
Abstract
Hypertrophic pachymeningitis (HP) is a rare disorder that causes thickening of the dura mater. Inflammatory lesions may be located in the cerebral or spinal dura mater or, less frequently, in both locations simultaneously. Numerous clinico-pathological entities cause thickening of the pachymeninges. Indeed, HP is a potential manifestation of many different diseases, but the diagnosis often remains uncertain. Cases in which the pachymeningitis has no known aetiology are termed “idiopathic” HP (IHP). Recently, it has been suggested that IgG4-related disease represents a subset of cases previously diagnosed as idiopathic hypertrophic pachymeningitis. Little is known regarding the pathogenic events of IHP. In a general theory, the inflammatory infiltrate, mainly consisting of B and T lymphocytes, activates fibroblasts and induces collagen deposition, leading to tissue hypertrophy and increased dural thickness. Clinical manifestations of IHP depend upon the location of the inflammatory lesions and compression of the adjacent nervous structures. Three central pathological features are lymphoplasmacytic infiltration, obliterative phlebitis, and storiform fibrosis. MRI is the examination of choice for the preliminary diagnosis of IHP. Histopathological examination of a biopsy specimen of the dura mater would finally confirm the diagnosis. The differential diagnosis for HP is broad and includes infections, autoimmune disorders, and neoplasia. Currently, there is no consensus about treatment for patients with IHP. There is a preference for glucocorticoid treatment on diagnosis followed by the addition of other immunosuppressive agents in the event of a recurrence. Rituximab is used in patients who did not respond to glucocorticoids or to conventional steroid-sparing agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypertrophic pachymeningitis is a rare disorder that causes localized or diffused thickening of the dura mater [1]. Inflammatory lesions may be located in the cerebral or spinal dura mater or, less frequently, in both locations simultaneously [2, 3].
Charcot and Joffrey were the first to describe the medullar version of idiopathic hypertrophic pachymeningitis (IHP) in 1869 [4]. In 1949, Naffziger and Stern reported, for the first time, the cranial variant of IHPM [ 5].
Numerous clinico-pathological entities cause thickening of the pachymeninges. Indeed, HP is a potential manifestation of many different conditions, including infectious diseases, autoimmune disorders, and malignant neoplasms. The diagnosis of pachymeningitis often remains elusive. Cases in which the pachymeningitis has no known aetiology are termed “idiopathic” hypertrophic pachymeningitis [6–15].
Recently, it has been suggested that IgG4-related sclerosing disease represents a subset of cases previously diagnosed as idiopathic hypertrophic pachymeningitis [16, 17]. Immunoglobulin G4 (IgG4)-related disease is a systemic syndrome that can affect virtually any organ, is characterized by sclerosing lesions and is usually associated with a raised serum IgG4 level [18], and the pancreas, salivary glands, and lacrimal glands are typically affected. Curiously, IgG4-RD in the brain parenchyma or spinal cord has never been reported, and central nervous system involvement has only been demonstrated in the form of hypophysitis.
The histopathological features described for IHP, the frequency and the type of organ involvement beyond the pachymeninges, and the demographic characteristics of IHP have many similarities with IgG4-related disease (IgG4-RD) [18]. Indeed, several case reports and a case series have implicated IgG4-RD as a cause of IHP [16, 19–21], but the relative frequency of IgG4-RD as the cause of pachymeningitis remains uncertain.
Epidemiology
Reviews of IHP suggest that men are affected more commonly than women by this condition and that patients typically present in the sixth or seventh decades of life [9]. No data on the incidence and prevalence of IHP have been reported to date for specific racial, geographic, or ethnic groups.
Both the tendency of IgG4-RHP to affect men and the predilection for individuals who are middle-aged or older are concordant with the known epidemiology of IgG4-RD [22].
Pathogenesis
Little is known regarding the pathogenic events of IgG4-RD and, consequently, of IgG4-RHP. In a general theory, the inflammatory infiltrate, mainly consisting of B and T lymphocytes, activates fibroblasts and induces collagen deposition, leading to tissue hypertrophy and increased dural thickness (Fig. 1) [23]. Recent data support the concept that IgG4-RD is an antigen-driven disease, involving collaboration between CD4 + T cells and activated IgG4 + B cells [24].

Pathogenic model of IgG4-related hypertrophic pachymeningitis: activated T-helper and T-regulatory cells produce interleukins (ILs) that recruit eosinophils and macrophages and activate fibroblasts. Interleukin 4 and IL-10 drive class switching of autoreactive B cells to IgG4 and IgE and induce the differentiation and expansion of IgG4+ plasma cells. Heavy chains are inserted from different IgG4 molecules and separate and recombine randomly (Fab-arm exchange), thereby generating asymmetric bispecific antibodies. pMHC indicates peptide major histocompatibility complex; TGFβ, transforming growth factor β; Th2, type 2 helper T cell; and Treg, regulatory helper T cell. From: Jama Neurol 2014;71:785–793
Two distinct processes could underlie the observed pathological features in IgG4-related disease. The first is the induction of a polarized CD4 + T cell population that activates innate immune cells, including macrophages, myofibroblasts, and fibroblasts, to drive fibrosis. The second is a process that might involve the generation of IgG4-secreting plasmablasts, plasma cells, and IgG4 antibodies [25].
One plausible model of pathogenesis is that in genetically susceptible individuals, some environmental insult, possibly an encounter with a specific microbe, triggers tissue damage and a break in immunological tolerance. A self-antigen-driven, polarized CD4 + T-helper response would induce a fibrotic pathological process at one or several sites. The reasons for the targeting of particular organs remain unclear. Within these organs, increased CD4 + T cells activate innate immune cells that secrete other cytokines and drive the pathology [25]. The signal for B-cell class switching to IgG4 remains unclear, but many cytokines that stimulate IgE production also promote IgG4 production. The immunoregulatory cytokine interleukin (IL)-10 is thought to divert classic Th2-type immunity away from IL-4- and IL-13-induced IgE responses in favour of IgG4 [26]. Indeed, IL-10 has been shown to trigger a modified Th2 response by inducing the differentiation of IgG4 + B cells. In the presence of IL-4, IL-10 directs B cells to undergo antibody class switching and secrete IgG4 (Fig. 2) [27].

Anatomic pathology of IgG4-related hypertrophic pachymeningitis: a the IgG4-related disease causes hypertrophic thickening of the dura mater (and likely leptomeninges) with mass effect on neighbouring structures. Healthy dura mater consists of dense fibrous connective tissue with only scattered fibroblasts (hematoxylin-eosin, original magnification ×200). b IgG4-related hypertrophic pachymeningitis disrupts this ordered structure and leads to a characteristic pattern of storiform fibrosis (hematoxylin-eosin, original magnification ×200). From: Jama Neurol 2014;71:785–793
The memory CD4 + T cells that orchestrate the disease are presumably sustained by antigen-presenting B cells, which would explain the clinical improvement after B-cell depletion [28, 29].
Clinical features
IgG4-related disease seldom, if ever, affects the brain parenchyma, but it could be one of the most common causes of hypertrophic pachymeningitis [30].
The clinical manifestations of IHP depend upon the location of the inflammatory lesions and compression of the adjacent nervous structures.
The location of the pathological lesions in the vertebral canal is manifested by radiculopathies, limb paresis, as well as sphincter muscle functioning disturbances [9].
Neurological symptoms of the intracranial variation of IHP depend on the location of the inflammatory process, which is most frequently located at the skull base. Thickening of the dura mater in the area of the anterior fossa skull base, cavernous sinuses area, and superior orbital fissure is frequently connected with the occurrence of pain behind the eyeball, deteriorated vision, and disturbances affecting the mobility of the eyeballs. These symptoms are often manifested in the form of Tolosa–Hunt syndrome [31, 32]. Local dural involvement of the periorbital areas, vestibular structures, and brainstem typically cause focal signs, such as visual or hearing impairment, cranial motor nerve palsies, and sensory alterations. When the inflammatory lesions are located in the cerebellar tentorium and at the posterior cranial fossa base, mainly in the clivus and foramen magnum area, lesions in cranial nerves VI to XII are observed along with cerebellar ataxia features [2, 6, 33–35].
In contrast, more diffuse symptoms, such as headache, neck stiffness, and seizures, occur if the meningeal inflammation spreads along the hemispheric and basal dura or the tentorium cerebelli.
Patients with IgG4-RHP may also present with signs of systemic disease, namely constitutional features, such as weight loss and malaise, thyroid dysfunction (e.g. from Riedel’s thyroiditis), abdominal pain caused by autoimmune pancreatitis, flank or back pain associated with retroperitoneal fibrosis or periaortitis, facial or neck swelling due to lacrimal or salivary gland enlargement, proptosis caused by orbital disease, and pulmonary manifestations, such as interstitial pneumonia or tracheobronchial stenosis. Constellations of several of these other forms of organ involvement strongly suggest IgG4-RD and offer the possibility of confirming the diagnosis underlying IHP without necessarily requiring a meningeal biopsy [23].
Histopathology
Histopathology is the key to the diagnosis of IgG4-RHP. Three central pathological features are lymphoplasmacytic infiltration, obliterative phlebitis, and storiform fibrosis (Fig. 2) [36].
The lymphocytes and plasma cells are polyclonal. Hematoxylin-eosin staining displays dense basophilic lymphocytic infiltrates comprising both T and B cells. T lymphocytes, primarily CD4 + cells, are identified prominently within IgG4-RD tissues and are arrayed diffusely throughout the tissues. In contrast, B cells tend to be located within lymphoid aggregates or even in frank germinal centres. Macrophages, and even histiocytes, are often observed. Eosinophils are also commonly present, and extreme examples can resemble eosinophilic organopathy, but neutrophilic infiltration is rare [36, 37].
Fibrosis is a histological prerequisite for the diagnosis. Some fibrosis is present in all cases, even in patients who present shortly after symptom onset. Storiform fibrosis, characterized by radially arranged collagen fibres that seem to weave through the tissue, typifies the unique pattern associated with IgG4-related disease (Fig. 1) [36, 37]. Fibrosis commonly predominates over a long disease course, and the histological features can become less specific in patients with long-standing disease. A review of biopsy samples taken earlier in the course, however, could document the progression of IgG4-related disease from a lymphoplasmacytic infiltrate to one characterized mainly by fibrosis.
The characteristic venous lesions, obliterative phlebitis is defined as the partial or complete obliteration of medium-sized veins [36, 37]. Obliterated veins sometimes can be identified as veins only through elastin staining (Fig. 1).
Diagnosis
Tissue biopsy is the gold standard for diagnosis in most settings. An international, multidisciplinary group of IgG4-RD experts described a consensus strategy for the pathological diagnosis of IgG4-RD, including IgG4-RHP [36]. The pathological criterion for the diagnosis of IgG4-RD consists of three characteristic histological features, including a dense lymphoplasmacytic infiltrate, fibrosis with storiform features, and obliterative phlebitis.
The consensus maintains that two of these three histologic features should be fulfilled for a diagnosis of IgG4-RD. When only one of these criteria is met, additional information, such as an increased serum IgG4 concentration, elevated IgG4 to IgG ratio within tissue, or multiorgan involvement with typical clinical manifestations of IgG4-RD in different organs, is required to support the diagnosis. A common feature of IgG4-RD is an increased IgG4 to IgG plasma cell ratio upon immunohistochemical evaluation. Previous studies have suggested that an IgG4 to IgG ratio greater than 40 % is indicative of IgG4-RD [20].
Radiology
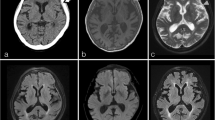
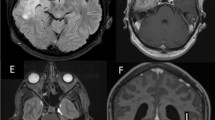
Imaging is an important part of the diagnostic approach in many organs. In CT scans and MRI studies, IgG4-RHP may appear either as a linear dural thickening or as a bulging mass (Fig. 3).

Radiological features of IgG4-related hypertrophic pachymeningitis. T1-weighted magnetic resonance (MR) imaging, showing hyperintense linear dural thickening (arrowheads) overlying the supratentorial hemispheres (a and b) and the tentorium cerebelli (c) as well as nodular pachymeningeal enhancement along the clivus (d and e). Dural thickening appears hypointense on the T2-weighted MR image (f, asterisks). Pachymeningitis involving the cranial nerves’ canals (arrowheads) shown by gadolinium-enhanced T1-weighted MR imaging (g) and positron emission tomography (h). Dural thickening is seen throughout the cervical spine with focal nodularity at the C2-3 and C6-7 levels (arrowheads) (i). From: Jama Neurol 2014;71:785–793
Nuclear imaging, in the form of positron emission tomography scans with fludeoxyglucose F 18, has potential for use in assessing the degree of active inflammation within the meninges and in identifying extracranial meningeal involvement, as well as disease, in other organs [38]. For the evaluation of intracranial meningeal lesions, carbon 11-labelled methionine is preferred because of its low uptake in the normal brain [39].
PET can also be helpful in monitoring disease activity after treatment [40].
Serology
Elevated serum IgG4 levels are consistent with IgG4-RD and are present in 70 to 90 % of patients with this condition. The dramatic serum IgG4 elevations observed in some patients with this condition correlate with multiorgan involvement. The serum IgG4 concentrations in the periphery may remain normal even as they increase in the central nervous system, particularly if the meninges are the only site of disease. The presence of an elevated serum IgG4 concentration in patients with IgG4-RHP generally implicates extrameningeal disease.
High serum IgG4 concentrations are neither sufficiently sensitive nor specific for diagnosis. Serum IgG4 concentrations are useful for screening, but are unreliable as a single diagnostic marker. Increased ratios of IgG4 to total IgG (>10 %) or IgG1 (>24 %) increase the diagnostic specificity, especially when IgG4 concentrations are only slightly raised [41].
Cerebrospinal fluid
Lumbar puncture provides essential information, the primary value of which is the exclusion of central nervous system infections and malignancies. Compared with healthy control individuals and patients with other forms of pachymeningitis, patients with IgG4-RHP demonstrated higher cerebro spinal fluid IgG4 concentrations.
Differential diagnosis
The differential diagnosis for HP is broad. The condition has been described in relation to specific infections, including Lyme disease, syphilis, Mycobacterium tuberculosis, fungal infections, cysticercosis, HTLV-1, and malignant external necrotizing otitis due to Pseudomonas. Similarly, it has been described in relation to autoimmune disorders, such as Wegener’s granulomatosis, rheumatoid arthritis, sarcoidosis, Behçet’s disease, Sjögren’s syndrome, and temporal arteritis. Finally, it can be related to neoplasia, such as carcinomatosis, lymphoma, meningioma en plaque [1, 42], and melanoma; to storage diseases after prolonged dialyses; and prolonged administration of medicines directly to the cerebrospinal fluid [6].
In that context, the diagnosis of IHP consists of excluding those disease entities whose treatment is of the causal type, which is of particular importance in the case of infectious and neoplastic diseases [1, 42, 43].
MRI is the examination of choice in the preliminary diagnostics of IHP. Histopathological examination of a biopsy specimen of the dura mater would confirm the diagnosis [2].
Prognosis
Mikawa et al. [44, 45] subdivided IHP into two groups: first those with inflammatory signs, including fever, increased erythrocyte sedimentation rate, leukocytosis, and increased (group P), and second, those without inflammatory signs (group N). They analysed and suggested that group P had a worse prognosis than group N.
In the literature, the clinical observation periods of the disease course are often several years long. Complete recoveries of IHP are rather rare. The disease is most often progressive with remissions and requires prolonged treatment. Death of patients with IHP is most often caused by obturation of the large sinuses of the dura by inflammatory infiltrations, as well as compression of the latter upon the structures of the hypothalamus and brainstem [2, 6, 33].
Treatment of IgG4-RHP
Glucocorticoids
Currently, there is no consensus regarding the treatment for patients with IgG4-RHP. A review of the published reports reveals a preference for glucocorticoid treatment upon diagnosis, followed by the addition of other immunosuppressive agents in the event of a recurrence [30]. A consensus guideline from Japan recommended treating IgG4-related autoimmune pancreatitis with prednisolone (0.6 mg/kg/d) for 4 weeks, followed by a 3- to 6-month taper to a maintenance dose of 2.5 to 5.0 mg/d for up to 3 years [46, 47]. The degree to which these recommendations can be extrapolated to IgG4-RHP is not clear, but the potential implications of severe neurologic dysfunction caused by IgG4-RHP justify beginning treatment in some patients with pulse methylprednisolone (e.g. 1 g of methylprednisolone a day for 3 days), followed by a glucocorticoid taper longer than those used in autoimmune pancreatitis [38]. The doses and duration of glucocorticoid therapy are highly empirical and not specified in detail in most published reports.
Conventional steroid-sparing agents
Steroid-sparing agents have been used following disease flares, including methotrexate (20 mg/wk), azathioprine (100–200 mg/d), mycophenolate mofetil (1000 mg twice daily), and cyclophosphamide (either oral [100 mg/d] or intravenous [1 g/m2/mo]). The efficacy of these treatments has never been confirmed to be independent of the effects of glucocorticoids, which are always used concomitantly. To our knowledge, no randomized clinical trials of conventional steroid-sparing agents have been performed [23].
B-cell depletion
The most promising steroid-sparing agent is rituximab, an anti-CD20 + B-cell-depleting agent that leads to both substantial reductions of serum IgG4 concentrations and clinical improvement [29].
Rituximab was initially used in patients who did not respond to glucocorticoids, conventional steroid-sparing agents, or both, under the assumption that B-cell depletion might ameliorate the condition putatively mediated by high serum concentrations of IgG4 [28, 48, 49]. The fundamental assumption underlying this approach now seems incorrect or at least not entirely true, but careful mechanistic studies of patients with IgG4-related disease treated with rituximab have led to several important observations and novel insights about the pathophysiology of this disorder. First, B-cell depletion targets the subset of plasma cells that produce IgG4 in IgG4-related disease [28, 29]. They seem to achieve this action by depleting all circulating CD20-positive cells (i.e. B cells), which interferes in turn with the repletion of the short-lived plasma cells making IgG4. In other words, the plasma cells generating IgG4 in IgG4-related disease are mainly of the short-lived type that naturally undergo apoptosis within weeks. Once these cells disappear as programmed, they cannot be repleted after rituximab administration because their precursors, CD20-positive B cells, are not available.
Second, IgG4-positive plasmablasts (positive for IgG4) seem to be a good biomarker for IgG4-related disease and are probably superior to serum IgG4 concentrations for the diagnosis and monitoring of disease activity [24, 50].
However, experience with B-cell depletion strategies in IgG4-RHP remains limited and has alternate results [30]. Indeed, as for other immunosuppressive agents, the overall efficacy of rituximab may depend on the stage of the fibrosis. In fact, dense, well established, end-stage, fibrosis is likely to be unresponsive to immunomodulation.
Conclusions
IHP is a rare, chronic inflammatory process that probably has an immunological aetiology. The most frequently encountered neurological manifestations include headaches and cranial and spinal nerves paresis. The diagnosis of IHP is based upon MRI examination and the histopathological assessment of the dura mater biopsy specimen. The treatment of choice for this increasingly often diagnosed disease is the administration of immunosuppressive substances, with steroids as the main choice.
Future treatment studies are likely to focus on B-cell depletion and the mechanistic impact of this intervention on both the clinical and immunological features of IgG4-RD. In that respect, IHP has become an interdisciplinary problem because its diagnosis and treatment requires not only radiologists, neurologists, and neurosurgeons but also specialists in otorhinolaryngology, ophthalmology, and internal medicine, including immunologists, allergologists, and rheumatologists.
Take home messages
-
Hypertrophic pachymeningitis is a rare disorder that causes thickening of the cerebral or spinal dura mater. Hypertrophic pachymeningitis is a potential manifestation of many different conditions, such as infectious diseases, autoimmune disorders, and malignant neoplasms, but the diagnosis in pachymeningitis often remains elusive. These cases are termed “idiopathic” hypertrophic pachymeningitis.
-
Recently, it has been suggested that IgG4-related disease represents a subset of cases previously diagnosed as idiopathic hypertrophic pachymeningitis. Little is known regarding the pathogenic events. It is probable that the inflammatory infiltrate, mainly consisting of B and T lymphocytes, activates fibroblasts and induces collagen deposition, leading to tissue hypertrophy and increased dural thickness. The clinical manifestations of idiopathic hypertrophic pachymeningitis depend upon the location of the inflammatory lesions and compression of the adjacent nervous structures.
-
Three central pathological features are lymphoplasmacytic infiltration, obliterative phlebitis, and storiform fibrosis. MRI is the examination of choice for the preliminary diagnosis of idiopathic hypertrophic pachymeningitis. Histopathological examination of a biopsy specimen of the dura mater would finally confirm the diagnosis. The disease is most often progressive with remissions and requires prolonged treatment.
-
There is a preference for glucocorticoid treatment upon diagnosis followed by the addition of other immunosuppressive agents in the event of a recurrence. Rituximab is used in patients who did not respond to glucocorticoids or to conventional steroid-sparing agents.
References
D’Andrea G, Trillò G, Celli P, Roperto R, Crispo F, Ferrante L. Idiopathic intracranial hypertrophic pachymeningitis: two case reports and review of the literature. Neurosurg Rev. 2004;27:199–204.
Charcot JM, Joffroy A. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere. Arch Physiol Norm Pathol. 1869;2:354–67.
de Deus-Silva L, Queiroz L, Zanardi V, Ghizoni E, Pereira H, Malveira GL, et al. Hypertrophic pachymeningitis: case report. Arq Neuropsiquiatr. 2003;61:107–11.
Hamada J, Yoshinaga Y, Korogi Y, Ushio Y. Idiopathic hypertrophic cranial pachymeningitis associated with a dural arteriovenous fistula involving the straight sinus: case report. Neurosurgery. 2000;47:1230–3.
Hatano N, Behari S, Nagatani T, Kimura M, Ooka K, Saito K, et al. Idiopathic hypertrophic cranial pachymeningitis: clinicoradiological spectrum and therapeutic options. Neurosurgery. 1999;45:1336–42.
Dumont AS, Clark AW, Sevick RJ, Myles ST. Idiopathic hypertrophic pachymeningitis: a report of two patients and review of the literature. Can J Neurol Sci. 2000;27:333–40.
Fan Y, Liao S, Yu J, Ling L, Hou Q, Xing S, et al. Idiopathic hypertrophic cranial pachymeningitis manifested by transient ischemic attack. Med Sci Monit. 2009;15:178–81.
Ito Z, Osawa Y, Matsuyama Y, Aoki T, Harada A, Ishiguro N. Recurrence of hypertrophic spinal pachymeningitis. Report of two cases and review of the literature. J Neurosurg Spine. 2006;4:509–13.
Kupersmith MJ, Martin V, Heller G, Shah A, Mitnick HJ. Idiopathic hypertrophic pachymeningitis. Neurology. 2004;62:686–94.
Lampropoulos CE, Zain M, Jan W, Nader-Sepahi A, Sabin IH, D’Cruz DP. Hypertrophic pachymeningitis and undifferentiated connective tissue disease: a case report and review of the literature. Clin Rheumatol. 2006;25:399–401.
Loy ST, Tan CW. Granulomatous meningitis. Singap Med J. 2009;50:371–3.
Pai S, Welsh CT, Patel S, Rumboldt Z. Idiopathic hypertrophic spinal pachymeningitis: report of two cases with typical MR imaging findings. AJNR Am J Neuroradiol. 2007;28:590–2.
Rojana-udomsart A, Pulkes T, Viranuwatti K, Laothamatas J, Phudhichareonrat S, Witoonpanich R. Idiopathic hypertrophic cranial pachymeningitis. J Clin Neurosci. 2008;15:465–9.
Sridhar K, Vasudevan MC. Idiopathic chronic hypertrophic pachymeningitis causing thoracic cord compression. Br J Neurosurg. 2004;18:515–7.
Takahashi H, Wada A, Yokoyama Y, Ishii M, Shibuya K, Suguro T. Idiopathic hypertrophic spinal pachymeningitis: a case report. J Orthop Surg (Hong Kong). 2010;18:113–7.
Chan SK, Cheuk W, Chan KT, Chan JK. IgG4-related sclerosing pachymeningitis: a previously unrecognized form of central nervous system involvement in IgG4-related sclerosing disease. Am J Surg Pathol. 2009;33:1249–52.
Shapiro KA, Bove RM, Volpicelli ER, Mallery RM, Stone JH. Relapsing course of immunoglobulin G4-related pachymeningitis. Neurology. 2012;79:604–6.
Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–51.
Kosakai A, Ito D, Yamada S, Ideta S, Ota Y, Suzuki N. A case of definite IgG4-related pachymeningitis. Neurology. 2010;75:1390–2.
Lindstrom KM, Cousar JB, Lopes MB. IgG4-related meningeal disease: clinico-pathological features and proposal for diagnostic criteria. Acta Neuropathol. 2010;120:765–76.
Riku S, Hashizume Y, Yoshida M, Riku Y. Is hypertrophic pachymeningitis a dural lesion of IgG4-related systemic disease? Rinsho Shinkeigaku. 2009;49:594–6.
Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathol. 2014;9:315–47.
Lu LX, Della-Torre E, Stone JH, Clark SW. IgG4-related hypertrophic pachymeningitis: clinical features, diagnostic criteria, and treatment. JAMA Neurol. 2014;71:785–93.
Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134:679–87.
Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385:1460–71.
Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY. IgE versus IgG4 production can be differentially regulated by IL-10. J Immunol. 1998;160:3555–61.
Satoguina JS, Weyand E, Larbi J, Hoerauf A. T regulatory-1 cells induce IgG4 production by B cells: role of IL-10. J Immunol. 2005;174:4718–26.
Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755–62.
Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91:57–66.
Wallace ZS, Carruthers MN, Khosroshahi A, Carruthers R, Shinagare S, Stemmer-Rachamimov A, et al. IgG4-related disease and hypertrophic pachymeningitis. Medicine (Baltimore). 2013;92:206–16.
Lee YC, Chueng YC, Hsu SW, Lui CC. Idiopathic hypertrophic cranial pachymeningitis: case report with 7 years of imaging follow-up. AJNR Am J Neuroradiol. 2003;24:119–23.
Matsumoto K, Natori Y, Hirokawa E. Hypertrophic pachymeningitis as a result of a retropharyngeal inflammatory pseudotumor: case report. Neurosurgery. 2002;51:1061–4.
Choi IS, Park SC, Jung YK, Lee SS. Combined therapy of corticosteroid and azathioprine in hypertrophic cranial pachymeningitis. Eur Neurol. 2000;44:193–8.
Naffziger HC, Stern WE. Chronic pachymeningitis: report of a case and review of the literature. Arch Neurol Psychiatry. 1949;62:383–411.
Nakazaki H, Tanaka T, Isoshima A, Hida T, Nakajima M, Abe T. Idiopathic hypertrophic cranial pachymeningitis with perifocal brain edema—case report. Neurol Med Chir (Tokyo). 2000;40:239–43.
Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–92.
Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812–9.
Della Torre E, Bozzolo EP, Passerini G, Doglioni C, Sabbadini MG. IgG4-related pachymeningitis: evidence of intrathecal IgG4 on cerebrospinal fluid analysis. Ann Intern Med. 2012;156:401–3.
Norikane T, Yamamoto Y, Okada M, Maeda Y, Aga F, Kawai N, et al. Hypertrophic cranial pachymeningitis with IgG4-positive plasma cells detected by C-11 methionine PET. Clin Nucl Med. 2012;37:108–9.
Ebbo M, Grados A, Guedj E, Gibert D, Colavolpe C, Zaidan M, et al. Usefulness of 2-[18F]-fluoro-2-deoxy-d-glucose-positron emission tomography/computed tomography for staging and evaluation of treatment response in IgG4-related disease: a retrospective multicenter study. Arthritis Care Res (Hoboken). 2014;66:86–96.
Boonstra K, Culver EL, de Buy Wenniger LM, van Heerde MJ, van Erpecum KJ, Poen AC, et al. Serum IgG4 and IgG1 for distinguishing IgG4-associated cholangitis from primary sclerosing cholangitis. Hepatology. 2014;59:1954–63.
Mamelak AN, Kelly WM, Davis RL, Rosenblum ML. Idiopathic hypertrophic cranial pachymeningitis: report of three cases. J Neurosurg. 1993;79:270–6.
Phanthumchinda K, Sinsawaiwong S, Hemachudha T, Yodnophaklao P. Idiopathic hypertrophic cranial pachymeningitis: an unusual cause of subacute and chronic headache. Headache. 1997;37:249–52.
Mikawa Y, Watanabe R, Hino Y, Hirano K. Hypertrophic spinal pachymeningitis. Spine. 1994;19:620–5.
Park SH, Whang CJ, Sohn M, Oh YC, Lee CH, Whang YJ. Idiopathic hypertrophic spinal pachymeningitis: a case report. J Korean Med Sci. 2001;16:683–8.
Yamakita N, Hanamoto T, Muraoka N, Ikeda T, Hirata T, Yasuda K, et al. Hypopituitarism and diabetes insipidus with localized hypertrophic pachymeningitis (Tolosa-Hunt syndrome) associated with Hashimoto thyroiditis. Am J Med Sci. 2004;327:38–43.
Kamisawa T, Okazaki K, Kawa S, Shimosegawa T, Tanaka M, Research Committee for Intractable Pancreatic Disease and Japan Pancreas Society. Japanese consensus guidelines for management of autoimmune pancreatitis, III: treatment and prognosis of AIP. J Gastroenterol. 2010;45:471–7.
Inoue D, Zen Y, Sato Y, Abo H, Demachi H, Uchiyama A, et al. IgG4-related perineural disease. Int J Rheumatol. 2012;2012:401890.
Carruthers MN, Khosroshahi A, Topazian M, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74:1171–7.
Wallace ZS, Mattoo H, Carruthers MN, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2014;74:190–5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
De Virgilio, A., de Vincentiis, M., Inghilleri, M. et al. Idiopathic hypertrophic pachymeningitis: an autoimmune IgG4-related disease. Immunol Res 65, 386–394 (2017). https://doi.org/10.1007/s12026-016-8863-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-016-8863-1