
Abstract
Charcot-Marie-Tooth (CMT) disease is a heterogeneous disorder of the peripheral nervous system that collectively affects approximately 1 in 2,500 individuals, thus making it the most common inherited neurologic disorder. X-linked inheritance may account for 10–20 % of CMT neuropathy. We report a Czech family with a 30-year-old woman affected by CMT since the age of 10 years, originally as an isolated case. Nerve conduction study (NCS) showed demyelinating neuropathy, and DNA testing revealed a novel heterozygous gap junction beta-1 protein (GJB1) mutation c.784_786delTA. The same mutation, but surprisingly in heterozygous state, was subsequently found in her subjectively healthy father and later also in one of her sisters but not in her two other sisters. NCS showed intermediate type of motor and sensory neuropathy in these two females manifesting heterozygotes and normal results in the other healthy sisters and one brother, all without the c.784_786delTA mutation. The father has a phenotype milder than his daughter and has only subclinical signs of CMT. The index female patient had normal karyotype 46, XX, and normal FISH for centromeric X chromosome. We concluded that the proband’s father is a heterozygote due to the somatic mosaicism for the GJB1 mutation in his leukocytes (detected by DNA sequencing) and also in his germ cells as confirmed by the unexpectedly different genotypes in his four daughters. Quantitative analysis revealed a mutated signal in 25:75 allele proportion of mutated to healthy allele in the mosaic father. This study has important consequences for genetic counseling and prognosis in CMTX1 families.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Charcot-Marie-Tooth (CMT) disease represents a genetically very heterogeneous group of inherited motor and sensory neuropathies. Classification of CMT continues to be traditionally based on forearm motor nerve conduction velocity (MNCV) that divides CMT into type 1 (demyelinating; MCNV, <38 m/s) and type 2 (axonal; MNCV, >38 m/s) and on the mode of inheritance. Patients with X-linked CMT due to mutations in the GJB1 gene typically have intermediate slowing of nerve conduction velocities which are higher than in most CMT1 patients and lower than in most CMT2 patients [1–3].
The X-linked form of CMT type 1 (CMTX1) is the second most common type of inherited neuropathy caused by mutations in the gap junction beta one (GJB1) gene coding connexin 32 mapped to chromosome Xq13.1 [4]. GJB1 is highly conserved among species and is expressed in many tissues [5].
CMTX1 follows X-linked dominant inheritance—the affected man typically transmits the abnormal X chromosome (with GJB1 mutation) to all his daughters but not to his sons. Thus, all daughters will be heterozygous for the GJB1 mutation and sons will not inherit the affected GJB1 gene and will consequently not be CMTX1 affected. Clinical symptoms of CMTX1 in men start earlier than in women with corresponding GJB1 mutations, and men are also more severely affected. CMTX1 phenotype in women heterozygotes may be variable, probably due to random X chromosome inactivation (lyonisation) [6]. Heterozygous state in men for an X-linked gene mutation may occur under special conditions, chromosomal aberrations, a partial duplication of the GJB1 gene chromosomal region but also germ-line mosaicism.
Although more than 300 different mutations in the GJB1 gene have been reported, de novo mutations and mosaicism were demonstrated in only a few studies [7–10]. Zlotogora [11] and Chial [12] described even a formation process of a mutation in mosaicism. Whereas de novo mutation develops as new mutation by chance in germ cells and in mosaicism, mutation occurs very early in a somatic cell before the separation to germinal cells and is therefore present both in somatic and germinal cells. In such a case, expressions of phenotype can be mild-to-severe, depending on the extent of mutated cells in the mosaic population.
In this study, we report somatic mosaicism of a novel c.784_786delTA mutation in the GJB1 gene. Mosaicism was discovered in a 64-year-old father of four daughters and one son. The daughters were discordant in their GJB1 genotype, and the father was surprisingly heterozygote for this mutation. In addition, his grandson proved to have hemizygous deletion of TA in the GJB1 gene.
Materials and methods
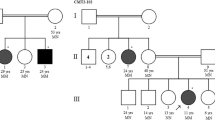
The examined family consisted of five affected and five unaffected members in three generations, all of whom provided written informed consent for the DNA analysis of hereditary neuropathies related CMT genes (in the case of children informed consent was provided by their parents as their legal representatives). The pedigree of the investigated family is showed in Fig. 1a.

The pedigree and sequencing results. a The pedigree showed the inheritance of a paternal X-linked mutation from father to his daughters. A man-to-man transmission absence of dominantly inherited CMT and testing of his daughters (half of them were affected) produced an explanation for paternal germ-line mosaicism. b Sequencing data presented detection of the c.784_786delTA mutation (pink box) in ten intercepted family members. Numbers in family members are laboratory designations of samples (year of birth); the female proband is marked by an arrow
Clinical diagnosis of CMT disease was based on the results of neurological examination (distal muscle weakness and wasting, pes cavus, and absence of deep tendon reflexes) and electrophysiological studies. Neurophysiological studies consisted of standard nerve conduction studies (NCS), including motor responses of the median, ulnar, tibial, and peroneal nerves with F-wave latencies, orthodromic median, ulnar, and sural nerve sensory action potential (SNAP).
Genomic DNA from blood (samples: 3585, 5278, 5277, 3584, 3533, 2505, 3363, 3480, 5238, 5355, and 3364) and saliva (only sample 3474) was extracted using standard protocols. The CMT1A duplication/hereditary neuropathy with liability to pressure palsies (HNPP) deletion were previously excluded using a set of 17 microsatellite markers according to Seeman et al. [13].
Fluorescent primers were used for amplification of X-linked microsatellite short tandem repeat (STR) markers (DXS6743, DXS8066, AFM263we1, AFM051tc3, PLP1CArep, and AFM191za11) flanking the GJB1 gene which were amplified separately and then analyzed on an ABI310 Genetic Analyzer (Applied Biosystems).
Sequencing of the GJB1 gene was performed in accordance with the Brožková et al. [8] study. Sequencing data files were inserted into Mutation Surveyor Software (SoftGenetics) that established the allele proportion of somatic mutation from a physical comparison of sequence traces using an anticorrelation algorithm with the results shown in an electropherogram.
The relative amount of mutant allele was also quantified by amplification of the GJB1 gene using fluorescent primers 5′-CACCTGAATACAAGCAGAATGAGA-3′ and 5′-CCTGGTATGTGGCATCAGC-3′. Amplification products, formed by allele with TA deletion (144 base pair) and by unaffected allele (146 base pair), were analyzed on an ABI310 Genetic Analyzer (Applied Biosystems).
Fluorescence in situ hybridization (FISH) using X-centromeric probes DXZ1 and LSI Steroid Sulfatase probes located in the Xp region from Vysis (Abbott Molecular) was performed. The karyotyping was examined by conventional Giemsa banding technology.
Results
The first clinical symptoms of CMT appeared in the female proband at the age of about 10 years with typical distal muscle weakness and the onset of atrophy more pronounced in the legs decreased or absent reflexes and feet deformities (Fig. 2). The female proband originally introduced her family history as unremarkable. Specifically, she declared her parents and sisters as CMT unaffected. However, the result of nerve conduction study and DNA testing of family members led to different findings later.

Lower limb weaknesses and atrophies are obvious in the affected proband (in upper line), but milder or absent in the mosaic father (in the lower line)
Routine nerve conduction study revealed an intermediate type of motor and sensory neuropathy in the female proband, a milder form of a similar type was later proven in the proband’s father and one of her sisters. The proband’s father has less abnormal electrophysiological values compared to his daughters at their younger age (summarized in Table 1). Abnormal values of NCS were shown in the proband’s young nephew. Normal results of NCS were in two of the father’s siblings in the next three proband’s siblings and her daughter.
The proband’s daughter was examined neurologically and electrophysiologically at the age of 10 years. Nerve conduction study showed normal values for motor and sensory nerves, and clinically, there were no atrophies, normal muscle strength, and present patellar and decreased Achilles tendon reflexes. Normal results of NCS are usual in young women with GJB1 mutation proof.
The proband’s nephew was 9 years old when he was examined. Boys with CMTX1 at this age usually just begin to have CMT symptoms. In his case, planovalgosity of the feet was observed and he was unable to walk on his heels but able to walk on tip toe. There were decreased tendon reflexes L2–L4 and absent reflexes L5–S2. Nerve conduction study showed abnormal values with decreased motor nerve conduction velocity at the peroneal nerve and also decreased sensory conduction velocity at the sural nerve (see Table 1).
Based on NCS results, genetic testing for CMT was performed. Initially, the CMT1A duplication/HNPP deletion was excluded in the female proband. DNA testing continued examination of the GJB1 gene by sequencing and revealed a novel heterozygous mutation c.784_786delTA. The same mutation was also found, but surprisingly in the heterozygous state in the proband’s father, who was originally declared as unaffected, and later in one of her sisters but not in the other two proband’s sisters (Fig. 1b). To exclude the possibility of a sample exchange, all the tests were repeated from new blood samples with the same results. The mutation c.784_786delTA was later also detected in heterozygous state in the proband’s daughter and in hemizygous state in the proband’s nephew.
Sequencing data were further evaluated by Mutation Surveyor Software where two samples were as standard; heterozygote with 50 % of allele proportion (female proband) and wild type with 0 % of allele proportion (unaffected proband’s sister). The proband’s father with somatic mosaicism has mutated allele proportion of 22 % calculated from genomic DNA derived from saliva.
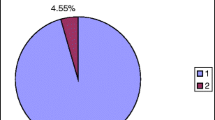
A similar mutation proportion of somatic mosaicism in the proband’s father was also confirmed by relative quantification of fluorescently labeled PCR products including the deleted region (Fig. 3). By simultaneous amplification of the deleted (2 bp shorter) and wild type allele, two fluorescent PCR products were obtained and the ratio between both peaks was calculated. The amount of cells carrying mutation in the proband’s father with mosaicism was represented by 27 % in DNA from saliva and by 24 % in DNA from blood. Both his daughters and the proband’s daughter had an allelic ratio 1:1 (50 % allele proportion for the mutation) which is consistent with normal heterozygous state. Affected allele in hemizygous state was detected only in the proband’s nephew.

Fragment analysis of the mutated region in the GJB1 gene. Two full alleles (peaks) indicated heterozygous state of GJB1 gene in samples 3533, 3363, and 3364; one peak in women demonstrates homozygous wild type X chromosomes in samples 5278, 3480, and 5238; one single allele of mutated X chromosome was obvious only in affected male sample 5355. Quantitative analysis in the case of male samples 3474 (saliva) and 3585 (peripheral blood) detected a proportion of mutated to healthy allele of approximately 1:3 of both used tissues
Subsequently, haplotype analysis using microsatellite STR markers was carried out among the family members (Fig. 4). Reconstructed haplotypes in the proband’s father and brother proved monoallelic for particular markers making a possible rearrangement, duplication of the critical region, very unlikely. Two of the proband’s sisters share the same X-linked haplotype with their father. The same haplotype of chromosome X was further detected in the proband’s daughter and nephew.

Haplotype analysis in the family core using X chromosome STR markers (introduced on the left side)
Genetic profiles by microsatellite STR markers in the proband’s father, which were obtained by independent DNA extractions from his saliva and also from peripheral blood, showed no differences.
Karyotyping in the female proband confirmed 46, XX, and FISH detected two signals of both chromosomes X. This further excluded the possibility that the unusual finding in this family could be caused by an aberration on the chromosomal level.
All these results in this family confirm that the proband’s father has somatic mosaicism. This somatic mosaicism was detected in DNA obtained from blood and saliva; germ-line mosaicism is demonstrated by the different genotypes of his daughters.
Discussion
Cases of X-linked disorders, in which mosaicism was demonstrated by molecular analysis, have already been reported. Only two cases of somatic mosaicism in CMTX1 disease have been reported so far, namely in studies by Kochanski et al. [9] and by Baker et al. [7] but none was documented completely as in our family. Our Czech family is therefore the best and most completely documented GJB1 somatic mosaicism so far. We present the novel mutation c.784_786delTA detected in five family members of three generations including a heterozygous man with somatic mosaicism in the oldest generation.
Kochanski et al. [9] described a CMTX1 mosaicism in a grandfather whose diagnosis became apparent through his 24-year-old grandson. The proband’s mother was minimally affected by sensory greater than motor involvement. Electrophysiological examination was not performed in the proband’s grandfather. Baker et al. [7] later referred a second case of CMTX1 mosaicism where a mutation was detected in one male proband with approximately one third affected cells in peripheral blood. Germ-line mosaicism was best confirmed in our family by different genotypes in the four daughters of the mosaic man. In the Kochanski et al. [9] study, the mutated allele was transmitted from mosaic patient to his only daughter. In the Baker et al. [7] study, the mutated allele was not transmitted to the offspring at all because the mosaic patient had only two unaffected daughters. Therefore, the germ-line mosaicism could be only expected there, but was not confirmed. In all three studies, somatic mosaicism was verified by sequencing of leukocytes-derived DNA. Quantification of mutation rate within somatic mosaicism was carried out by denaturing high-pressure liquid chromatography only in the Baker et al. [7] study. We quantified allele with mutation c.784_786delTA from two somatic tissues, saliva (ectodermal origin) and peripheral blood (mesodermal origin), and the occurrence of mutation proportion in both was approximately identical. We also demonstrated that the mutation c.784_786delTA is present in ectodermal as well as in mesodermal cells. Mutation had to occur before differentiation to germ layers in early embryogenesis which coincides with the Youssoufian and Pyeritz [14] report.
In early embryogenesis, the majority of cells in blastocyst contribute to extraembryonic tissue and only a few cells within the inner cell mass are committed to form the embryo. The dividing cells that give rise to the embryo intermingle extensively before the final tissue allocation. Three to four cells are directed toward germ cell lineage before the appearance of somatic cell lineages; somatic lineages are derived from at least eight founder cells. Thus, mutations arising at later stages of development may spare the germ cells and exclusively affect a portion of somatic cells [12, 14]. Zlotogora [11] investigated in her study a mosaicism in the germ cells which were diagnosed in parents without detectable mutation because of the birth of more than one affected descendent. The mutation was also present in somatic cells over 50 % of the examined cases. They reached a decision that mutation, occurring after a few divisions of the respective type of cell, must be present in a relatively high percentage within somatic or germ-line mosaicism. Hence, she suggested a possible relationship between the risk of a mutation arising and the number of cell divisions. The frequency of the two type events should be approximately in a similar range.
The clinical phenotype resulting from somatic mosaicism varies depending on the mutation proportion or threshold in the particular tissue. The degree of somatic mosaicism is often variable in different tissues. Therefore, the examination of several different tissues may increase the chance to detect somatic mosaicism. We analyzed DNA from blood and salivary buccal cells in the proband’s father. The presence of the mutated allele was verified by two methods, sequencing and fluorescent fragment analysis with similar results in both tissues with approximately one quarter of cells within peripheral blood and saliva.
Generally, CMTX1 is characterized by a more severe phenotypic expression in men than women of the same age. The age of symptom onset is always earlier in hemizygous men than in heterozygous women. At least half of affected men have recognized symptoms in the first two decades, whereas less than a third of women note symptoms by this age. CMTX1 men also have lower MNCV than CMTX1women [15, 16]. These prevalent characteristics of CMTX1 are not true in our family with mosaicism. In our family, the clinical symptoms were first expressed in the heterozygous female proband when she was about 10 years old whereas the symptoms in her father were detected at the age of 60 and only due to the diagnosis in the proband (his daughter). In addition, electrophysiological studies showed that MNCV was higher in the older father (with mosaicism) than in his affected heterozygous daughters.
Identification of somatic mosaicism in CMTX1 disease may be crucial for clinical prognosis of CMTX1 in mosaic patients and for correct genetic counseling in prenatal diagnosis.
Abbreviations
- CMTX1:
-
X-linked Charcot-Marie-Tooth type 1 disease
- GJB1:
-
Gap junction beta-1 protein
- HNPP:
-
Hereditary neuropathy with liability to pressure palsies
- NCS:
-
Nerve conduction study
- STR:
-
Short tandem repeat
References
Nicholson G, Nash J (1993) Intermediate nerve conduction velocities define X-linked Charcot-Marie-Tooth neuropathy families. Neurology 43:2558–2564. doi:10.1385/NMM:8:1-2:123
Kleopa KA, Scherer SS (2006) Molecular genetics of X-linked Charcot-Marie-Tooth disease. Neuromolecular Med 8:107–22. doi:10.1385/NMM:8:1-2:107
Mandlich P, Grandis M, Geroldi A, Acquaviva M, Varese A, Gulli R, Ciotti P, Bellone E (2008) Gab junction beta 1 (GJB1) gene mutation in Italian patients with X-linked Charcot-Marie-Tooth disease. J Hum Genet 53:529–533. doi:10.1007/s10038-008-0280-4
Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262:2039–2042. doi:10.1126/science.8266101
Bone LJ, Deschenes SM, Balice-Gordon RJ, Fischbeck KH, Scherer SS (1997) Connexin 32 and X-linked Charcot-Marie-Tooth disease. Neurobiol Dis 4:221–244. doi:10.1006/nbdi.1997.0152
Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ (1998) Connexin 32-null mice develop a demyelinating peripheral neuropathy. Glia 24:8–20. doi:10.1002/(SICI)1098-1136(199809)24:1<8::AID-GLIA2>3.0.CO;2–3
Baker SK, Reith CC, Ainsworth PJ (2008) Novel 95G > A (R32K) somatic mosaic connexin 32 mutation. Muscle Nerve 38(5):1510–4. doi:10.1002/mus.21145
Brožková D, Mazanec R, Haberlová J, Sakmaryová I, Šubrt I, Seeman P (2010) Six New Gap Junction Beta 1 gene mutations and their phenotypic expression in Czech patients with Charcot-Marie-Tooth disease. Genet Testing Mol Biomarkers 14(1):1–5. doi:10.1089=gtmb.2009.0093
Kochanski A, Nowakowski A, Kawulak M, Kabzińska D, Hausmanowa-Petrusewicz I (2004) Somatic mosaicism in Charcot-Marie-Tooth type X disease. Neurology 27(62):336–337. doi:10.1212/01.WNL.0000103441.52563.02
Li QH, Liu KX, Feng JL, Zeng AY, Li H, Wu L, Tang YG, Chen ML, Lin XH, Jiang JZ (2010) A new mutation in the GJB1 gene of a Chinese family with Charcot-Marie-Tooth disease associated with vocal cord paresis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 27(5):497–500. doi:10.3760/cma.j.issn.1003-9406.2010.05.005
Zlotogora J (1998) Germ-line mosaicism. Hum Genet 102(4):381–6. doi:10.1007/s004390050708
Chial H (2008) Somatic mosaicism and chromosomal disorders. Nat Educ 1:1. http://www.nature.com/scitable/topicpage/somatic-mosaicism-and-chromosomal-disorders-867
Seeman P, Mazanec R, Zidar J, Hrusáková S, Čtvrtečková M, Rautenstrauss B (2000) Charcot-Marie-Tooth disease type A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP): reliable detection of the CMT1A duplication and HNPP deletion using 8 microsatellite markers in 2 multiplex PCRs. Int J Mo Med 6:421–426
Youssoufian H, Pyeritz RE (2002) Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet 3:748–758. doi:10.1038/nrg906
Birouk N, LeGuern E, Maisonobe T, Rouger H, Gouider R, Tardieu S, Gugenheim M, Routon MC, Léger JM, Agid Y, Brice A, Bouche P (1998) X-linked Charcot-Marie-Tooth disease with connexin 32 mutations: clinical and electrophysiologic study. Neurology 50(4):1074–1082. doi:10.1212/WNL.50.4.1074
Hahn AF, Bolton CF, White CM, Brown WF, Tuuhe SE, Tan CC, Ainworth PJ (1999) Genotype/phenotype correlations in X-linked dominant Charcot-Marie-Tooth disease. Ann N Y Acad Sci 8883:366–383. doi:10.1111/j.1749-6632.1999.tb08598.x
Acknowledgments
This study is supported by IGA MH CR No: NT 11521–4.
Ethical standards
The experiments presented in this manuscript are in accordance with the law of the Czech Republic.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Borgulová, I., Mazanec, R., Sakmaryová, I. et al. Mosaicism for GJB1 mutation causes milder Charcot-Marie-Tooth X1 phenotype in a heterozygous man than in a manifesting heterozygous woman. Neurogenetics 14, 189–195 (2013). https://doi.org/10.1007/s10048-013-0368-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-013-0368-7