
Abstract
Pediatric low-grade gliomas (PLGGs) have relatively favorable prognosis and some resectable PLGGs, such as cerebellar pilocytic astrocytoma, can be cured by surgery alone. However, many PLGG cases are unresectable and some of them undergo tumor progression. Therefore, a multidisciplinary approach is necessary to treat PLGG patients. Recent genomic analysis revealed a broad genomic landscape underlying PLGG. Notably, the majority of PLGGs present MAPK pathway-associated genomic alterations and MAPK signaling-dependent tumor progression. Following preclinical evidence, many clinical trials based on molecular target therapy have been conducted on PLGG patients, some of whom exhibited durable response to target therapy. Here, we provide an overview of PLGG genetics and the evidence supporting the application of molecular target therapy in these patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pediatric low-grade gliomas (PLGGs) are clinically and genetically distinct from adult LGG [63]. PLGGs include pilocytic astrocytoma (PA), dysembryoplastic neuroepithelial tumor (DNET), ganglioglioma (GG), pleomorphic astrocytoma (PXA), subependymal giant cell astrocytoma (SEGA), diffuse astrocytoma (DA) and diffuse oligodendroglial tumors (d-OT), and other tumors [70]. Recent large-scale genomic analysis uncovered the genetic landscape of pediatric brain tumors, including PLGG. Importantly, there is a tight relationship between specific genetic alterations and anatomical tumor location in PLGG. Several clinical trials, based on molecular target therapy, are ongoing in PLGG patients. Herein, we summarize the genetic characterization of PLGG and the relevant molecular therapeutic targets potentially suitable for precision medicine.
Genetic characterization of PLGG
Somatic gene abnormalities
MAPK signaling pathway
Although, histologically, PLGGs comprise different tumors, the vast majority of PLGGs are caused by various genetic alterations affecting the MAPK pathway (Fig. 1) [53]. In particular, genetic alterations to promote MAPK pathway activation are found in almost the totality of PA cases [29, 56, 75].

Major oncogenic pathways (MAPK pathway and PI3K/AKT/mTOR pathway) in pediatric low-grade gliomas (PLGG). Blue fonts. Major oncogenic alterations in PLGG. Red fonts. Proposed molecular therapeutic inhibitors for suppressing major oncogenic pathways
KIAA1549: BRAF fusion
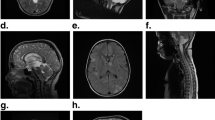
A 2-Mb tandem duplication of 7q34, encompassing the BRAF gene, was commonly detected in PA cases [11]. Importantly, younger patients with infratentorial posterior fossa PA tend to display a high frequency of the BRAF fusion [23, 30]. In the tandem duplication, the N-terminal end of the KIAA1549 protein replaces the N-terminal regulatory region of BRAF, while the BRAF kinase domain is retained, resulting in constitutive activation of the MAPK pathway [30, 75]. Other BRAF fusions, involving FAM131B, RNF130, CLCN6, MKRN1, GNA11, QKI, FZR1, and MACF1, were also identified. These events also induce loss of the N-terminal regulatory region of the BRAF protein and constitutive activation of the kinase domain. The BRAF fusions arise from various genetic mechanisms, including deletions and translocations in PA [29, 75]. In addition to BRAF fusions, BRAF small insertions, activating BRAF kinase signaling, were also observed in a small proportion of PA [29]. BRAF fusions are almost exclusive to PA [70]. PAs carrying the KIAA1549:BRAF fusion occur in almost all anatomical locations, but most frequently in the cerebellum (Fig. 2A) [10].

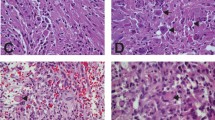
a Axial (left) and sagittal (right) contrast enhancing MR imaging indicating contrast-enhanced lesion with cyst in cerebellum. Hematoxylin and eosin staining indicating pseudo-oligodendroglial pattern. b Axial (left) and sagittal (right) contrast enhancing MR imaging indicating weak contrast-enhanced lesion in suprasellar lesion. Hematoxylin and eosin staining showing typical biphasic pattern with Rosenthal fibers (arrows). These tumors are histologically diagnosed as pilocytic astrocytoma, but harboring distinct genetic alteration. Magnification, × 400
BRAF V600E mutation
PLGGs bearing the BRAFV600E mutation comprise a heterogeneous group of tumors with divergent histology, location, cooperating genetic alterations, and outcomes [31]. The BRAFV600E status alone is not a sufficient diagnostic or prognostic biomarker of PLGG [31]. A strong association between the BRAFV600E mutation and tumor location was observed in PA (WHO grade I). More specifically, 20% of extra-cerebellar PA were found positive for the BRAFV600E mutation, whereas only 2% cerebellar PA harbored the mutation [65, 70].
The BRAFV600E mutation was observed in approximately 65% of PXA cases (WHO grade II and grade III) [14, 64], and is highly correlated with age. PXA patients carrying the BRAFV600E mutation were mainly observed in young subjects (mean age; 18 years), while PXA tumors related to wild-type BRAF display a later onset (mean age; 38 years) [64]. The BRAFV600E mutation was also found in 18–50% of GG (WHO grade I), 30–51% of DNET (WHO grade I), and 43% of SEGA (WHO grade I), respectively [8, 42, 55, 65].
FGFR1 mutations, fusions, and kinase domain duplications
Recent large-scale genetic analysis uncovered recurrent FGFR1 somatic mutations (p.N546K, p.K656E), FGFR1–TACC1 fusions, and a novel internal tandem FGFR1 kinase domain duplication in patients with PA and DNET [29, 56, 76]. Abnormalities in FGFR1 are frequent in PLGGs with oligodendroglial phenotype and composed of primary of oligodendrocyte-like cells, being present in 82% of DNETs and 40% of d-OT, respectively [56].
Fusions of the NTRK gene family
NTRK receptor transcript fusion is a very rare event in adult and pediatric tumors (0.3%), but relatively frequent in pediatric melanoma (11.1%) and PLGG (2.5%), as well as pediatric high-grade glioma (PHGG, 5.3%) [50]. Among NTRK family members, NTRK2 fusions (QKI-NTRK2 and NACC2-NTRK2 fusions) were identified in PA [29]. The NTRK gene fusions are characterized by various 5′partners with a dimerization domain that presumably leads to constitutive dimerization and kinase activation in PA [29, 75].
Homozygous CDKN2A deletion
CDKN2A is a tumor-suppressor gene and key regulator of cell cycle. A CDKN2A homozygous deletion inactivates cell cycle regulation and promotes malignant cancer progression. Consistently, loss of CDKN2A (p16) in immortalized human astrocytes abrogated the senescent features [26]. Although activated BRAF alone is not sufficient for tumorigenesis, the combination of BRAF activation and CDKN2A loss results in cell transformation [61], indicating a critical role for CDKN2A homozygous deletion in tumor progression. Indeed, BRAFV600E mutation and CDKN2A deletion were found to be early genetic events in PLGG with malignant transformation [46]. In BRAFV600E mutant PLGGs, CDKN2A deletion was rated as a poor independent prognostic factor [40]. CDKN2A homozygous deletion was found in 25% of the BRAFV600E mutant PLGGs [40], 60% of PXA, and 16.7% of BRAFV600E mutant GG. However, the number of available studies is relatively small [55, 72].
Amplification and/or rearrangement of MYB/MYBL1 and MYB–QKI fusion
A triple alteration consisting of MYB truncation, increased expression through enhancer hijacking, and loss of QKI tumor suppressor function was identified in a proportion of PLGGs with astrocytic phenotype (diffuse astrocytic and angiocentric glioma). An MYB–QKI fusion characterizes almost all angiocentric gliomas (87%), while MYB–ESR1 fusion and QKI rearrangement was also identified in a subset of angiocentric gliomas [56]. In contrast, MYB or MYBL1 rearrangement was detected in 41% of DA [56]. MYB/MYBL1 alterations were co-occured with BRAF alteration in a subset of DA [56, 57, 75].
ROS1 or ALK fusions
ALK or ROS1 rearrangements, which activate RAS/MAPK, PI3K, and JAK/STAT pathways, have been identified in a subset of PHGG and PLGG [9, 28, 48, 52]. These genetic events were found in infants and younger children.
Germline gene abnormalities
TSC1/TSC2
Tuberous sclerosis complex (TSC) is an autosomal dominant neurocutaneous disorder caused by mutations in either hamartin-encoding TSC1 or tuberin-encoding TSC2. In the central nervous system, TSC is characterized by the development of SEGA, subcortical heterotopic nodule, and cortical tuber [47]. SEGAs belong to the group of astrocytic neoplasms, even though they are also expressed in neurons. The majority of SEGAs exhibit loss of heterozygosity in the same gene in which the mutation of TSC1 or TSC2 was identified [3], while no evidence of BRAFV600E or other BRAF mutations was found in these tumors.
Neurofibromatosis type I (NF1)
The NF1 gene encodes an RAS GTPase-activating protein known as neurofibromin and is one of several genes that affect RAS–MAPK signaling [58]. The somatic NF1 loss occurred by frameshift mutation, loss of heterozygosity, and methylation and no additional gene alteration was identified in NF-1 associated PA, indicating NF1 loss is likely sufficient for PA formation [22]. About 20% of NF1 patients develop PAs [47], most of which are located in the supratentorial midline (Fig. 2B) [70].
Molecular target therapy for pediatric low-grade tumors
Target for BRAF V600E mutation in pediatric gliomas
A large clinical study indicated that patients with BRAFV600E mutant PLGG exhibited poorer prognosis compared to patients with wild-type BRAF. Moreover, conventional therapies were scarcely effective in BRAFV600E mutant PLGG patients [40]. Thus, novel therapeutic strategies are needed for BRAFV600E mutant gliomas.
Since BRAFV600E triggers the constitutive activation of MAPK signaling pathway (Fig. 1), the latter has been proposed as a therapeutic target. An early preclinical study demonstrated that BRAF knockdown and pharmacological BRAF inhibition induce p-ERK inactivation and antiproliferative activity in BRAFV600E-expressing, but not in wild-type BRAF-expressing malignant gliomas. The first-generation BRAF inhibitor, PLX4720, extended the overall survival in a BRAFV600E mutant orthotopic xenograft model, indicating that BRAFV600E is a promising therapeutic target in BRAFV600E-expressing brain tumors [49].
Burger et al. demonstrated a durable response to dabrafenib (a first-generation BRAF inhibitor) in three patients with disseminated leptomeningeal BRAFV600E-positive glioma. Of note, one patient was stable for up to 27 months. In addition, they established a patient-derived cell line and found that dabrafenib reduced the cell density and inhibited p-ERK [5], indicating that drug sensitivity is through on-target effect. A phase-1/2 clinical trial demonstrated that the objective response rate of dabrafenib is 38% in PLGG [13]. Another clinical trial for pediatric BRAFV600E mutant relapsed/refractory PLGG demonstrated that dabrafenib was also effective (44% overall response rate) and tolerable (clinicaltrials.gov, NCT01677741). Another first-generation BRAF inhibitor, vemurafenib (a PLX4720 analog), controlled tumor progression in patients with recurrent PXA [7, 43]. In addition, a vemurafenib-based regimen decreased tumor size in patients with brain stem GG expressing the BRAFV600E mutation [62]. In another study, all six BRAFV600E-mutant PLGG patients favorably responded to BRAF inhibitors [40]. The VE-BASKET study, focusing on BRAFV600E-mutant non-melanoma cancers, demonstrated the characterization of 24 patients with glioma received vemurafenib until disease progression. The objective response rate was 42.9% (3/7) in PXA, 9.1% (1/11) in malignant diffuse glioma, and 33.3% (2/6) in other tumors, including one pilocytic astrocytoma [32]. Another phase-2 basket study also indicated a 75% (3/4) response after vemurafenib monotherapy in PXA patients with the BRAFV600E mutation [25].
Unfortunately, resistance to the BRAF inhibitor is frequently observed due to MAPK pathway reactivation, which is mediated by RAF-independent activation of MEK and ERK [69]. Additionally, acquired gene alterations conferring drug resistance, including NRAS, BRAF, MEK1, and MEK2, were identified after treatment with BRAF inhibitors [71]. In contrast, combined BRAF/MEK inhibition was reported to block p-ERK signaling and prevent the acquisition of resistance in melanoma cell lines [54]. In addition, a phase-1/2 clinical trial demonstrated that a combination of dabrafenib and trametinib (MEK inhibitor) prolonged progression-free survival (PFS) in patients with metastatic melanoma [44]. Various phase-3 studies demonstrated improved overall survival after treatment of naïve unresectable or metastatic melanoma patients with BRAFV600E mutation [45, 60]. Similar combination effects were also observed in other solid cancers, including colorectal cancer [12].
Consistently, whereas monotherapy with a BRAF inhibitor results in transient MEK-ERK inhibition in BRAFV600E mutant glioma cell lines, combined BRAF/MEK inhibition prevented MAPK reactivation, resulting in enhanced antitumor effects, both in vitro and in vivo [77]. Moreover, in a BRAFV600E mutant-expressing glioma cell line, combined BRAF/MEK inhibition was found to be more effective compared to the combination between BRAF and mTOR inhibitors (everolimus) or that between an MEK1/2 inhibitor (AZD6244) and everolimus [51]. A higher efficacy of the combined BRAF/MEK inhibition was also observed in a BRAFV600E mutant-expressing and CDKN2A-deficient syngenic mice glioma model [21], which is considered a high-risk group in BRAFV600E mutant PLGG [40]. Brown et al. reported a clinical–radiological response in two cases of anaplastic PXA treated with a combination of BRAF and MEK inhibitors [4]. Another group described the results of BRAF/MEK inhibitor treatment of two patients with BRAFV600E-positive tumors (one anaplastic PXA and one glioblastoma (GBM)) after conventional treatment. Accordingly, the anaplastic PXA case exhibited a partial response for 14 months, although progression was observed thereafter. On the other hand, the GBM patient was stable after 16 months of treatment [66]. In a phase-2 open-label trial (clinicaltrials.gov, NCT02034110), patients with the BRAFV600E mutation received dabrafenib plus trametinib until unacceptable toxicity, disease progression, or death. In that study, 37 high-grade glioma (HGG) patients were enrolled, 31 of whom were evaluable for response. The overall response rate was 26% (8/31), including one complete response. Of note, five of eight (62.5%) responding patients exhibited a response duration of more than 12 months. A global, open-label phase-2 study (clinicaltrials.gov, NCT02684058), evaluating dabrafenib in combination with trametinib in pediatric patients with BRAFV600E-mutant HGG, is currently ongoing, as well as a phase-1/2 study employing the trametinib/dabrafenib combination in PLGG (clinicaltrials.gov, NCT02124772). In addition to MAPK pathway reactivation, it has been demonstrated that resistance to BRAF inhibitors may occur through activation of the EGFR signaling pathway in glioma [74]. Therefore, a combination of BRAF inhibition and EGFR target therapy might be an alternative therapeutic option for BRAFV600E-mutant glioma.
Target for KIAA1549-BRAF fusion gliomas
Importantly, a paradoxical activation of MAPK signaling after treatment with PLX4720 (first-generation BRAF inhibitor) was reported in KIAA1549-BRAF fusion cells [68]. Similarly, sorafenib, a multi-kinase inhibitor targeting BRAF, VEGFR, PDGFR, and c-kit induced MAPK-dependent stimulation of tumor growth, mostly in recurrent/progressive PLGGs carrying the BRAF1549-KIAA fusion [33]. To overcome this issue, a second-generation BRAF inhibitor was developed and found to prevent the paradoxical MAPK activation [68]. However, another study reported that after treatment with the second-generation BRAF inhibitor, PLX8394, inhibitor-resistant cells emerged because of PI3K/AKT/mTOR pathway activation in cells stably expressing the KIAA1549-BRAF fusion [27], further emphasizing the difficulty in controlling KIAA1549-BRAF fusion tumors using a single BRAF inhibitor.
In addition to the BRAF inhibitor, the MEK1/2 inhibitor demonstrated antitumor effect in a BRAFV600E mutant xenograft model [38]. A phase-1 Pediatric Brain Tumor Consortium study indicated that selumetinib (AZD6244, MEK1/2 inhibitor) displays promising antitumor activity in PLGG [1]. Currently, selumetinib effects are under examination in young patients with recurrent or refractory LGG (clinicaltrials.gov, NCT01089101). Preliminary results indicated partial response in 29% (2/7) of BRAFV600E mutant tumors and 33% (6/18) of tumors carrying the BRAF-KIAA1549 fusion.
A preclinical study demonstrated that the KIAA1549-BRAF fusion regulates neuroglial cell growth in an mTOR-dependent manner [34], implying that targeting of mTOR and downstream pathways may be another suitable therapeutic strategy for KIAA1549-BRAF fusion tumors. Notably, KIAA1549-BRAF-containing cells were sensitive to the MEK1/2 inhibitor, AZD6244, and the combination of AZD6244 and everolimus-induced enhanced effect, when compared with BRAF wild-type cells [51]. AZD6244 and everolimus reduced p-ERK and p-S6, respectively, and combination treatment enhanced suppression in both signaling [51]. Additionally, an acquired PI3K/AKT/mTOR pathway-mediated resistance to the MEK inhibitor emerged in KIAA1549-BRAF fusion cells, whereas the combination treatment of MEK and mTOR inhibitors enhanced cell toxicity in vitro and in vivo [27].
NF1 mutation targeting in pediatric gliomas
The NF-1 gene encodes neurofibromin, a negative regulator of Ras activity, and loss of NF-1 leads to Ras dysregulation and activation of downstream pathways, including MAPK and PI3K pathway, as well as tumorigenesis [59]. Initial preclinical experiment demonstrated that an MEK inhibitor sensitized an NF-1-deficient acute myeloid leukemia model [41]. Of note, the MEK1/2 inhibitor, selumetinib, decreased tumor size in 71% (17/24) of NF-1-related pediatric plexiform neurofibromas [16]. Therefore, Ras–MAPK signaling may be a novel therapeutic target in gliomas carrying the NF-1 mutation. See et al. demonstrated that NF-1 deficiency is associated with MEK inhibitor sensitivity in GBM cell lines and an MEK inhibitor (PD0325901) suppressed NF-1-deficient GBM orthotopic xenograft formation. In addition, they found that inhibition of PI3K/AKT/mTOR signaling enhanced the sensitivity to MEK inhibition [67]. In the NCT01089101 trial, 10 of 25 (40%) NF-1-associated LGGs under treatment with selumetinib achieved partial response, with a 2-year PFS of 96%. Further large-scale studies are required to establish the efficacy of MEK inhibition in NF-1-deficient PLGG.
In addition, it has been demonstrated that the NF-1-related mTOR pathway regulates glial cell growth and gliomagenesis [2]. Kaul and colleagues reported that the growth of NF-1-deficient optic glioma is suppressed by both AKT- or MEK-mediated mTOR inhibition in vitro and in vivo [35]. Moreover, rapamycin suppressed the proliferation of NF-1+/-GFAPCKO mice optic glioma [24]. Interestingly, when an mTOR inhibitor and the tyrosine kinase inhibitor, erlotinib, were tested on recurrent PLGG, two NF-1 patients had disease control for more than 1 year [73]. A phase-2 study employing everolimus for recurrent or progressive low-grade gliomas, with or without NF-1 deficiency, is currently recruiting patients (clinicaltrials.gov, NCT01734512).
Other molecular targets in pediatric gliomas
A breakthrough in molecular target therapy has been the employment of everolimus for tuberous sclerosis-associated SEGA. SEGAs are the most common brain tumors in TSC and occur in 5–20% of these patients. Mutations in TSC1 (hamartin) or TSC2 (tuberin) upregulate the mTOR complex 1, leading to cell proliferation in SEGA, raising the possibility that mTOR inhibitor may be a specific therapeutic agent. Several case reports demonstrated that rapamycin caused the regression of TSC-related astrocytomas [19, 37]. Importantly, large clinical trials (ClinicalTrials. gov number, NCT00789828, NCT00411619, and NCT01713946) indicated that everolimus reduced tumor volume and seizure frequency in patients with TSC-associated SEGA [18, 20, 39]. Additionally, treatment with everolimus provided long-term clinical benefit to patients with SEGA [17].
Furthermore, recurrent genetic alterations, such as those involving NTRK, FGFR1, and MYB, were identified in PLGG and are promising therapeutic targets [29, 56, 76]. Receptor tyrosine kinase tropomyosin-related A, B, and C (TRKA, TRKB, and TRKC) are encoded by NTR1, NTRK2, and NTRK3, respectively. These NTRK oncogenic fusions lead to downstream activation of signaling pathway, including the PI3K pathway and MAPK pathway [36].
Various inhibitors targeting TRK family members have been developed and tested in clinical trials. In 2018, the U.S Food and Drug Administration (FDA) approved the use of larotrectinib for solid tumors containing NTRK gene fusions. Importantly, larotrectinib led to dramatic tumor regression in a pediatric HGG patient with ETV6-NTRK3 fusion [78]. Clinical trials testing larotrectinib for NTRK fusion-positive solid tumors, including brain tumors, are currently ongoing (NCT02576431). Further studies are required to assess the therapeutic potential of TRK inhibition in PLGG.
Recurrent FGFR1 mutations (hotspot residues at N546 and K656), as well as PTPN11 mutations, were identified in a subset of PAs [29]. Moreover, duplication of the FGFR1 tyrosine kinase domain and FGFR1-TACC fusion were identified in PLGG [76]. These mutations were found to be mutually exclusive with RAF/RAS alterations and also result in MAPK pathway activation [29]. Anti-FGF/FGFR targeting drugs have been introduced and are being tested in clinical trials [6, 15].
In summary, large-scale genomic analyses uncovered the genetic landscape of pediatric low-grade gliomas, which are distinct from adult gliomas. Since most of the PLGGs harbor gene alterations that activate the MAPK pathway, the efforts to develop a therapeutic strategy targeting this pathway are intensifying. The results of several ongoing clinical trials are expected to improve PLGG patients’ outcome by novel molecular target therapies.
Abbreviations
- PLGG:
-
Pediatric low-grade glioma
- PA:
-
Pilocytic astrocytoma
- PXA:
-
Pleomorphic xanthoastrocytoma
- DNET:
-
Dysembryoplastic neuroepithelial tumor
- GG:
-
Ganglioglioma
- TSC:
-
Tuberous sclerosis complex
- SEGA:
-
Subependymal giant cell astrocytoma
- DA:
-
Diffuse astrocytoma
- d-OT:
-
Diffuse oligodendroglial tumor
References
Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, Fangusaro J, Phillips J, Perry A, Turner D, Prados M, Packer RJ, Qaddoumi I, Gururangan S, Pollack IF, Goldman S, Doyle LA, Stewart CF, Boyett JM, Kun LE, Fouladi M (2017) A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol 19:1135–1144
Banerjee S, Crouse NR, Emnett RJ, Gianino SM, Gutmann DH (2011) Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc Natl Acad Sci USA 108:15996–16001
Bongaarts A, Giannikou K, Reinten RJ, Anink JJ, Mills JD, Jansen FE, Spliet GMW, den Dunnen WFA, Coras R, Blumcke I, Paulus W, Scholl T, Feucht M, Kotulska K, Jozwiak S, Buccoliero AM, Caporalini C, Giordano F, Genitori L, Soylemezoglu F, Pimentel J, Nellist M, Schouten-van Meeteren AYN, Nag A, Muhlebner A, Kwiatkowski DJ, Aronica E (2017) Subependymal giant cell astrocytomas in tuberous sclerosis complex have consistent TSC1/TSC2 biallelic inactivation, and no BRAF mutations. Oncotarget 8:95516–95529
Brown NF, Carter T, Kitchen N, Mulholland P (2017) Dabrafenib and trametinib in BRAFV600E mutated glioma. CNS Oncol 6:291–296
Burger MC, Ronellenfitsch MW, Lorenz NI, Wagner M, Voss M, Capper D, Tzaridis T, Herrlinger U, Steinbach JP, Stoffels G, Langen KJ, Brandts C, Senft C, Harter PN, Bahr O (2017) Dabrafenib in patients with recurrent, BRAF V600E mutated malignant glioma and leptomeningeal disease. Oncol Rep 38:3291–3296
Chae YK, Ranganath K, Hammerman PS, Vaklavas C, Mohindra N, Kalyan A, Matsangou M, Costa R, Carneiro B, Villaflor VM, Cristofanilli M, Giles FJ (2017) Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget 8:16052–16074
Chamberlain MC (2013) Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neurooncol 114:237–240
Chappe C, Padovani L, Scavarda D, Forest F, Nanni-Metellus I, Loundou A, Mercurio S, Fina F, Lena G, Colin C, Figarella-Branger D (2013) Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol 23:574–583
Cocce MC, Mardin BR, Bens S, Stutz AM, Lubieniecki F, Vater I, Korbel JO, Siebert R, Alonso CN, Gallego MS (2016) Identification of ZCCHC8 as fusion partner of ROS1 in a case of congenital glioblastoma multiforme with a t(6;12)(q21;q24.3). Genes Chromosomes Cancer 55:677–687
Collins VP, Jones DT, Giannini C (2015) Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathologica 129:775–788
Collins VP, Jones DT, Giannini C (2015) Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129:775–788
Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, Hamid O, Messersmith WA, Daud A, Kurzrock R, Pierobon M, Sun P, Cunningham E, Little S, Orford K, Motwani M, Bai Y, Patel K, Venook AP, Kopetz S (2015) Combined BRAF and MEK Inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol 33:4023–4031
Dabrafenib Effective in Pediatric Glioma (2017) Cancer Discov 7:OF5
Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, Batchelor TT, Ligon KL, Iafrate AJ, Ligon AH (2011) BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PloS One 6:e17948
Dieci MV, Arnedos M, Andre F, Soria JC (2013) Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov 3:264–279
Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE, Rizvi TA, Wu J, Ershler R, Wolters P, Therrien J, Glod J, Belasco JB, Schorry E, Brofferio A, Starosta AJ, Gillespie A, Doyle AL, Ratner N, Widemann BC (2016) Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med 375:2550–2560
Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, Witt O, Kohrman MH, Flamini JR, Wu JY, Curatolo P, de Vries PJ, Berkowitz N, Anak O, Niolat J, Jozwiak S (2014) Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol 15:1513–1520
Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, Witt O, Kohrman MH, Flamini JR, Wu JY, Curatolo P, de Vries PJ, Whittemore VH, Thiele EA, Ford JP, Shah G, Cauwel H, Lebwohl D, Sahmoud T, Jozwiak S (2013) Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 381:125–132
Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR (2006) Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 59:490–498
French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz N, Voi M, Peyrard S, Pelov D, Franz DN (2016) Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388:2153–2163
Grossauer S, Koeck K, Murphy NE, Meyers ID, Daynac M, Truffaux N, Truong AY, Nicolaides TP, McMahon M, Berger MS, Phillips JJ, James CD, Petritsch CK (2016) Concurrent MEK targeted therapy prevents MAPK pathway reactivation during BRAFV600E targeted inhibition in a novel syngeneic murine glioma model. Oncotarget 7:75839–75853
Gutmann DH, McLellan MD, Hussain I, Wallis JW, Fulton LL, Fulton RS, Magrini V, Demeter R, Wylie T, Kandoth C, Leonard JR, Guha A, Miller CA, Ding L, Mardis ER (2013) Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res 23:431–439
Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, Alon N, Kahn D, Fried I, Scheinemann K, Tsangaris E, Dirks P, Tressler R, Bouffet E, Jabado N, Tabori U (2011) BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 17:4790–4798
Hegedus B, Banerjee D, Yeh TH, Rothermich S, Perry A, Rubin JB, Garbow JR, Gutmann DH (2008) Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res 68:1520–1528
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, Gervais R, Elez-Fernandez ME, Italiano A, Hofheinz RD, Hidalgo M, Chan E, Schuler M, Lasserre SF, Makrutzki M, Sirzen F, Veronese ML, Tabernero J, Baselga J (2015) Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373:726–736
Jacob K, Quang-Khuong DA, Jones DT, Witt H, Lambert S, Albrecht S, Witt O, Vezina C, Shirinian M, Faury D, Garami M, Hauser P, Klekner A, Bognar L, Farmer JP, Montes JL, Atkinson J, Hawkins C, Korshunov A, Collins VP, Pfister SM, Tabori U, Jabado N (2011) Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res 17:4650–4660
Jain P, Silva A, Han HJ, Lang SS, Zhu Y, Boucher K, Smith TE, Vakil A, Diviney P, Choudhari N, Raman P, Busch CM, Delaney T, Yang X, Olow AK, Mueller S, Haas-Kogan D, Fox E, Storm PB, Resnick AC, Waanders AJ (2017) Overcoming resistance to single-agent therapy for oncogenic BRAF gene fusions via combinatorial targeting of MAPK and PI3K/mTOR signaling pathways. Oncotarget 8:84697–84713
Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, Daniel S, Covert M, Frampton GM, Hsu S, Lesser GJ, Stogner-Underwood K, Mott RT, Rush SZ, Stanke JJ, Dahiya S, Sun J, Reddy P, Chalmers ZR, Erlich R, Chudnovsky Y, Fabrizio D, Schrock AB, Ali S, Miller V, Stephens PJ, Ross J, Crawford JR, Ramkissoon SH (2017) Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 22:1478–1490
Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, Fontebasso AM, Stutz AM, Hutter S, Zuckermann M, Sturm D, Gronych J, Lasitschka B, Schmidt S, Seker-Cin H, Witt H, Sultan M, Ralser M, Northcott PA, Hovestadt V, Bender S, Pfaff E, Stark S, Faury D, Schwartzentruber J, Majewski J, Weber UD, Zapatka M, Raeder B, Schlesner M, Worth CL, Bartholomae CC, von Kalle C, Imbusch CD, Radomski S, Lawerenz C, van Sluis P, Koster J, Volckmann R, Versteeg R, Lehrach H, Monoranu C, Winkler B, Unterberg A, Herold-Mende C, Milde T, Kulozik AE, Ebinger M, Schuhmann MU, Cho YJ, Pomeroy SL, von Deimling A, Witt O, Taylor MD, Wolf S, Karajannis MA, Eberhart CG, Scheurlen W, Hasselblatt M, Ligon KL, Kieran MW, Korbel JO, Yaspo ML, Brors B, Felsberg J, Reifenberger G, Collins VP, Jabado N, Eils R, Lichter P, Pfister SM, International Cancer Genome Consortium PedBrain Tumor PedBrain Tumor P (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927–932
Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677
Jones DTW, Witt O, Pfister SM (2018) BRAF V600E status alone is not sufficient as a prognostic biomarker in pediatric low-grade glioma. J Clin Oncol 36:96
Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, Keedy V, Bielle F, Hofheinz RD, Joly F, Blay JY, Chau I, Puzanov I, Raje NS, Wolf J, DeAngelis LM, Makrutzki M, Riehl T, Pitcher B, Baselga J, Hyman DM (2018) BRAF inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET study. J Clin Oncol 36:3477 (JCO2018789990)
Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, Harter DH, Goldberg JD, Hochman T, Merkelson A, Bloom MC, Sievert AJ, Resnick AC, Dhall G, Jones DT, Korshunov A, Pfister SM, Eberhart CG, Zagzag D, Allen JC (2014) Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neurooncology 16:1408–1416
Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH (2012) Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev 26:2561–2566
Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH (2015) Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neurooncology 17:843–853
Kheder ES, Hong DS (2018) Emerging targeted therapy for tumors with NTRK fusion proteins. Clin Cancer Res 24:5807–5814
Koenig MK, Butler IJ, Northrup H (2008) Regression of subependymal giant cell astrocytoma with rapamycin in tuberous sclerosis complex. J Child Neurol 23:1238–1239
Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, Carol H, Lock R, Phelps D, Kang MH, Reynolds CP, Maris JM, Billups C, Smith MA (2010) Initial testing (stage 1) of AZD6244 (ARRY-142886) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 55:668–677
Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN (2010) Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 363:1801–1811
Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, Guerreiro Stucklin A, Zhukova N, Arnoldo A, Ryall S, Ling C, McKeown T, Loukides J, Cruz O, de Torres C, Ho CY, Packer RJ, Tatevossian R, Qaddoumi I, Harreld JH, Dalton JD, Mulcahy-Levy J, Foreman N, Karajannis MA, Wang S, Snuderl M, Nageswara Rao A, Giannini C, Kieran M, Ligon KL, Garre ML, Nozza P, Mascelli S, Raso A, Mueller S, Nicolaides T, Silva K, Perbet R, Vasiljevic A, Faure Conter C, Frappaz D, Leary S, Crane C, Chan A, Ng HK, Shi ZF, Mao Y, Finch E, Eisenstat D, Wilson B, Carret AS, Hauser P, Sumerauer D, Krskova L, Larouche V, Fleming A, Zelcer S, Jabado N, Rutka JT, Dirks P, Taylor MD, Chen S, Bartels U, Huang A, Ellison DW, Bouffet E, Hawkins C, Tabori U (2017) Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol 35:2934–2941
Lauchle JO, Kim D, Le DT, Akagi K, Crone M, Krisman K, Warner K, Bonifas JM, Li Q, Coakley KM, Diaz-Flores E, Gorman M, Przybranowski S, Tran M, Kogan SC, Roose JP, Copeland NG, Jenkins NA, Parada L, Wolff L, Sebolt-Leopold J, Shannon K (2009) Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature 461:411–414
Lee D, Cho YH, Kang SY, Yoon N, Sung CO, Suh YL (2015) BRAF V600E mutations are frequent in dysembryoplastic neuroepithelial tumors and subependymal giant cell astrocytomas. J Surg Oncol 111:359–364
Lee EQ, Ruland S, LeBoeuf NR, Wen PY, Santagata S (2016) Successful treatment of a progressive BRAF V600E-mutated anaplastic pleomorphic xanthoastrocytoma with vemurafenib monotherapy. J Clin Oncol 34:e87–e89
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Casey M, Ouellet D, Martin AM, Le N, Patel K, Flaherty K (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371:1877–1888
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion-Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Swann S, Legos JJ, Jin F, Mookerjee B, Flaherty K (2015) Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386:444–451
Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, Stavropoulos J, Alon N, Pole JD, Ray PN, Navickiene V, Mangerel J, Remke M, Buczkowicz P, Ramaswamy V, Guerreiro Stucklin A, Li M, Young EJ, Zhang C, Castelo-Branco P, Bakry D, Laughlin S, Shlien A, Chan J, Ligon KL, Rutka JT, Dirks PB, Taylor MD, Greenberg M, Malkin D, Huang A, Bouffet E, Hawkins CE, Tabori U (2015) BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33:1015–1022
Mizuguchi M, Takashima S (2001) Neuropathology of tuberous sclerosis. Brain Dev 23:508–515
Nakano Y, Tomiyama A, Kohno T, Yoshida A, Yamasaki K, Ozawa T, Fukuoka K, Fukushima H, Inoue T, Hara J, Sakamoto H, Ichimura K (2019) Identification of a novel KLC1-ROS1 fusion in a case of pediatric low-grade localized glioma. Brain Tumor Pathol 36:14–19
Nicolaides TP, Li H, Solomon DA, Hariono S, Hashizume R, Barkovich K, Baker SJ, Paugh BS, Jones C, Forshew T, Hindley GF, Hodgson JG, Kim JS, Rowitch DH, Weiss WA, Waldman TA, James CD (2011) Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res 17:7595–7604
Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R (2018) Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol 1:1–20
Olow A, Mueller S, Yang X, Hashizume R, Meyerowitz J, Weiss W, Resnick AC, Waanders AJ, Stalpers LJ, Berger MS, Gupta N, James CD, Petritsch CK, Haas-Kogan DA (2016) BRAF Status in personalizing treatment approaches for pediatric gliomas. Clin Cancer Res 22:5312–5321
Olsen TK, Panagopoulos I, Meling TR, Micci F, Gorunova L, Thorsen J, Due-Tonnessen B, Scheie D, Lund-Iversen M, Krossnes B, Saxhaug C, Heim S, Brandal P (2015) Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neurooncology 17:1365–1373
Packer RJ, Pfister S, Bouffet E, Avery R, Bandopadhayay P, Bornhorst M, Bowers DC, Ellison D, Fangusaro J, Foreman N, Fouladi M, Gajjar A, Haas-Kogan D, Hawkins C, Ho CY, Hwang E, Jabado N, Kilburn LB, Lassaletta A, Ligon KL, Massimino M, Meeteren SV, Mueller S, Nicolaides T, Perilongo G, Tabori U, Vezina G, Warren K, Witt O, Zhu Y, Jones DT, Kieran M (2017) Pediatric low-grade gliomas: implications of the biologic era. Neurooncology 19:750–761
Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, Messina JL, Flaherty KT, Smalley KS (2010) Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 102:1724–1730
Pekmezci M, Villanueva-Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP, Bastian BC, Chamyan G, Maher OM, Khatib Z, Kleinschmidt-DeMasters BK, Samuel D, Mueller S, Banerjee A, Clarke JL, Cooney T, Torkildson J, Gupta N, Theodosopoulos P, Chang EF, Berger M, Bollen AW, Perry A, Tihan T, Solomon DA (2018) The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6:47
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, Tang B, Haupfear K, Punchihewa C, Easton J, Mulder H, Boggs K, Shao Y, Rusch M, Becksfort J, Gupta P, Wang S, Lee RP, Brat D, Peter Collins V, Dahiya S, George D, Konomos W, Kurian KM, McFadden K, Serafini LN, Nickols H, Perry A, Shurtleff S, Gajjar A, Boop FA, Klimo PD Jr, Mardis ER, Wilson RK, Baker SJ, Zhang J, Wu G, Downing JR, Tatevossian RG, Ellison DW (2016) Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131:833–845
Ramkissoon LA, Horowitz PM, Craig JM, Ramkissoon SH, Rich BE, Schumacher SE, McKenna A, Lawrence MS, Bergthold G, Brastianos PK, Tabak B, Ducar MD, Van Hummelen P, MacConaill LE, Pouissant-Young T, Cho YJ, Taha H, Mahmoud M, Bowers DC, Margraf L, Tabori U, Hawkins C, Packer RJ, Hill DA, Pomeroy SL, Eberhart CG, Dunn IF, Goumnerova L, Getz G, Chan JA, Santagata S, Hahn WC, Stiles CD, Ligon AH, Kieran MW, Beroukhim R, Ligon KL (2013) Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci USA 110:8188–8193
Ratner N, Miller SJ (2015) A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer 15:290–301
Ricker CA, Pan Y, Gutmann DH, Keller C (2016) Challenges in drug discovery for neurofibromatosis type 1-associated low-grade glioma. Front Oncol 6:259
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372:30–39
Robinson JP, VanBrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL (2010) Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene 29:335–344
Rush S, Foreman N, Liu A (2013) Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol 31:e159–e160
Ryall S, Tabori U, Hawkins C (2017) A comprehensive review of paediatric low-grade diffuse glioma: pathology, molecular genetics and treatment. Brain Tumor Pathol 34:51–61
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathologica 121:397–405
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M, Louis DN, Korshunov A, Pfister S, Hartmann C, Paulus W, Reifenberger G, von Deimling A (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405
Schreck KC, Guajardo A, Lin DDM, Eberhart CG, Grossman SA (2018) Concurrent BRAF/MEK inhibitors in BRAF V600-mutant high-grade primary brain tumors. J Natl Compr Cancer Netw 16:343–347
See WL, Tan IL, Mukherjee J, Nicolaides T, Pieper RO (2012) Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res 72:3350–3359
Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, Kellet M, Storm PB, Resnick AC (2013) Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA 110:5957–5962
Solit DB, Rosen N (2011) Resistance to BRAF inhibition in melanomas. N Engl J Med 364:772–774
Sturm D, Pfister SM, Jones DTW (2017) Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. J Clin Oncol 35:2370–2377
Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, Cibulskis K, Farlow D, Zimmer L, Hillen U, Gutzmer R, Goldinger SM, Ugurel S, Gogas HJ, Egberts F, Berking C, Trefzer U, Loquai C, Weide B, Hassel JC, Gabriel SB, Carter SL, Getz G, Garraway LA, Schadendorf D, Dermatologic Cooperative Oncology Group of G (2014) The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 4:94–109
Weber R, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B, Becker A, Weber-Mangal S, Jauch A, Radlwimmer B (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14 ARF/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26:1088
Yalon M, Rood B, MacDonald TJ, McCowage G, Kane R, Constantini S, Packer RJ (2013) A feasibility and efficacy study of rapamycin and erlotinib for recurrent pediatric low-grade glioma (LGG). Pediatr Blood Cancer 60:71–76
Yao TW, Zhang J, Prados M, Weiss WA, James CD, Nicolaides T (2015) EGFR blockade prevents glioma escape from BRAFV600E targeted therapy. Oncotarget 6:21993–22005
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45:602
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, Boop FA, Lu C, Kandoth C, Ding L, Lee R, Huether R, Chen X, Hedlund E, Nagahawatte P, Rusch M, Boggs K, Cheng J, Becksfort J, Ma J, Song G, Li Y, Wei L, Wang J, Shurtleff S, Easton J, Zhao D, Fulton RS, Fulton LL, Dooling DJ, Vadodaria B, Mulder HL, Tang C, Ochoa K, Mullighan CG, Gajjar A, Kriwacki R, Sheer D, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Baker SJ, Ellison DW, St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome P (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45:602–612
Zhang J, Yao TW, Hashizume R, Hariono S, Barkovich KJ, Fan QW, Prados M, James CD, Weiss WA, Nicolaides T (2017) Combined BRAF(V600E) and MEK blockade for BRAF(V600E)-mutant gliomas. J Neurooncol 131:495–505
Ziegler DS, Wong M, Mayoh C, Kumar A, Tsoli M, Mould E, Tyrrell V, Khuong-Quang DA, Pinese M, Gayevskiy V, Cohn RJ, Lau LMS, Reynolds M, Cox MC, Gifford A, Rodriguez M, Cowley MJ, Ekert PG, Marshall GM, Haber M (2018) Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br J Cancer 119:693–696
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tateishi, K., Nakamura, T. & Yamamoto, T. Molecular genetics and therapeutic targets of pediatric low-grade gliomas. Brain Tumor Pathol 36, 74–83 (2019). https://doi.org/10.1007/s10014-019-00340-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10014-019-00340-3