
Abstract
Background
Hypertrophic pachymeningitis (HP) is characterized by cranial and/or spinal thickening of the dura mater with or without associated inflammation. Neuroimaging studies reveal dura mater thickening and focal or diffuse contrast enhancement. It is described in association with trauma, infections, tumors, autoimmune/inflammatory diseases, and cerebrospinal fluid hypotension syndrome, with some cases remaining idiopathic.
Methods
A retrospective study was conducted with patients’ identification through a key terms search within MRI reports in the period of July 2008 to September 2015. Clinical files, MRI, laboratory, and pathology data were reviewed.
Results
Fifty-three patients were identified and 20 were excluded because they did not meet the inclusion criteria. Of the 33 included, 19 were female, with a mean age at symptoms onset of 51.2 ± 17.6 years. The most common presenting symptoms were headache and cranial nerves palsy, followed by seizures, delirium, lumbar pain, cognitive decline, motor deficit, and language impairment. In 17 patients, a neoplastic etiology was identified; in eight, inflammatory/autoimmune; in six, infectious; and two were classified as idiopathic. Of the eight patients with inflammatory/autoimmune etiology, four had possible IgG4-related disease (IgG4-RD) and the remaining had granulomatosis with polyangiitis, sarcoidosis, rheumatoid arthritis, and Tolosa-Hunt syndrome. Treatment was directed according to the underlying etiology.
Discussion
In the described series, a female predominance was identified, with symptoms’ onset in the 5th decade. Although headache was the most common symptom, clinical presentation was varied, emphasizing the role of MRI in HP diagnosis. The underlying etiologies were diverse, with only a few cases remaining idiopathic, also reflecting the contribution of the recently described IgG4-RD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Hypertrophic pachymeningitis (HP) is characterized by a localized or diffuse thickening of the dura mater, with or without associated inflammation [1], affecting predominantly the brain and cervical medulla. First described in 1869 by Charcot and Joffroy in its medullary form, it was only 40 years later that its brain involvement was acknowledged [2, 3].
Thickening of the dura mater may cause mechanical compression of vascular structures and nerves as well as meningeal inflammation [1, 4], ultimately leading to intracranial hypertension, cranial nerve palsy, and spinal cord dysfunction [2, 5]. Besides cranial nerve palsy (particularly optic, oculomotor, and vestibulocochlear), headache is also one of the most frequent presenting symptoms [1, 4,5,6]. Brain magnetic resonance imaging (MRI) is crucial for the diagnosis, usually disclosing thickening and contrast enhancement of the affected dura mater [5]. Clinical presentation is independent of the underlying etiology, with associated features and systemic involvement being determinant in the etiological diagnosis. Initially described in association with trauma, infections, and tumors [2], HP has been also reported in cerebrospinal fluid (CSF) hypotension syndrome [7] and several autoimmune and inflammatory diseases, with the most recently identified being IgG4-related diseases (IgG4-RD) [8,9,10,11,12,13,14,15,16,17]. Also described in plasma cells dyscrasias as POEMS syndrome [18, 19], although here, the histopathological process seems different from other etiologies and the term POEMS-associated pachymeningeal involvement was suggested [20]. Some patients, even after an exhaustive investigation, remain without a definite diagnosis, referred as idiopathic HP.
There are no European data on the prevalence of HP. The first epidemiological survey was conducted in Japan [5] and an average prevalence of 0.949/100.000 was identified. It remains a diagnostic challenge with only a few large series available and scarce data on the relative incidence of the different etiologies.
The primary aim of this study was to perform a clinical characterization of a cohort with neurological symptoms and typical HP features in MRI. The secondary aim was to determine the relative frequency of the different HP etiologies in our study population.
Material and methods
A retrospective clinical study was conducted at Centro Hospitalar do Porto, a tertiary and university hospital in the North of Portugal.
The study period was set from July 2008 until September 2015. Patients were identified through an electronic search of the terms “hypertrophic pachymeningitis” or “pachymeningeal thickening” within the MRI reports database.
Demographic, clinical, imaging, and laboratorial data were collected from the clinical files, through a structured protocol. Demographic data included the date of birth and gender, while clinical data encompassed age at neurological presentation, symptoms characterization, treatment, and outcome. MRI sequences reviewed were: T1, T2, diffusion, fluid attenuation inversion recovery, gradient echo, and T1 with gadolinium enhancement. Whenever available, further data included serum and CSF analysis as well as pathology studies. Symptoms onset was classified as acute (≤ 7 days), subacute (> 8 and < 30 days), and progressive (≥ 30 days). Patients with identified dural thickening who did not present neurological symptoms were considered incidental and were excluded from the analysis. Cases were classified according to etiology into neoplastic, inflammatory/autoimmune, infectious, and idiopathic. Statistical analysis was performed using SPSS software, 25th edition®. Chi-square test (χ2) was used for comparing categorical variables and Kruskal-Wallis H test for comparing multiple continuous variables. Post hoc analysis with pairwise comparisons was performed using Dunn’s procedure with a Bonferroni correction for multiple comparisons.
The study was approved by the Centro Hospitalar do Porto Ethical Committee. Due to its retrospective and observational nature, informed consents were not considered necessary.
Results
From the 56,301 MRI performed in this period, the search terms were identified in 63, corresponding to 53 patients, of which 20 were excluded due to the absence of neurological symptoms. In the excluded patients, pachymeningeal thickening was an incidental finding in 12, attributed to meningeal fibrosis/scarring after neurosurgery in five, and related to a previous lumbar puncture in three. Of the 33 included, 19 were female (57.6%) with a mean age at neurological presentation of 51.2 ± 17.6 years and a mean diagnostic delay of 7.6 ± 17.8 months.
Symptoms onset was acute in nine patients, subacute in 13, and progressive in 11. Twelve (36.4%) patients presented headache, nine (27.3%) cranial nerves palsy, four (12.1%) seizures, two (6.1%) delirium, two (6.1%) lumbar pain, two (6.1%) cognitive impairment, one (3%) motor deficit, and one (3%) language impairment. Headache was commonly described as a pressure, localized, continuous, with progressive worsening and refractory to medication. Cranial neuropathies were mononeuritis in seven and multineuritis in two; being the most affected cranial nerves, the optic, oculomotor, trigeminal, abducens, and facial (each involved in two patients), followed by the trochlear, vestibulocochlear, and glossopharyngeal (each affected in one patient). MRI revealed focal HP in 31 patients and diffuse in two. The remaining study was performed in accordance with the clinical suspicion, with basic serum studies (hemogram, routine chemistry with renal and liver enzymes, and ionogram) performed in all, serum immunology in 14, CSF analysis in 16, and pathology in 19. The relative frequencies of the identified etiological groups were 51.5% neoplastic (17 patients), 24.2% inflammatory/autoimmune (eight), 18.2% infectious (six), and 6.1% idiopathic (two).
The clinical, imaging, and pathological characteristics of patients from the different etiological categories are summarized in Table 1, with the inflammatory/autoimmune group exhaustively detailed in Table 2.
Headache, seizures, and cranial nerve palsy were the most common presentations in the neoplastic group (n = 17). Neuropathology studies were obtained in all patients and treatment comprehended surgery, radiotherapy, and chemotherapy. At follow-up, seven patients died and six presented minor to moderate deficits.
In the inflammatory/autoimmune group (n = 8), patients presented with cranial nerve palsy, headache, cognitive impairment, and lumbar pain with medullary symptoms (paraparesis and lower limbs paresthesia). MRI disclosed focal and diffuse HP (Fig. 1) and two patients underwent meningeal biopsy. Neuropathology studies revealed in one (Fig. 2) features suggestive of rheumatoid arthritis (due to the clinical background despite the absence of rheumatoid nodules) and in the other an inflammatory infiltrate of lymphoplasmacytic predominance without lesions of vasculitis or granulomas [a patient later diagnosed with granulomatosis with polyangiitis (GPA)]. Treatment was based on steroids associated with other immunomodulators. One patient with IgG4-RD had a relapse managed with steroid dose increase. At follow-up, patients were asymptomatic or presented minor neurological deficits, but one died due to unrelated causes (lung cancer).

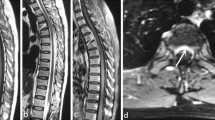
MRI of inflammatory HP patients on Table 2. a, b Spinal dural thickening with hypointensity in T2 and gadolinium enhancement (arrows, pt. 1). c Diffuse dural thickening in a patient with sarcoidosis (pt 3). d Nodular dural thickening in the left cavernous sinus, simulating a meningioma (arrow, pt. 5). e, f. Frontoparietal unilateral meningeal thickening in rheumatoid arthritis related HP (pt 4)

Histopathological findings in a case of rheumatoid arthritis related HP (pt 4—Table 2). a Dural thickening. b, c Leptomeningeal lymphohistiocytic infiltrate with d rare giant multinucleated cells
In the infectious category (n = 6), the most common presentation was headache. The infectious agents were identified through CSF culture, although lumbar puncture was not performed in one patient due to signs of increased intracranial pressure and an infectious agent was not isolated in blood cultures. Antibiotic, antiviral, or antituberculosis treatments were directed to the isolated agent. Half of the patients fully recovered and the other half died or rested with major neurological deficits. There was no difference between symptoms onset and treatment onset between these two groups.
The two patients in idiopathic group presented headache and oculomotor nerve palsy. One was treated with steroids and the other did not receive any treatment. In both, there was complete symptoms recovery.
Statistical analysis was performed between the different etiological groups (Table 3). Due to a low number of cases in the idiopathic category, these patients were grouped together with the inflammatory to allow for statistical analysis. No significant differences were found for gender [χ2(2) = 0.922, p = 0.631] or age at onset [χ2(2) = 0.436, p = 0.804] between etiologies. We found a statistically significant difference in median time to diagnosis between PH etiologies (χ2(2) = 15.155, p = 0.001). Post hoc analysis revealed statistically significant differences in median time to diagnosis between the inflammatory and infectious groups (median = 10.8 and 0.1 months, respectively; p = 0.001) and between the inflammatory and neoplastic groups (median = 10.8 and 1.4 months, respectively; p = 0.013), but not between the infectious and neoplastic groups (p = 0.313). To allow for statistical analysis, clinical outcomes were dichotomized between “asymptomatic” and “neurological sequelae/death,” notwithstanding, no significant differences were found between etiological groups [χ2(2) = 2.496, p = 0.287].
Discussion
Full comprehension of HP is still challenging, with a limited number of epidemiological studies and large series published. The broad range of underlying etiologies poses an additional effort when comparing different series since not all etiological groups are considered in most of them.
General overview of the cohort
All patients with symptoms and MRI evidence of HP were included in the analysis, which directly influenced the final results. There was an overall predominance of the female gender, with symptoms’ onset in the 5th decade. The most frequent etiological group was neoplastic, identified in 51.5% of patients. When considering the non-neoplastic group (48.5%), an inflammatory/autoimmune etiology was most frequently observed (24.2%), followed by infectious (18.2%) and idiopathic (6.1%). The latter group represented only 12.5% of the non-neoplastic subgroup, which is considerably less than the 44% [5] and 50% [22] reported in two large series. Age at symptoms’ onset age in accordance with the previous series [5, 22], while gender distribution varied with some authors also reporting a female predominance [18], but others describing a higher male frequency [5]. Not surprisingly, female predominance was most notorious in the inflammatory/autoimmune group although the average onset age was higher (49.8 ± 12.4 years) than what is commonly observed in other inflammatory/autoimmune neurological diseases. Diagnostic delay was in accordance with the onset type, with those presenting an acute onset, as the infectious group, revealing the shortest delay (only a few days), while the ones with a progressive onset, as idiopathic and inflammatory groups, revealing the longest delay (several months). The mean time to diagnosis was statistically significant between different PH etiologies.
Clinical presentation and diagnosis
Headache and cranial nerves palsies were the most frequent initial symptoms, present in 63.6% of our patients. Previous studies also reported headache as the most common initial symptom of HP (35.2 [5] and 40.9% [22]) and cranial nerve involvement as the most frequent neurological finding (62.3 [5] and 54.5% [22]). Headache may reflect meningeal inflammation and rarely intracranial hypertension, with its lateralization being directly related to the distribution of the dura mater hypertrophy [6]. In our series, only two patients with headache presented signs of increased intracranial pressure, one from the neoplastic and the other from the infectious group. Cranial nerves palsy is usually due to compression by the thickened dura mater [4], a relation that was also clear in our patients. The remaining clinical presentations (motor or sensory deficits, seizures, and dementia) usually reflects not only compression by meningeal thickening but also the presence of vascular infarctions caused by vessel compression [4, 5]. None of our patients with these other presentations had vascular infarctions related to meningeal thickening.
The broad spectrum of symptoms renders an exclusively clinical diagnosis very difficult, emphasizing the role of brain MRI. Our methodology relayed on patients’ selection through MRI, increasing its importance, but other authors also supported its value in HP diagnosis [22]. It is notoriously more sensitive than computed tomography, revealing an isointense or hypointense signal in T1 sequences and hypointense in T2 with hyperintense margins and uniform enhancement [5]. Only two (7%) patients presented diffuse HP on MRI, being one secondary to neurosarcoidosis and the other, idiopathic. Previous series reported diffuse HP in 21.4% [23], 23.4% [5], and 63.6% [22] of their cases, being more common in idiopathic [22] and GPA [23] patients, while focal HP was observed in IgG4-RD [23] and neurosarcoidosis [22] patients. Notwithstanding, the reduced number of cases per etiology restricts the generalization of these findings. Clinical presentation and brain MRI further directed subsequent investigation, commonly involving serum and CSF analysis. Histopathological studies remain crucial for patients without a definite etiology despite an extensive investigation. In our series, neuropathology was vital in the neoplastic group, in establishing a diagnosis, and allowing an adequate treatment strategy. Unfortunately, aside from this group, it was only performed in two patients from the inflammatory/autoimmune group. It could certainly be of major relevance in the two idiopathic HP, but the good clinical outcome prevented us from performing an invasive exam.
Autoimmune/inflammatory and idiopathic cases
HP association with the inflammatory/autoimmune diseases described here has been previously reported [11,12,13], including the most recent etiological entity of IgG4-RD [10, 21, 24,25,26,27]. Moreover, several authors suggest that the pathophysiology of idiopathic HP is a non-specific chronic non-chemokine inflammatory process, being also regarded as autoimmune [1, 4].
Two of our patients currently diagnosed with possible IgG4-RD had been initially classified as idiopathic, a finding also reported by other authors [10]. One of our idiopathic patients had normal serum and CSF IgG4 levels, while the other was not tested due to spontaneous resolution, being discharged from the outpatient clinic previous to the description of the disease. In IgG4-RD, IgG4 serum levels are high, occurring its deposition in several tissues inducing local fibrosis [23,24,25]. Our IgG4-RD patients (n = 4) presented cranial nerve palsies, headache, lumbar pain with medullary symptoms, and cognitive impairment. In none of them, an extra-neurological involvement was identified, although one was also diagnosed with systemic lupus erythematosus (SLE) and antiphospholipid syndrome (APS). IgG4-RD diagnosis hinges on the presence of both specific histopathology features and an increased number of IgG4 plasma cells (or IgG4/IgG ratio) in the affected tissue, in conjunction with close clinicopathologic correlation [25]. The three histopathological cardinal features are predominant lymphoplasmacytic infiltrate with IgG4-positive plasma cells, storiform fibrosis, and obliterative phlebitis [23, 28]. Due to lack of histopathological examination, only a possible diagnosis of IgG4-related disease was established in our patients, according to the currently accepted criteria [26]. Interestingly, one of our IgG4-RD cases also had a positive serum antineutrophil cytoplasmic antibodies (ANCA) without other features of ANCA-related disease, a co-occurrence previously described [21] that may represent a distinct subgroup of inflammatory HP.
Treatment and prognosis
There are no randomized clinical trials specific to HP treatment, being the existing information based on expert opinion or case series. In the inflammatory/autoimmune and idiopathic groups, steroids were the mainstay treatment, used in eight out of ten patients. The most consensual regimen described is prednisolone at a dose of 0.6–1 mg/kg/day for 2–4 weeks followed by 3–6 months of titration until the lowest maintenance dose [1, 4]. In the presence of severe neurological deficits, initial administration of methylprednisolone 1 g/day for 3 days is advised, followed by titration as described [1, 4, 29]. In our series, adjuvant therapy was used as steroid-sparing agent in half of the patients of the inflammatory/autoimmune group. Adjuvant agents are used with relative success and usually reserved for steroid unresponsive patients or as steroid-sparing agents [4, 17, 28, 29]. MRI is considered the mainstay exam to monitor treatment success [1]. Prognosis in these two groups was relatively good, with 90% of patients being asymptomatic or presenting minor neurological deficits.
When considering IgG4-RD patients (n = 4), all were treated with steroids, except one whose symptoms spontaneously subsided. The patient with SLE and APS needed mycophenolate mofetil as a steroid-sparing agent. Three were in remission and one had a relapse, managed by increasing the steroid dose. Although a good steroid response is observed most frequently; refractory cases have been reported with adjuvant treatments being needed [24].
In infectious HP, treatment was direct against the causal agent. Half the patients (n = 3) fully recovered but the other half died or rested with major neurological deficits. So far, only a few case reports of infectious etiology were published, limiting further comparisons. Prompt treatment against the causal agent seems to be the most important prognostic factor, although that was not observed with all our patients [28, 29].
The neoplastic group was managed with resective surgery, radiotherapy, and chemotherapy. Surgical treatment may also be performed in non-neoplastic HP patients refractory to medical treatment, when dura mater thickening causes mass effect or in those with symptomatic hydrocephalus in need for ventriculoperitoneal shunt [4]. Not surprisingly, in our series, this group presented the worst prognosis with 41.2% of deaths and only 24% of asymptomatic patients. Since this etiology is excluded from larger published series, a comparison was not possible.
In closing, we would like to acknowledge the limitations of the current study. By the nature of a retrospective review, we were dependent upon the accuracy and completeness of the existing medical records. Also, being a single-center study, our cohort is limited, which limited the statistical analysis between the different etiological categories. Notwithstanding, we sought to overcome these limitations by identifying consecutive cases through the brain and medullar MRI and include all patients, despite the underlying etiology.
Information on HP is largely based on case reports, with few large series published [5, 22, 23]. With this retrospective 7-year survey, we wish to further contribute into the epidemiological and clinical characterization of this entity.
References
Kupersmith MJ, Martin V, Heller G, Shah A, Mitnick HJ (2004) Idiopathic hypertrophic pachymeningitis. Neurology 62(5):686–694
Kim JH, Park Y, Chin DK (2011) Idiopathic hypertrophic spinal pachymeningitis: report of two cases and review of the literature. J Korean Neurosurg Soc 50(4):392–395. https://doi.org/10.3340/jkns.2011.50.4.392
Mott FW (1909) A case of localized syphilitic pachymeningitis cerebri. Arch Neurol Psychiatr 4:63–69
Sylaja PN, Cherian P, Das CK, Radhakrishnan VV, Radhakrishnan K (2002) Idiopathic hypertrophic cranial pachymeningitis. Neurol India 50(1):53–59
Yonekawa T, Murai H, Utsuki S, Matsushita T, Masaki K, Isobe N, Yamasaki R, Yoshida M, Kusunoki S, Sakata K, Fujii K, Kira JI (2014) A nationwide survey of hypertrophic pachymeningitis in Japan. J Neurol Neurosurg Psychiatry 85(7):732–739. https://doi.org/10.1136/jnnp-2013-306410
Wang YJ, Fuh JL, Lirng JF, Lu SR, Wang SJ (2004) Headache profile in patients with idiopathic hypertrophic cranial pachymeningitis. Headache 44(9):916–923
Honma S, Fukazama T, Hamada K, Hamada T, Tashiro K (1996) MRI changes in spontaneous intracranial hypotension. Rinsho Shinkeigaku 36(7):912–915
Joelson E, Ruthrauff B, Ali F, Lindeman N, Sharp FR (2000) Multifocal dural enhancement associated with temporal arteritis. Arch Neurol 57(1):119–122
Muthukumar N, Senthilbabu S, Usharani K (2005) Idiopathic hypertrophic cranial pachymeningitis masquerading as Tolosa-Hunt syndrome. J Clin Neurosci 12(5):589–592
Lu LX, Della-Torre E, Stone JH, Clark SW (2014) IgG4-related hypertrophic pachymeningitis: clinical features, diagnostic criteria, and treatment. JAMA Neurol 71(6):785–793. https://doi.org/10.1001/jamaneurol.2014.243
Tan HJ, Raymond A, Phadke PP, Rozman Z (2004) Rheumatoid pachymeningitis. Singap Med J 45(7):337–339
Just SA, Knudsen J, Nielsen MK, Junker P (2011) Wegener’s granulomatosis presenting with pachymeningitis: clinical and imaging remission by rituximab. ISRN Rheumatol 608942:1–4. https://doi.org/10.5402/2011/608942
Ranoux D, Devaux B, Lamy C, Mear JY, Roux FX, Mas JL (1992) Meningeal sarcoidosis, pseudo-meningioma, and pachymeningitis of the convexity. J Neurol Neurosurg Psychiatry 55(4):300–303
Li JY, Lai PH, Lam HC, Lu LY, Cheng HH, Lee JK (1999) Hypertrophic cranial pachymeningitis and lymphocytic hypophysitis in Sjögren syndrome. Neurology 52:420–423
Noel N, Drier A, Wechsler B, et al. (2014) Neurological manifestations of Behçet’s disease. Rev Med Interne 35(2):112–20. https://doi.org/10.1016/j.revmed.2013.10.332
Yu WL, Bhatia KSS, Wang K (2012) Hypertrophic Pachymeningitis as the first manifestation of systemic lupus erythematosus. Hong Kong J Radiol 15:119–122
Bosman T, Simonin C, Launay D, Caron S, Destée A, Defebvre L (2008) Idiopathic hypertrophic cranial pachymeningitis treated by oral methotrexate: a case report and review of literature. Rheumatol Int 28(7):713–718. https://doi.org/10.1007/s00296-007-0504-5
Hori T, Tsuboi Y, Okubo R, Hirooka M, Yamada T (1999) Crow-Fukase syndrome associated with Castleman disease showing hypertrophic cranial pachymeningitis and bilateral internal carotid artery occlusion. Rinsho Shinkeigaku 39(4):456–460
Watanabe M, Ushiyama O, Matsui M, Kakigi R, Kuroda Y (1993) A case of Crow-Fukase syndrome associated with chronic pachymeningitis. Rinsho Shinkeigaku 33(4):422–426
Briani C, Fedrigo M, Manara R, Castellani C, Zambello R, Citton V, Campagnolo M, Torre CD, Lucchetta M, Orvieto E, Rotilio A, Marangoni S, Magi S, Pareyson D, Florio I, Pegoraro E, Thiene G, Battistin L, Adami F, Angelini A (2012) Pachymeningeal involvement in POEMS syndrome: MRI and histopathological study. J Neurol Neurosurg Psychiatry 83(1):33–37. https://doi.org/10.1136/jnnp-2011-300047
Massey J (2017) IgG4-related hypertrophic pachymeningitis coexpressing antineutrophil cytoplasmic antibodies. Neurol Neuroimmunol Neuroinflamm 4(3):e341. https://doi.org/10.1212/NXI.0000000000000341
Wallace ZS, Carruthers MN, Khosroshahi A, Carruthers R, Shinagare S, Stemmer-Rachamimov A, Deshpande V, Stone JH (2013) IgG4-related disease and hypertrophic pachymeningitis. Medicine 92:206–216. https://doi.org/10.1097/MD.0b013e31829cce35
Hahn LD, Fulbright R, Baehring JM (2016) Hypertrophic pachymeningitis. J Neurol Sci 367:278–283. https://doi.org/10.1016/j.jns.2016.06.024
Williams T, Marta M, Giovannoni G (2015) IgG4-related disease: a rare but treatable cause of refractory intracranial hypertension. Pract Neurol 0:1–5. https://doi.org/10.1136/practneurol-2015-001275
Baptista B, Casian A, Gunawardena H, D’Cruz D, Rice CM (2017) Neurological manifestations of IgG4-related disease. Curr Treat Options Neurol 19(4):14. https://doi.org/10.1007/s11940-017-0450-9.
Deshpande V, Zen Y, Chan JK et al (2012) Consensus statement on the pathology of IgG4-related disease. Mod Pathol 25:1181–1192. https://doi.org/10.1038/modpathol.2012.72
Makino S, Tanaka Y (2013) A case of hypertrophic pachymeningitis with elevated serum IgG4. J Clin Exp Ophthalmol 4:1. https://doi.org/10.4172/2155-9570.1000260
Thurtell MJ, Keed AB, Yan M, Gottlieb T, Spies JM, Halmagyi GM (2007) Tuberculous cranial pachymeningitis. Neurology 68:298–300
Nadgir DB, Ramdas R, Kulkarni RV, Oak PJ, Shah AB (2003) Cavernous sinus syndrome due to syphilitic pachymeningitis. Neurol India 51(2):289–290
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cação, G., Calejo, M., Alves, J.E. et al. Clinical features of hypertrophic pachymeningitis in a center survey. Neurol Sci 40, 543–551 (2019). https://doi.org/10.1007/s10072-018-3689-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-018-3689-3