
Abstract
Introduction/objectives
Systemic lupus erythematosus (SLE) was an autoimmune disease with a large variety of clinical manifestations and involving many organs. Its exact etiology was unclear, and studies had shown that T cells may play an important role. In this study, we wished to study the regulatory mechanism of circRNA in the T cells from SLE patients.
Method
GSE84655 was retrieved from the GEO database, and the corresponding probe name was converted into an international standard circRNA name by using the practical extraction and report language. The differentially expressed circRNAs (DECs) were analyzed by using R software. Subsequently, we used multiple bioinformatics methods to obtain the target miRNAs of circRNAs and the downstream mRNAs of miRNAs. Finally, a circRNA–miRNA–mRNA regulatory network was constructed and visualized by using Cytoscape 3.6.1 software.
Results
There were a total of 29 DECs that had been identified, including 2 upregulated circRNAs and 27 downregulated circRNAs. After a lot of in-depth analysis, we finally obtained a circRNA–miRNA–mRNA regulatory network consisting of 8 DECs (hsa_circ_0006770, hsa_circ_0002904, hsa_circ_0034044, hsa_circ_0023685, hsa_circ_0049271, hsa_circ_0074491, hsa_circ_0074559, and hsa_circ_0023461), 4 overlap miRNAs (hsa-miR-326, hsa-miR-569, hsa-miR-638, and hsa-miR-1246), and 13 target mRNAs (EPHB3, USH1G,UBE4A, DCAF7, TBL1XR1, SLC27A4, SMO, NAA30, RSBN1, PLAG1, SOX2, GPATCH11, and DYRK1A).
Conclusions
This study could provide a novel insight into the role of circRNA and the circRNA–miRNA–mRNA regulation network in the SLE. However, it also needed to be verified by subsequent experiments and clinical studies.
Key Points • There were 29 DECs (2 up and 27 down) between T cells of SLE and health control. • Hsa-miR-338-3p, hsa-miR-767-3p, and hsa-miR-1827 were the most frequent miRNAs. • We obtained a circRNA–miRNA–mRNA regulatory network for SLE. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is a chronic, multisystemic, complex autoimmune disease, and it can affect multiple tissues and organs and displays with varying clinical manifestations [1]. Similar to other autoimmune diseases, SLE is also more prevalent in the female groups than male groups, with a ratio exceeding 9: 1 [2]. Although the exact etiology of SLE had not been clearly elucidated, there were numerous studies that had shown that genetic, environmental, endocrine, and other factors may play very important roles in the occurrence and development of SLE [3]. T cells can participate in the development of SLE not only by affecting other immune cells through direct contact, but also by secreting pro-inflammatory cytokines and directly acting on the targeted tissues [4].
Circular RNA (circRNA) is a special non-coding RNA which does not have 5′ end caps and 3′ end poly (A) tails and forming a circular structure with covalent bonds [5]. The circRNA was first discovered by Sanger et al. in the higher plant virus RNAs in 1976 [6]. In the following decades, only a few circRNAs were found occasionally; however, with the development of high-throughput sequencing technology and bioinformatics technology, scholars had discovered a large number of circRNAs, which were widely distributed in eukaryotic cells, and thousands of circRNAs had been found in human cells [7]. CircRNA is rich in microRNA (miRNA) binding sites, which can act as a miRNA sponge by binding to miRNA; then, it can prevent the binding between miRNAs and its target mRNAs and indirectly regulating the downstream expression of miRNA target genes [8]. This indicated that circRNA may affect and regulate the human diseases by regulating the disease-related miRNAs [7]. Meanwhile, some latest studies had shown that circRNA was related to many diseases, such as neurological disorders [9], coronary artery disease [10], cancers [11], and SLE [3] etc.
In this study, we aimed to study the regulatory mechanism of circRNA in the SLE T cells, especially the circRNA–miRNA–mRNA regulatory network. We identified differentially expressed circRNAs (DECs) in the T cells between patients with SLE and healthy controls by using the limma package of the R language. Subsequently, we used bioinformatics prediction methods to find the target miRNAs and mRNAs. Our results would provide novel information on the role of circRNA in SLE and provide a theoretical basis for the mechanism of circRNA, miRNA, and mRNA interactions in SLE.
Materials and methods
Microarray data
Gene expression profile dataset GSE84655 was retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) [12]. The annotation platform was GPL21825: 074301 Arraystar Human CircRNA microarray V2. The circRNA expression profiles included 6 SLE T cells and 3 healthy controls T cells.
Differentially expression analysis
The series of matrix file(s) and platform file(s) of GSE84655 was downloaded, and then we converted the corresponding probe name into an international standard circRNA name by using the practical extraction and report language (Perl). The DECs were analyzed by using the Bioconductor limma package of R software. The criterion for DECs were adjusted P value < 0.05 and |log2fold-change (FC) | ≥ 1.
Prediction of circRNAs target miRNAs
Studies had shown that there were multiple miRNAs binding sites in the circRNAs, and the circRNAs can regulate gene expression through sponge interaction with the target miRNAs. In this study, we used the Circular RNA Interactome (https://circinteractome.nia.nih.gov/) [13] to predict the circRNAs target miRNAs.
SLE-related miRNAs
Human microRNA Disease Database (HMDD), which had collected about 1206 miRNA genes and 893 diseases from 19,280 papers, was a collection of experimentally confirmed diseases related to miRNAs [14]. In this study, we utilized the HMDD database to search for the SLE-related miRNAs. Subsequently, we used the Venny 2.1.0 online database (http://bioinfogp.cnb.csic.es/tools/venny/index.html) to analyze the intersection of DECs target miRNAs and SLE-related miRNAs. If the above analyzed DECs were interacting with these SLE-related miRNAs, they would be more related to SLE.
Prediction of miRNA target genes
The target genes of the overlap DECs target miRNAs and SLE-related miRNAs were predicted based on 3 different miRNAs target gene databases, including miRTarBase [15], TargetScan [16], and miRDB databases [17]. Only those genes which were confirmed by all the 3 databases would be considered as the true candidate target mRNAs.
Construction of the circRNA–miRNA–mRNA regulatory network
The DECs, the overlap miRNAs, and the target mRNAs of overlap miRNAs were used to construct a regulatory network and visualized by using Cytoscape 3.6.1 software [18].
Results
The differentially expressed circRNAs between SLE patients and healthy controls
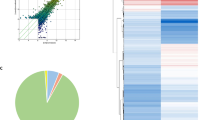
A flow chart displaying the screening process is shown in Fig. 1. In this study, GSE84655 data was analyzed by the R software, and the criterion for DECs was adjusted P value < 0.05 and |log2fold-change (FC) | ≥ 1; there were a total of 29 DECs identified (Fig. 2). Among these DECs, 2 circRNAs were upregulated and 27 circRNAs were downregulated (Table 1).

A flow chart displaying the screening process in this study

Heat map of DECs between the SLEs and Health controls. Colors from green to red mean increasing expression of DECs between the SLEs and health controls. DECs, differentially expressed circRNAs
The circRNA–microRNA interactions in SLE
There are many miRNA binding sites on the circRNA; thus, it can act as a miRNA sponge. An important biological function of circRNA is that it can prevent the interaction between miRNA and its target mRNA, and then regulating the miRNA downstream target genes. Therefore, we use the Circular RNA Interactome database to predict the target miRNA molecules related to DECs. The results showed that DECs can regulate a large number of target miRNAs (Supplement Table 1). By assessing the frequency of miRNAs that targeted these DECs, we found that the 3 most frequent miRNAs were hsa-miR-338-3p that interacted with 9 DECs, hsa-miR-767-3p that interacted with 9 DECs, and hsa-miR-1827 that interacted with 8 DECs (Table 2).
Overlap of DECs predict target miRNAs and HMDD disease-related miRNAs
The Human microRNA Disease Database (HMDD) contained various disease-related miRNAs, and the relationship between these miRNA molecules and diseases had been experimentally confirmed. And we obtained 87 SLE-related miRNAs from the HMDD database (Supplement Table 2). If the DECs had an interaction with these SLE-related miRNAs, the DECs may more likely have a relationship with SLE. So we used the Venn diagram to get the intersection between the DECs predict target miRNA and SLE-related miRNAs, as shown in Fig. 3; there were 18 overlap miRNAs between these two kinds of miRNAs. And these miRNAs were associated with 21 DECs (Fig. 4), and the specific details can be found in Table 3.

Venn diagram analysis of DEC predict target miRNAs and SLE-related miRNAs. The blue circle represents the DEC predict target miRNAs, and the yellow circle represents the SLE-related miRNAs. The intersection of the two circles represents the overlapping miRNAs between the two kinds of miRNAs, and the specific miRNA names are listed in the right box. DECs, differentially expressed circRNAs

The overlap miRNAs and DECs. The blue nodes represent the overlap miRNAs and the green and pink nodes represent the up- and downregulated circRNAs, respectively
Construction of the circRNA–miRNA–mRNA regulatory network
There were only 4 overlap miRNAs (hsa-miR-326, hsa-miR-569 hsa-miR-638, and hsa-miR-1246) that had the same target mRNAs in the 3 different miRNA target databases (Table 4). Finally, we obtained a circRNA–miRNA–mRNA regulatory network consisting of 8 DECs, 4 overlap miRNAs, and 13 target mRNAs (Fig. 5).

The circRNA–miRNA–mRNA regulatory network. The pink nodes represent the circRNAs, the blue nodes represent the overlap miRNAs, and the gray nodes represent the mRNAs
Discussion
SLE was an autoimmune disease with very complex pathogenesis mechanisms and clinical manifestations [19]. The exact cause of SLE was not yet clear, but it had been proved that the immune system dysregulation was involved in [7]. For its circular structure, circRNA can avoid the influence of RNA exonuclease and it was more stable than the linear RNA [10]. More and more scholars had started to focus on the relationship between circRNA and disease pathogenesis. Researchers found that circRNA was linked to many diseases, such as neurological disorders [9], coronary artery disease [10], cancers [11], etc. In addition, circRNA had also been found to be linked to a variety of autoimmune diseases, including multiple sclerosis [20], rheumatoid arthritis [21], and SLE [3]. Some researchers had also performed microarray analysis of circRNA in the plasma or peripheral blood mononuclear cell (PBMC) of SLE patients and found that there were a large number of differentially expressed circRNA molecules in the plasma or PBMC of SLE patients, and these molecules might play an important role in the early diagnosis and development of SLE [7, 8, 19, 22, 23]. Li et al. also briefly analyzed the GSE84655 data, but they mainly discussed the function of hsa_circ_0045272 in SLE and did not perform an in-depth analysis and interpretation of the circRNA–miRNA–mRNA network [3]. In this study, a variety of bioinformatics methods were used to compare the expression of circRNA in T cells of SLE and health control populations and to construct a circRNA–miRNA–mRNA regulatory network that may be related to SLE.
CircRNA contained miRNA binding sites and could act as a competitive endogenous RNA (ceRNA), which bind to the miRNA and acted as a miRNA sponge in the cell, which in turn released the inhibitory effect of miRNA on the target gene and upregulated the expression level of the target gene [3]. In this study, we used the Circular RNA Interactome database to predict the circRNAs target miRNA molecules. The more DECs that the miRNA could bind to, the more likely it was involved in SLE pathogenesis. By assessing the frequency of these DECs target miRNAs, the top 3 miRNAs were hsa-miR-338-3p, hsa-miR-767-3p, and hsa-miR-1827. Previous studies had shown that these 3 miRNAs were mainly related to tumors and our study was the first to explore their relationship with SLE [24,25,26].
The miRNA was an evolutionarily conserved small molecule with a length of 18 to 25 nucleotides [23]. A series of studies had confirmed that miRNAs were associated with various diseases, such as cancer, diabetes, heart disease, and autoimmune disease etc. [27,28,29,30]. We extracted 87 SLE-related miRNAs from HMDD database. If the DECs we analyzed were interacting with these reported SLE-related miRNAs, they were more likely to be SLE-related circRNAs. There were 21 DECs associated with 18 SLE-related miRNAs. Next, we predicted the target genes of these 18 miRNAs. Only 4 miRNAs had the same target genes in the 3 distinct miRNA target gene prediction databases. Finally, we obtained a circRNA–miRNA–mRNA network consisting of 8 DECs (hsa_circ_0006770, hsa_circ_0002904, hsa_circ_0034044, hsa_circ_0023685, hsa_circ_0049271, hsa_circ_0074491, hsa_circ_0074559, and hsa_circ_0023461), 4 overlap miRNAs (hsa-miR-326, hsa-miR-569, hsa-miR-638, and hsa-miR-1246), and 13 target mRNAs (EPHB3, USH1G, UBE4A, DCAF7, TBL1XR1, SLC27A4, SMO, NAA30, RSBN1, PLAG1, SOX2, GPATCH11, and DYRK1A). For the 8 DECs and 13 target mRNAs, there were no relevant studies reporting that they were linked to SLE before. Sharaf-Eldin et al. found that the serum expression level of hsa-miR-326 was significantly downregulated in the neuropsychiatric systemic lupus erythematosus, and it could be a new diagnostic biomarker [31]. However, Chen et al. used the miRNA expression profiles to analyze the differential expression of miRNAs in PBMCs from SLE patients and normal controls; they found the expression level of hsa-miR-326 was significantly overexpressed in the SLE patients [32]. Hikami et al. found that there was a polymorphism site (rs1057233) in the 3-untranslated region of SPI1, which just located in the hsa-miR-569 target binding site, and the rs1057233 was associated with the susceptibility to SLE [33]. Steen et al. used high-throughput chip technology to compare the expression levels of miRNAs in plasma of SLE patients and normal controls and found that hsa-miR-638 significantly increased in plasma of SLE patients [34]. Meanwhile, Ishibe et al. also used high-throughput chip technology to compare the expression levels of miRNAs in the plasma of SLE patients and normal controls and found that hsa-miR-1246 significantly decreased in plasma of SLE patients [35].
In summary, we first obtained 29 DECs by comparing the expression of circRNAs in the T cells from patients with SLE and health controls. Subsequently, we used a variety of different bioinformatics methods to obtain 18 confirmed SLE-related miRNAs and 21 corresponding DECs. Finally, we constructed a circRNA–miRNA–miRNA network consisting of 8 DECs, 4 miRNAs, and 13 mRNAs. This study could be available for some help for the diagnosis, treatment, and prognosis of SLE patients. However, it also needed to be verified by subsequent experiments and clinical studies.
Data Availability
Not applicable.
Abbreviations
- SLE:
-
Systemic lupus erythematosus
- DECs:
-
Differentially expressed circRNAs
- circRNA:
-
Circular RNA
- miRNA:
-
MicroRNA
- HMDD:
-
Human microRNA Disease Database
- PBMC:
-
Peripheral blood mononuclear cell
References
Zucchi D, Elefante E, Calabresi E, Signorini V, Bortoluzzi A, Tani C (2019) One year in review 2019: systemic lupus erythematosus. Clin Exp Rheumatol 37(5):715–722
Nusbaum JS, Mirza I, Shum J, Freilich RW, Cohen RE, Pillinger MH, Izmirly PM, Buyon JP (2020) Sex differences in systemic lupus erythematosus: epidemiology, clinical considerations, and disease pathogenesis. Mayo Clin Proc 95(2):384–394. https://doi.org/10.1016/j.mayocp.2019.09.012
Li LJ, Zhu ZW, Zhao W, Tao SS, Li BZ, Xu SZ, Wang JB, Zhang MY, Wu J, Leng RX, Fan YG, Pan HF, Ye DQ (2018) Circular RNA expression profile and potential function of hsa_circ_0045272 in systemic lupus erythematosus. Immunology 155(1):137–149. https://doi.org/10.1111/imm.12940
Sharabi A, Tsokos GC (2020) T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nat Rev Rheumatol 16(2):100–112. https://doi.org/10.1038/s41584-019-0356-x
Li S, Zhang J, Tan X, Deng J, Li Y, Piao Y, Li C, Yang W, Mo W, Sun J, Sun F, Han T, Wang J, Kuang W, Li C (2019) Microarray expression profile of circular RNAs and mRNAs in children with systemic lupus erythematosus. Clin Rheumatol 38(5):1339–1350. https://doi.org/10.1007/s10067-018-4392-8
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK (1976) Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A 73(11):3852–3856. https://doi.org/10.1073/pnas.73.11.3852
Li H, Li K, Lai W, Li X, Wang H, Yang J, Chu S, Wang H, Kang C, Qiu Y (2018) Comprehensive circular RNA profiles in plasma reveals that circular RNAs can be used as novel biomarkers for systemic lupus erythematosus. Clin Chim Acta 480:17–25. https://doi.org/10.1016/j.cca.2018.01.026
Miao Q, Zhong Z, Jiang Z, Lin Y, Ni B, Yang W, Tang J (2019) RNA-seq of circular RNAs identified circPTPN22 as a potential new activity indicator in systemic lupus erythematosus. Lupus 28(4):520–528. https://doi.org/10.1177/0961203319830493
Floris G, Zhang L, Follesa P, Sun T (2017) Regulatory role of circular RNAs and neurological disorders. Mol Neurobiol 54(7):5156–5165. https://doi.org/10.1007/s12035-016-0055-4
Wang L, Shen C, Wang Y, Zou T, Zhu H, Lu X, Li L, Yang B, Chen J, Chen S, Lu X, Gu D (2019) Identification of circular RNA Hsa_circ_0001879 and Hsa_circ_0004104 as novel biomarkers for coronary artery disease. Atherosclerosis 286:88–96. https://doi.org/10.1016/j.atherosclerosis.2019.05.006
Lei M, Zheng G, Ning Q, Zheng J, Dong D (2020) Translation and functional roles of circular RNAs in human cancer. Mol Cancer 19(1):30. https://doi.org/10.1186/s12943-020-1135-7
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A (2013) NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res 41(Database issue):D991–D995. https://doi.org/10.1093/nar/gks1193
Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M (2016) CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol 13(1):34–42. https://doi.org/10.1080/15476286.2015.1128065
Huang Z, Shi J, Gao Y, Cui C, Zhang S, Li J, Zhou Y, Cui Q (2019) HMDD v3.0: a database for experimentally supported human microRNA-disease associations. Nucleic Acids Res 47(D1):D1013–D1017. https://doi.org/10.1093/nar/gky1010
Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, Tang Y, Chen YG, Jin CN, Yu Y, Xu JT, Li YM, Cai XX, Zhou ZY, Chen XH, Pei YY, Hu L, Su JJ, Cui SD, Wang F, Xie YY, Ding SY, Luo MF, Chou CH, Chang NW, Chen KW, Cheng YH, Wan XH, Hsu WL, Lee TY, Wei FX, Huang HD (2020) miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res 48(D1):D148–D154. https://doi.org/10.1093/nar/gkz896
Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4. https://doi.org/10.7554/eLife.05005
Chen Y, Wang X (2020) miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 48(D1):D127–D131. https://doi.org/10.1093/nar/gkz757
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504. https://doi.org/10.1101/gr.1239303
Guo G, Wang H, Ye L, Shi X, Yan K, Lin K, Huang Q, Li B, Lin Q, Zhu L, Xue X, Zhang H (2019) Hsa_circ_0000479 as a novel diagnostic biomarker of systemic lupus erythematosus. Front Immunol 10:2281. https://doi.org/10.3389/fimmu.2019.02281
Zurawska A, Mycko MP, Selmaj KW (2019) Circular RNAs as a novel layer of regulatory mechanism in multiple sclerosis. J Neuroimmunol 334:576971. https://doi.org/10.1016/j.jneuroim.2019.576971
Wang J, Yan S, Yang J, Lu H, Xu D, Wang Z (2019) Non-coding RNAs in rheumatoid arthritis: from bench to bedside. Front Immunol 10:3129. https://doi.org/10.3389/fimmu.2019.03129
Luo Q, Zhang L, Li X, Fu B, Guo Y, Huang Z, Li J (2019) Identification of circular RNAs hsa_circ_0044235 and hsa_circ_0068367 as novel biomarkers for systemic lupus erythematosus. Int J Mol Med 44(4):1462–1472. https://doi.org/10.3892/ijmm.2019.4302
Zhang MY, Wang JB, Zhu ZW, Li LJ, Liu RS, Yang XK, Leng RX, Li XM, Pan HF, Ye DQ (2018) Differentially expressed circular RNAs in systemic lupus erythematosus and their clinical significance. Biomed Pharmacother 107:1720–1727. https://doi.org/10.1016/j.biopha.2018.08.161
Xue Q, Sun K, Deng HJ, Lei ST, Dong JQ, Li GX (2014) MicroRNA-338-3p inhibits colorectal carcinoma cell invasion and migration by targeting smoothened. Jpn J Clin Oncol 44(1):13–21. https://doi.org/10.1093/jjco/hyt181
Gao L, Xiong DD, He RQ, Lai ZF, Liu LM, Huang ZG, Yang X, Wu HY, Yang LH, Ma J, Li SH, Lin P, Yang H, Luo DZ, Chen G, Dang YW (2019) Identifying TF-miRNA-mRNA regulatory modules in nitidine chloride treated HCC xenograft of nude mice. Am J Transl Res 11(12):7503–7522
Fasihi A, Bahram MS, Atashi A, Nasiri S (2018) Introduction of hsa-miR-103a and hsa-miR-1827 and hsa-miR-137 as new regulators of Wnt signaling pathway and their relation to colorectal carcinoma. J Cell Biochem 119(7):5104–5117. https://doi.org/10.1002/jcb.26357
Alexandri C, Daniel A, Bruylants G, Demeestere I (2020) The role of microRNAs in ovarian function and the transition toward novel therapeutic strategies in fertility preservation: from bench to future clinical application. Hum Reprod Update 26:174–196. https://doi.org/10.1093/humupd/dmz039
Jimenez-Lucena R, Camargo A, Alcala-Diaz JF, Romero-Baldonado C, Luque RM, van Ommen B, Delgado-Lista J, Ordovas JM, Perez-Martinez P, Rangel-Zuniga OA, Lopez-Miranda J (2018) A plasma circulating miRNAs profile predicts type 2 diabetes mellitus and prediabetes: from the CORDIOPREV study. Exp Mol Med 50(12):168–112. https://doi.org/10.1038/s12276-018-0194-y
Foinquinos A, Batkai S, Genschel C, Viereck J, Rump S, Gyongyosi M, Traxler D, Riesenhuber M, Spannbauer A, Lukovic D, Weber N, Zlabinger K, Hasimbegovic E, Winkler J, Fiedler J, Dangwal S, Fischer M, de la Roche J, Wojciechowski D, Kraft T, Garamvolgyi R, Neitzel S, Chatterjee S, Yin X, Bar C, Mayr M, Xiao K, Thum T (2020) Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat Commun 11(1):633. https://doi.org/10.1038/s41467-020-14349-2
Salvi V, Gianello V, Tiberio L, Sozzani S, Bosisio D (2019) Cytokine targeting by miRNAs in autoimmune diseases. Front Immunol 10:15. https://doi.org/10.3389/fimmu.2019.00015
Sharaf-Eldin WE, Kishk NA, Gad YZ, Hassan H, Ali MAM, Zaki MS, Mohamed MR, Essawi ML (2017) Extracellular miR-145, miR-223 and miR-326 expression signature allow for differential diagnosis of immune-mediated neuroinflammatory diseases. J Neurol Sci 383:188–198. https://doi.org/10.1016/j.jns.2017.11.014
Chen JQ, Papp G, Poliska S, Szabo K, Tarr T, Balint BL, Szodoray P, Zeher M (2017) MicroRNA expression profiles identify disease-specific alterations in systemic lupus erythematosus and primary Sjogren’s syndrome. PLoS One 12(3):e0174585. https://doi.org/10.1371/journal.pone.0174585
Hikami K, Kawasaki A, Ito I, Koga M, Ito S, Hayashi T, Matsumoto I, Tsutsumi A, Kusaoi M, Takasaki Y, Hashimoto H, Arinami T, Sumida T, Tsuchiya N (2011) Association of a functional polymorphism in the 3′-untranslated region of SPI1 with systemic lupus erythematosus. Arthritis Rheum 63(3):755–763. https://doi.org/10.1002/art.30188
Steen SO, Iversen LV, Carlsen AL, Burton M, Nielsen CT, Jacobsen S, Heegaard NH (2015) The circulating cell-free microRNA profile in systemic sclerosis is distinct from both healthy controls and systemic lupus erythematosus. J Rheumatol 42(2):214–221. https://doi.org/10.3899/jrheum.140502
Ishibe Y, Kusaoi M, Murayama G, Nemoto T, Kon T, Ogasawara M, Kempe K, Yamaji K, Tamura N (2018) Changes in the expression of circulating microRNAs in systemic lupus erythematosus patient blood plasma after passing through a plasma adsorption membrane. Ther Apher Dial 22(3):278–289. https://doi.org/10.1111/1744-9987.12695
Funding
The work was supported by NSFC (Natural Science Foundation of China) Grant U1605223 to Dr. Guixiu Shi.
Author information
Authors and Affiliations
Contributions
Junhui Zhang: analyze the data and write a first draft; Liu Yuan, revised paper; Shi Guixiu, overall guidance and revised paper.
Corresponding authors
Ethics declarations
Disclosures
None.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, J., Liu, Y. & Shi, G. The circRNA–miRNA–mRNA regulatory network in systemic lupus erythematosus. Clin Rheumatol 40, 331–339 (2021). https://doi.org/10.1007/s10067-020-05212-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-020-05212-2