padula V, Schenone A, Suter U (2005) Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot–Marie–Tooth disease. J Cell Biol 170(7):1067–1078
Pedrola L, Espert A, Wu X, Claramunt R, Shy ME, Palau F (2005) GDAP1, the protein causing Charcot–Marie–Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum Mol Genet 14(8):1087–1094
Cassereau J, Chevrollier A, Gueguen N, Desquiret V, Verny C, Nicolas G, Dubas F, Amati-Bonneau P, Reynier P, Bonneau D, Procaccio V (2011) Mitochondrial dysfunction and pathophysiology of Charcot–Marie–Tooth disease involving GDAP1 mutations. Exp Neurol 227(1):31–41
van Paassen BW, Bronk M, Verhamme C, van Ruissen F, Baas F, van Spaendonck-Zwarts KY, de Visser M (2017) Pseudodominant inheritance pattern in a family with CMT2 caused by GDAP1 mutations. J Peripher Nerv Syst 22(4):464–467
Masingue M, Perrot J, Carlier RY, Piguet-Lacroix G, Latour P, Stojkovic T (2018) WES homozygosity mapping in a recessive form of Charcot–Marie–Tooth neuropathy reveals intronic GDAP1 variant leading to a premature stop codon. Neurogenetics 19(2):67–76
Acknowledgements
We acknowledge the Iran National Science Foundation for funding the research and thank the patients and their family members for participating in the study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khani, M., Taheri, H., Shamshiri, H. et al. Deep geno- and phenotyping in two consanguineous families with CMT2 reveals HADHA as an unusual disease-causing gene and an intronic variant in GDAP1 as an unusual mutation. J Neurol 268, 640–650 (2021). https://doi.org/10.1007/s00415-020-10171-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10171-4