
Abstract
Somatic mutations of isocitrate dehydrogenase 1 and 2 (IDH1/2) are strongly associated with pathological subtypes, genetic profiles, and clinical features in gliomas. The IDH1/2 status is currently regarded as one of the most important molecular markers in gliomas and should be assessed accurately and robustly. However, the methods used for IDH1/2 testing are not fully standardized. The purpose of this paper is to review the clinical significance of IDH1/2 mutations and the methods used for IDH1/2 testing. The optimal method for IDH1/2 testing varies depending on a number of factors, including the purpose, sample types, sample number, or laboratory equipment. It is therefore important to acknowledge the advantages and disadvantages of each method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Somatic mutations of isocitrate dehydrogenase 1 (IDH1, 2q34) were first identified in a small subset of glioblastomas [1, 2]. A large number of studies soon followed, reporting that IDH1 mutations actually occur in the majority of lower grade gliomas [2–6]. Later, mutations of isocitrate dehydrogenase 2 (IDH2, 15q26) were also reported in a small number of gliomas [3, 4]. IDH1/2 mutations are currently regarded as the earliest event in gliomagenesis and one of the most significant genetic alterations in glioma biology [4]. IDH1/2 mutations also seem to be clinically significant based on their relationship with pathology, other genetic changes, and clinical presentation. A rapidly growing number of published studies constantly provide new information about IDH1/2, and IDH1/2 status is almost routinely evaluated in laboratories. However, the methods for IDH1/2 testing are not fully standardized. The purpose of this report is to review the clinical significance of IDH1/2 mutation and the methods used for testing these mutations.
Genetic aspect of IDH1/2 mutations
Mutational pattern of IDH1/2 in gliomas (Fig. 1)
All reported IDH1/2 mutations are missense mutations. They are almost always hemizygous and mutually exclusive to each other. IDH1 and IDH2 are homologous enzymes localized in the cytosol and mitochondria, respectively. IDH1/2 catalyze the conversion of isocitrate to α-ketoglutarate using NADP+ as a cofactor. The IDH1/2 mutations in gliomas result in the reduced ability to produce α-ketoglutarate and the acquisition of abnormal function to convert α-ketoglutarate into (R)-2-hydroxyglutarate ((R)-2HG) by using NADPH as a cofactor [7, 8]. (R)-2HG is considered to be a major oncometabolite, causing various biological effects on IDH1/2 mutated tumors. IDH1/2 mutations are therefore considered to be gain-of-function alterations [7].

a Frequency of each mutation in IDH1/2. The ratio was calculated from the pooled data from eight independent studies, including the data on a total of 3,029 glioma cases [3, 6, 9–14]. The left circular chart represents the frequency of IDH1 R132H mutation and non-R132H IDH1/2 mutations. The right chart represents the frequency of each type of non-R132H IDH1/2 mutation. b The nucleotide transitions in IDH1/2. All the mutations except R132V are point mutations
The frequency of each mutation type varies according to the reports. The frequency of each mutation in a pooled data from 8 independent studies, describing the mutation types are shown in Fig. 1a [3, 6, 9–14]. The most common IDH1/2 mutation in gliomas is c.395G>A transition in IDH1, which replaces the arginine with a histidine at codon 132 (R132H) (Fig. 1b). Other less common mutations also occur at codon 132 in IDH1 or codon 172 in IDH2. The second most frequent mutations are R132C in IDH1 and R172K in IDH2 (2.8 % of all mutations, respectively). Other mutations include R132S, R132G, and R132L in IDH1 and R172M, R172W, and R172S in IDH2. Extremely rare mutations include R132P, R132V, or those affecting R49, G97, and R100 in IDH1 and R172T in IDH2 ([15, 16] and references therein). The mutations affecting R140 in IDH2 are among the most common mutations in myeloproliferative neoplasms; however, these are not observed in gliomas [8, 17].
Thus, the great majority of IDH1/2 mutations involve codon 132 in IDH1 or codon 172 in IDH2, and approximately 90 % of them are R132H in IDH1.
The relationship with other genetic alterations
IDH1/2 mutations show a distinct pattern with other genetic alteration characteristics in diffuse gliomas.
Genetic changes
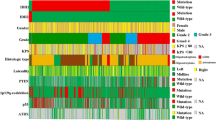
IDH1/2 mutations are frequently observed in grade II–III gliomas, as described above, and are mostly associated with either TP53 mutations or total 1p19q loss [4] (Fig. 2). TP53 mutations are typically observed in grade II–III astrocytomas with IDH1/2 mutations, and ATRX mutations are commonly observed in these populations [18]. Total 1p19q loss almost always coexists with IDH1/2 mutations and TERT promoter mutations. This combination is typically observed in oligodendroglial tumors [9, 19]. TP53 mutations and total 1p19q loss are mutually exclusive [4]. Similarly, ATRX and TERT promoter mutations also show mutually exclusive patterns [12, 19]. Thus, the combination of TP53-ATRX mutations or total 1p19q loss-TERT mutations is the hallmark of astrocytic or oligodendroglial tumors harboring IDH1/2 mutations, respectively.

Relationships among IDH1/2 mutations, other genetic alterations, and histological subtypes. Each column represents individual tumors. IDH1/2 mutations are strongly associated with TP53 mutation or total 1p19q loss. Most IDH1/2 mutated tumors show MGMT methylation. The data for the cases from National Cancer Center (Tokyo, Japan) are extracted and modified from the study by Arita et al. [9]. DA diffuse astrocytoma, AA anaplastic astrocytoma, GBM primary glioblastoma, sGBM secondary glioblastoma, OL oligodendroglioma, AO anaplastic astrocytoma, OA oligoastrocytoma, AOA anaplastic astrocytoma
Epigenetic changes
Another important feature in IDH1/2 mutated tumors is the epigenetic changes, which are some of the most fundamental alterations induced by these mutations. IDH1/2 mutations have strongly been associated with the glioma-CpG island methylator phenotype (G-CIMP) and aberrant histone methylation [20, 21]. The mechanisms involved in the changes in DNA methylation and histone methylation status caused by IDH1/2 mutations have been extensively discussed in several reviews [16, 22]. Thus, we only briefly summarized them in this review.
G-CIMP
G-CIMP was identified as a phenomenon in which DNA methylation in the CpG islands is increased genome-wide in a subset of gliomas [20]. It is well documented that G-CIMP is tightly associated with the presence of IDH1/2 mutations and relative absence of typical copy-number alterations normally observed in glioblastomas, including EGFR amplification and chromosomal arm 10q loss. The tumors with G-CIMP present gene expression profiles of the proneural type [20]. DNA hypermethylation in these loci is considered to downregulate the expression of target genes, some of which may act as a tumor suppressor.
Aberrant (R)-2HG production in IDH1/2 mutated tumors may inhibit α-ketoglutarate-dependent dioxygenase family, including ten–eleven-translocation (TET) [23]. The TET family catalyzes the conversion of 5-methylcytosine (5mC) to 5-hydroxymethycytosine (5hmC), which is a critical step in demethylating methylcytosine in the CpG dinucleotides, the main target of DNA methylation [24]. Inhibition of TET2 by (R)-2HG may possibly contribute to global DNA methylation in G-CIMP [21].
Histone modification
In IDH1/2 mutated tumors, trimethylation at lysine residues of histone H3, including H3K9, H3K27, and H3K79 are increased [23]. Histone methylation is regulated by histone methyltransferase and demethylases and may affect gene transcription by altering chromatin structures. A Fe(II) and α-ketoglutarate-dependent subset of histone demethylase (e.g., lysine (K)-specific demethylase 6A (KDM6A)) is inhibited by (R)-2HG in IDH1/2 mutated tumors, resulting in global alterations of histone demethylation and gene expression [23].
MGMT
CpG island methylation in O6-methylguanine-DNA methyltransferase (MGMT) has also been associated with IDH1/2 mutations [25, 26]. MGMT is a DNA-repair protein that removes alkyl adducts from O6 position of guanine. Increased activity of MGMT reduces the chemosensitivity of alkylating agents, including temozolomide because O6 position of guanine is the main target of DNA alkylation. MGMT methylation results in reduced MGMT expression and may lead to better response to temozolomide in glioblastoma [27]. MGMT methylation is invariably observed in IDH1/2 mutated tumors [26]. On the other hand, about half of glioblastomas harbor MGMT methylation, regardless of the IDH1/2 status (Fig. 2).
Clinical value
The relationship with pathology
In the pooled data from the eight reports (see above), IDH1/2 mutations were observed frequently in grade II–III astrocytomas and oligodendrogliomas (53–83 %) as well as in secondary glioblastomas (54 %), but rarely in primary glioblastomas (6.3 %). IDH1/2 mutations are highly specific to diffuse gliomas among CNS tumors. Other neuroepithelial tumors, including pilocytic astrocytoma, ependymoma, and ganglioglioma, rarely harbor IDH1/2 mutations [15]. Non-neoplastic lesions mimicking gliomas never present these mutations [28]. The presence of IDH1/2 mutations is a strong evidence of diffuse gliomas even in such cases (discussed further below).
Prognostic value
Several studies have suggested that patients with IDH1/2 mutated tumor show longer survival than those with IDH1/2 wild-type tumor, in most entities of gliomas [29].
The favorable prognosis of IDH1/2 mutated glioblastomas was first reported in the pioneering study about IDH1 [1], which was then followed by several other studies [3, 4, 14, 25, 30, 31]. IDH1/2 mutations are generally regarded as a positive prognostic factor in glioblastomas. The overall survival in IDH1/2 mutated cases is about twice longer than that of IDH1/2 wild-type cases (24–31 vs. 9.9–15 months) [3, 25, 30]. Using a multivariate analysis, some studies have shown that IDH1/2 status is an independent prognostic factor in glioblastomas [25], while others failed to reproduce this finding [31]. The small population of IDH1/2 mutated glioblastomas might cause this controversy.
Several studies have reported that IDH1/2 status is also a prognostic factor in grade III gliomas [14, 25, 32]. Some reports even demonstrated the positive prognostic value of IDH1/2 mutations in each subtype: anaplastic astrocytomas [3] or anaplastic oligodendroglial tumors [33].
The prognostic value of IDH1/2 status in grade II gliomas remains under debate. Some reports associated the presence of IDH1/2 mutations with better prognosis [25, 34–37], while others did not [14, 38]. Sun et al. [39] investigated the prognostic value of IDH1/2 status through a meta-analysis of ten previous studies and found that IDH1/2 mutation was associated with longer survival in grade II gliomas. They pointed out several problems in the interpretation of currently available data, which include mixed cohorts of astrocytomas and oligodendroglial tumors, the close relationship with other prognostic factors, different methodology to evaluate IDH1/2 status, and most importantly the lack of standard treatment in grade II gliomas. There are also other studies investigating the prognostic value in each subtype (i.e., astrocytomas or oligodendrogliomas); however, they remain inconclusive [14, 34, 35].
Overall, the independent value of IDH1/2 as a biomarker remains somewhat controversial. This can be attributed to confounding factors or the study design as pointed out by Sun et al. [39] in their analysis of grade II gliomas. IDH1/2 mutations are closely related to other prognostic/predictive factors, including patient age, MGMT status, or 1p19q copy number. Limited cohort size, different treatment, and the heterogeneity of tumor subtypes in each study may also result in conflicting results. Nonetheless, the IDH1/2 status adds valuable information to the WHO grades in predicting the clinical course, and it should be considered as a stratification factor in clinical trials of gliomas [40].
Molecular classification and IDH1/2
One of the aims of the current WHO classification is to predict the clinical outcome of the patients harboring the tumor [41]. Nonetheless, the current diagnostic system poses the limitation that one type of tumor can include biologically and clinically different subsets of tumors. The molecular classification is expected to refine the current diagnostic system [42]. IDH1/2 mutations present a strong association with the histological types and clinical outcome as described above. Therefore, these mutations are among the most promising markers.
The existence of IDH1/2 mutations strongly supports the diagnosis of grade II–III gliomas. Intriguingly, Hartmann and colleagues reported that patients with IDH1 wild-type anaplastic astrocytomas exhibited shorter survival than those with IDH1-mutated glioblastomas [43]. Underestimation of the tumor grades in histological diagnosis may lead to this observation along with the prognostic impact of the IDH1 mutation itself. Malignant gliomas are histologically heterogeneous, and missampling can lead to undergrading [43]. Their findings argue for the significant value of IDH1/2 mutation in the molecular classification combined with the current histological classification.
Combination of IDH1/2 and other genetic status may aid in further predicting the subtypes of gliomas, because IDH1/2 mutations show strong relationships with either the combination of TP53-ATRX mutations or total 1p19q loss-TERT mutation in astrocytic or oligodendroglial tumors, as described above.
The molecular diagnosis seems promising; however, it poses some significant limitations. The evaluation for molecular markers often needs DNA analysis, which requires expensive equipment and reduces the feasibility of the method in clinical use. Another issue is that standardization of the testing for each marker is needed. For example, MGMT methylation status is evaluated using various methods, including methylation-specific PCR (MSP) or pyrosequencing. However, these tests have not yet been standardized. Availability of molecular markers requires the accessibility of detection methods and their standardization.
IDH1/2 detection
IDH1/2 mutations undoubtedly divide diffuse gliomas into two groups, which have distinct biological and clinical features, as described above. IDH1/2 mutations also have a significant impact on glioma diagnosis. Their highly specific distribution in grade II–III diffuse gliomas among CNS tumors also indicates that the presence of IDH1/2 mutation is almost sufficient for the diagnosis of diffuse glioma, although the absence of these mutations does not exclude the diagnosis of gliomas [28]. Equivocal microscopic diagnosis can be derived from various factors, including small sample size (i.e., obtained by needle biopsy), sampling site (i.e., from infiltrative zone apart from the tumor core), or sample quality. The IDH1/2 status may provide clinically important information in such cases. IDH1/2 status therefore needs to be evaluated accurately.
IDH1/2 testing includes various methods targeting DNA sequence, mutant protein, or aberrant increase in (R)-2HG levels. However, IDH1/2 testing should be easy to be incorporated into daily diagnostic practice. Sanger sequencing and immunohistochemistry (IHC) are conventionally applied for the assessment of IDH1/2 status. Each assay has advantages and limitations (Table 1). Some of the currently available methods for IDH1/2 testing are reviewed below.
Sanger sequencing
Sanger sequencing is the gold standard for detecting IDH1/2 mutations, and most of the published data are based on this method [1–3]. This technique can detect all types of IDH1/2 mutations and, if the mutation is detected, the result is reliable.
However, this technique requires sophisticated equipment and trained personnel [44]. Complicated procedures including DNA extraction, polymerase chain reaction (PCR), or purification of PCR products need to be optimized. The difference in the equipment or procedure may yield inconsistent results between laboratories [45].
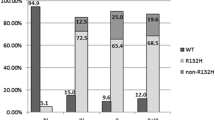
Another caveat is that the results of this method largely depend on the tumor cell content of the samples. The source of non-neoplastic DNA includes adjacent normal brain, infiltrating lymphocytes, and microglia or endothelial cells, which may dilute mutant alleles and cause false-negative results [15]. At least 20 % of the mutant allele is required for detection by Sanger sequencing in our analysis, evaluating the sensitivity of IDH1/2 detection [46] (Fig. 3a). Diagnosis for small samples obtained from tumor margin is challenging, but clinically important. The critical limitation of this method is its relatively low sensitivity, which might lead to missing out IDH1/2 mutations.

a Sanger sequencing. Chromatograms of Sanger sequencing for IDH1 in representative cases. The three cases are all anaplastic astrocytoma cases. Case 1 presents a wild-type IDH1, while cases 2 and 3 present the R132H mutant of IDH1. In case 3, a peak of adenine (green) indicates the c.395G>A transition. In case 2, a peak indicating the mutation is too low to be distinguished from other nonspecific peaks. b Immunohistochemistry. Immunohistochemical staining using anti-IDH1 R132H mutant antibody. The cytoplasm of tumor cells with IDH R132H is strongly stained, while endothelial cells are not stained. (Original magnification ×200). c Pyosequencing. Pyrograms for IDH1 in representative cases. The arrows indicate the specific peaks for the R132H mutants. The cases and analyzed samples are identical to those used for Sanger sequencing. The quantitative analysis of pyrosequencing reported that the frequency of R132H mutant alleles in each case was 0, 16, and 45 %, respectively. In case 2, the R132H mutation is apparent in pyrograms, although the result of Sanger sequencing is inconclusive
Immunohistochemistry (IHC)
IHC is universally performed in clinical practice, and the IHC-based mutation detection is one of the most accessible technologies.
The specific antibodies for IDH1 R132H mutation, monoclonal antibody (mAb) H09 and Imab-1, are well characterized and commonly used [47, 48]. The cytoplasm of tumor cells with IDH1 R132H mutation is strongly stained, while tumor cells without this mutation are not stained (Fig. 3b). These antibodies are highly specific to the mutant protein, and residual brain tissues, including reactive glia, endothelial cells, or blood cells are not stained [47–49]. A weak diffuse background staining and a strong granular cytoplasmic staining of macrophages are also observed in IDH1 wild-type tumors. Meningiomas and schwannomas can show nonspecific positive-stained fibers. These patterns can be easily recognized and distinguished from the true-positive staining [49]. Based on these criteria, the specificity for IDH1 R132H mutation is considered to be nearly 100 % [50]. A cross-reactivity for R132L mutant has been reported [44]; however, this would not cause misdiagnosis.
This method is highly accurate. It can detect tumor cells with IDH1/2 mutation in even tissues containing 6–9 % mutant allele [46, 51]. It has been claimed that the antibody can stain single cells reliably even in the infiltration zone and that it enables differentiation of tumor cells from reactive glia [50]. The results of IHC using IDH1 R132H-specific antibodies are consistent across laboratories, even though different staining procedures were used [45].
The obvious limitation is that these antibodies cannot detect non-R132H mutations in IDH1/2 that correspond to approximately 10 % of all the IDH1/2 mutations [50]. Antibodies specific for other IDH1/2 mutations than R132H have also been developed and some of them are commercially available for IHC [52, 53]. A multispecific anti-mutated IDH1/2 antibody recognizing a subset of IDH1 and IDH2 mutations has also been reported [54]. The reactivity of this antibody varies depending on the methodology used, IHC, enzyme-linked immunosorbent assay (ELISA), and/or Western blotting. These approaches, especially the use of the multispecific anti-mutated IDH1/2 antibody, need to be further validated for clinical application.
Pyrosequencing
The principle of this method is a sequencing-by-synthesis analysis based on the real-time detection of nucleotide incorporation by DNA polymerase [55]. In practice, a DNA template amplified by PCR is hybridized to a primer for pyrosequencing after purification, followed by pyrosequencing reactions using a pyrosequencer. Pyrosequencing reactions consist of four reactions: a DNA polymerase reaction, a sulfyrase reaction, a luciferase reaction, and nucleotide degradation by apyrase. Pyrosequencing reagents include the enzymes and the substrates for these reactions as well as adenosine 5′ phosphosulfate (APS) and luciferin (the four enzyme system) [56]. After adding the substrates and enzymes to the templates, each deoxynucleotide triphosphate (dNTP) is added to the samples stepwise, as programmed by the operator (dispensation order). If the injected dNTP is complementary to the template, the dNTP is incorporated by the DNA polymerase and the pyrophosphate is released. The concentration of ATP is then increased through the conversion of pyrophosphate and APS to ATP by the ATP sulfyrase, followed by the luciferase reaction. The light emitted by the luciferase reaction is quantitatively detected by a charge-coupled device (CCD) camera and represented as a peak at each nucleotide dispensation in the pyrogram. The unincorporated dNTP is rapidly degraded by the apyrase. The signal strength of the pyrosequencing reaction is proportional to the amount of pyrophosphate released and dNTP incorporated; hence the allele dosage. Therefore, the signal strength can be decreased when different sequences are mixed (i.e., heterozygous mutations) and also proportionally increased in a sequence containing homopolymers (a continuous stretch of the same nucleotide such as TT or CCC). The allele frequencies contained in the sample are automatically calculated from the signal strength by using a software developed for pyrosequencing analysis [56].
Several studies have applied pyrosequencing for IDH1 testing [46, 57–59], some of which have validated the advantages of this technique over Sanger sequencing.
This method is highly sensitive. The minimum detectable frequency of the mutant allele is 5–7 % for pyrosequencing [57, 58], while Sanger sequencing sensitivity is at least 20 % for reliable detection (Fig. 3a, c) [15, 46]. Fragmented DNA template from the formalin-fixed paraffin-embedded (FFPE) specimen can be utilized for analysis [57, 59], as pyrosequencing allows the use of DNA templates under 100 bp [46].
Pyrosequencing requires expensive equipment and is available only in limited centers and laboratories. The robustness of pyrosequencing depends on the assay design, including the primers and the dispensation order [46].
Melting curve analysis
Melting curve analysis is a technique that allows the generation of a melting temperature profile of the double-stranded DNA. The melting temperature is unique to each nucleotide sequence, and even a single nucleotide substitution could alter the melting temperature. Thus, a missense mutation can be accurately detected by measuring the melting curve. Two different variants of this method were reported as an application for IDH1/2 testing: fluorescent melting curve analysis (FMCA) and high-resolution DNA melting (HRM) [60–62].
In studies using FMCA, the PCR product of the target sequence is hybridized with a pair of fluorescent probes designed to complement the sequence, including mutation hotspot or its adjacent sequence [61, 62]. A real-time PCR system detects the fluorescent change derived by the denaturation of the probes during the gradual heating. In a mutant sequence, lower temperature is required for denaturation, unlike that in a wild-type sequence, because the probe imperfectly binds the mutant sequence. These differences appear in the patterns of the melting curve. A sample with only wild-type alleles shows a single peak in the melting curve, while a sample containing a mutant allele shows an additional peak at lower temperature [62]. This technique is also reported to be highly sensitive and rapid in detecting IDH1/2 status even in FFPE tissue samples; the entire duration of this assay is about 80 min, and the minimum amount of mutated allele for the detection is 10 % [62].
A fluorescent dye intercalating double-stranded DNA is used in HRM. The amplified template with a saturating fluorescent dye is first denatured by heating, and then annealing is performed at the lower temperature. The fluorescence of the double-stranded DNA reduces by the gradual heating in a melting curve analysis. The samples containing mutant sequences show different melting curves because of the formation of heteroduplexed DNA after the denaturation phase; a lower melting temperature is observed [63]. The detection limit of this assay after conventional PCR amplification is similar to that of Sanger sequencing (25 %) [60].
These methods detect all types of mutations by detecting the melting temperature specific to each mutation [60, 62]. The disadvantage is an unsatisfactory sensitivity, but the sensitivity can be improved by combining co-amplification at lower denaturation temperature PCR (COLD-PCR) (reviewed below). COLD-PCR HRM and FMCA assays allowed the detection of 2 or 1 % mutant allele, respectively [60, 61]. Another disadvantage is that these methods require expensive equipment, which is usually used for another purpose in clinical settings [62].
Co-amplification at lower denaturation temperature PCR (COLD-PCR)
COLD-PCR is a method used to amplify a specific allele with mutation selectively. COLD-PCR itself is not an independent technique for DNA analysis, and this technique is combined with another method. This method increases the sensitivity to detect a mutant allele contained in a sample [64].
The novelty of COLD-PCR is the use of critical denaturation temperature (Tc), which is lower than the standard denaturation temperature. At the Tc, the mismatched DNA formed by mutant and wild-type sequences is denatured, while the homo-duplex DNA of the mutant or wild-type sequences remains double stranded. Primer annealing and DNA extension by DNA polymerase follow the denaturation at the Tc in this procedure. Since the mismatched DNA formed by the mutant and wild-type sequences is selectively denatured, mutant sequences at low concentration are selectively amplified. This method improves the sensitivity of Sanger sequencing, pyrosequencing, or melting curve analysis [64]. Some studies have reported highly sensitive assays for IDH1 mutations by combining COLD-PCR with melting curve analysis [60, 61].
Molecular imaging
Another approach to detect IDH1/2 mutations is molecular imaging. The accumulated (R)-2HG in IDH1/2 mutated tumors has been considered as a good target for this approach, because this aberrant metabolite is only present at low levels in the normal tissue [65]. Recent studies have shown successful in vivo detection of (R)-2HG using magnetic resonance spectroscopy (MRS) [65–67]. The challenging point of this approach in vivo is that the (R)-2HG spectrum overlaps with that of other metabolites, including glutamate, glutamine, N-acetyl-l-aspartate (NAA), or gamma-aminobutyric acid (GABA) [65–67]. These studies have claimed that they overcame this by different MRS sequence optimized in each study (reviewed elsewhere [65]). The signal of (R)-2HG is present in background noises and further improvement will be needed for the robust detection in clinical use. These preliminary reports necessitate further MRS validation and standardization in clinical cases. Nonetheless, this approach potentially poses several advantages over the other IDH1/2 testing methods. This approach enables noninvasive and quantitative analysis of (R)-2HG, and hence IDH1/2 status. The concentration of (R)-2HG possibly reflects the tumor status, including cellularity [66]. Identification of (R)-2HG hotspots might also provide information that may help the planning of targeted biopsy [65]. Moreover, the most significant value of this method is the availability for repetitive and temporal measurement during the clinical course. Temporal testing using MRS might enable the dynamic monitoring of the effects of therapeutic agents, including inhibitors of mutant IDH1/2 in the future [68].
Which method should we use for IDH1/2 testing?
Several studies have compared the sensitivity and robustness of each method. In general, IHC using mutation-specific antibodies, pyrosequencing, or melting curve analysis has a higher sensitivity than Sanger sequencing [46, 49, 57, 62]. The high sensitivity, as reported in each method, is however not always required for all situations, if sufficient tissue specimen is adequately obtained. The optimal method for IDH1/2 testing varies depending on various factors, including the purpose, sample types (FFPE or frozen tissue), sample number (high throughput analysis is required or not), or laboratory equipment [46].
IHC is sensitive, robust, and accessible. Therefore, this technique is recommended for initial screening [15]. If IDH1/2 status is critical for a clinical decision, another method should be used in cases showing negative results in IHC, to avoid missing non-R132H mutations. The high throughput techniques, including pyrosequencing and melting curve analysis, are suitable for large-scale studies in which DNA samples are already prepared [46]. Sanger sequencing provides accurate information, if specimens with sufficient tumor cell content are obtained.
Conclusion
As reviewed in this report, IDH1/2 mutations have distinct relationships with clinical features, pathology, or other genetic/epigenetic alterations. IDH1/2 status is currently among the most important molecular markers of gliomas. The significance of IDH1/2 status argues for further improvement and standardization of IDH1/2 testing methods for practical use. The development of an accurate and robust method for molecular markers is a prerequisite for the establishment of molecular classification of gliomas in the future.
References
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 11:341–347
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A, Rodolfo M, Scarpa A, Felicioni L, Buttitta F, Malatesta S, Marchetti A, Bardelli A (2009) IDH1 mutations at residue p. R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30:7–11
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, Jeuken JW, Wesseling P, Reifenberger G, von Deimling A (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, Weinstock DM, Sharp KA, Thompson CB (2012) Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 31:2491–2498
Arita H, Narita Y, Fukushima S, Tateishi K, Matsushita Y, Yoshida A, Miyakita Y, Ohno M, Collins VP, Kawahara N, Shibui S, Ichimura K (2013) Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol 126:267–276
Sonoda Y, Kumabe T, Nakamura T, Saito R, Kanamori M, Yamashita Y, Suzuki H, Tominaga T (2009) Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci 100:1996–1998
Qi ST, Yu L, Lu YT, Ou YH, Li ZY, Wu LX, Yao F (2011) IDH mutations occur frequently in Chinese glioma patients and predict longer survival but not response to concomitant chemoradiotherapy in anaplastic gliomas. Oncol Rep 26:1479–1485
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477
Zhang CB, Bao ZS, Wang HJ, Yan W, Liu YW, Li MY, Zhang W, Chen L, Jiang T (2014) Correlation of IDH1/2 mutation with clinicopathologic factors and prognosis in anaplastic gliomas: a report of 203 patients from China. J Cancer Res Clin Oncol 140:45–51
Mukasa A, Takayanagi S, Saito K, Shibahara J, Tabei Y, Furuya K, Ide T, Narita Y, Nishikawa R, Ueki K, Saito N (2012) Significance of IDH mutations varies with tumor histology, grade, and genetics in Japanese glioma patients. Cancer Sci 103:587–592
von Deimling A, Korshunov A, Hartmann C (2011) The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol 21:74–87
Horbinski C (2013) What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol 125:621–636
Green A, Beer P (2010) Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 362:369–370
Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, Fleming A, Hadjadj D, Schwartzentruber J, Majewski J, Dong Z, Siegel P, Albrecht S, Croul S, Jones DT, Kool M, Tonjes M, Reifenberger G, Faury D, Zadeh G, Pfister S, Jabado N (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124:615–625
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, Hruban RH, Jallo GI, Mandahl N, Meeker AK, Mertens F, Netto GJ, Rasheed BA, Riggins GJ, Rosenquist TA, Schiffman M, Shih Ie M, Theodorescu D, Torbenson MS, Velculescu VE, Wang TL, Wentzensen N, Wood LD, Zhang M, McLendon RE, Bigner DD, Kinzler KW, Vogelstein B, Papadopoulos N, Yan H (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 110:6021–6026
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483
Ichimura K (2012) Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol 29:131–139
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935
Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154
Mulholland S, Pearson DM, Hamoudi RA, Malley DS, Smith CM, Weaver JM, Jones DT, Kocialkowski S, Backlund LM, Collins VP, Ichimura K (2012) MGMT CpG island is invariably methylated in adult astrocytic and oligodendroglial tumors with IDH1 or IDH2 mutations. Int J Cancer 131:1104–1113
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003
Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN (2009) Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol 68:1319–1325
Weller M, Stupp R, Hegi ME, van den Bent M, Tonn JC, Sanson M, Wick W, Reifenberger G (2012) Personalized care in neuro-oncology coming of age: why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol 14(Suppl 4):iv100–iv108
Nobusawa S, Watanabe T, Kleihues P, Ohgaki H (2009) IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 15:6002–6007
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, Westphal M, Schackert G, Simon M, Tonn JC, Heese O, Krex D, Nikkhah G, Pietsch T, Wiestler O, Reifenberger G, von Deimling A, Loeffler M (2009) Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27:5743–5750
Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, Sabel MC, Koeppen S, Ketter R, Meyermann R, Rapp M, Meisner C, Kortmann RD, Pietsch T, Wiestler OD, Ernemann U, Bamberg M, Reifenberger G, von Deimling A, Weller M (2009) NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Enting RH, French PJ, Dinjens WN, Vecht CJ, Allgeier A, Lacombe D, Gorlia T, Hoang-Xuan K (2013) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31:344–350
Okita Y, Narita Y, Miyakita Y, Ohno M, Matsushita Y, Fukushima S, Sumi M, Ichimura K, Kayama T, Shibui S (2012) IDH1/2 mutation is a prognostic marker for survival and predicts response to chemotherapy for grade II gliomas concomitantly treated with radiation therapy. Int J Oncol 41:1325–1336
Hartmann C, Hentschel B, Tatagiba M, Schramm J, Schnell O, Seidel C, Stein R, Reifenberger G, Pietsch T, von Deimling A, Loeffler M, Weller M (2011) Molecular markers in low-grade gliomas: predictive or prognostic? Clin Cancer Res 17:4588–4599
Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, Paris S, Boisselier B, Idbaih A, Laigle-Donadey F, Hoang-Xuan K, Sanson M, Delattre JY (2010) IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 75:1560–1566
Metellus P, Coulibaly B, Colin C, de Paula AM, Vasiljevic A, Taieb D, Barlier A, Boisselier B, Mokhtari K, Wang XW, Loundou A, Chapon F, Pineau S, Ouafik L, Chinot O, Figarella-Branger D (2010) Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol 120:719–729
Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, Sure U, Wrede K, Nakazato Y, Tanaka Y, Vital A, Mariani L, Stawski R, Watanabe T, De Girolami U, Kleihues P, Ohgaki H (2010) Molecular classification of low-grade diffuse gliomas. Am J Pathol 177:2708–2714
Sun H, Yin L, Li S, Han S, Song G, Liu N, Yan C (2013) Prognostic significance of IDH mutation in adult low-grade gliomas: a meta-analysis. J Neurooncol 113:277–284
van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, Frenay M, Tijssen CC, Lacombe D, Idbaih A, van Marion R, Kros JM, Dinjens WN, Gorlia T, Sanson M (2010) IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization For Research And Treatment Of Cancer Brain Tumor Group. Clin Cancer Res 16:1597–1604
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
van den Bent MJ (2010) Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol 120:297–304
Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, Westphal M, Schackert G, Meyermann R, Pietsch T, Reifenberger G, Weller M, Loeffler M, von Deimling A (2010) Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120:707–718
Agarwal S, Sharma MC, Jha P, Pathak P, Suri V, Sarkar C, Chosdol K, Suri A, Kale SS, Mahapatra AK (2013) Comparative study of IDH1 mutations in gliomas by immunohistochemistry and DNA sequencing. Neuro Oncol 15:718–726
van den Bent MJ, Hartmann C, Preusser M, Strobel T, Dubbink HJ, Kros JM, von Deimling A, Boisselier B, Sanson M, Halling KC, Diefes KL, Aldape K, Giannini C (2013) Interlaboratory comparison of IDH mutation detection. J Neurooncol 112:173–178
Arita H, Narita Y, Matsushita Y, Fukushima S, Yoshida A, Takami H, Miyakita Y, Ohno M, Shibui S, Ichimura K (2014) Development of a robust and sensitive pyrosequencing assay for the detection of IDH1/2 mutations in gliomas. Brain Tumor Pathol (in press)
Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601
Kato Y, Jin G, Kuan CT, McLendon RE, Yan H, Bigner DD (2009) A monoclonal antibody IMab-1 specifically recognizes IDH1R132H, the most common glioma-derived mutation. Biochem Biophys Res Commun 390:547–551
Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, Hartmann C, von Deimling A (2010) Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254
Capper D, Sahm F, Hartmann C, Meyermann R, von Deimling A, Schittenhelm J (2010) Application of mutant IDH1 antibody to differentiate diffuse glioma from nonneoplastic central nervous system lesions and therapy-induced changes. Am J Surg Pathol 34:1199–1204
Takano S, Tian W, Matsuda M, Yamamoto T, Ishikawa E, Kaneko MK, Yamazaki K, Kato Y, Matsumura A (2011) Detection of IDH1 mutation in human gliomas: comparison of immunohistochemistry and sequencing. Brain Tumor Pathol 28:115–123
Kaneko MK, Morita S, Tsujimoto Y, Yanagiya R, Nasu K, Sasaki H, Hozumi Y, Goto K, Natsume A, Watanabe M, Kumabe T, Takano S, Kato Y (2013) Establishment of novel monoclonal antibodies KMab-1 and MMab-1 specific for IDH2 mutations. Biochem Biophys Res Commun 432:40–45
Kaneko MK, Tian W, Takano S, Suzuki H, Sawa Y, Hozumi Y, Goto K, Yamazaki K, Kitanaka C, Kato Y (2011) Establishment of a novel monoclonal antibody SMab-1 specific for IDH1-R132S mutation. Biochem Biophys Res Commun 406:608–613
Kato Kaneko M, Ogasawara S, Kato Y (2013) Establishment of a multi-specific monoclonal antibody MsMab-1 recognizing both IDH1 and IDH2 mutations. Tohoku J Exp Med 230:103–109
Ronaghi M, Uhlen M, Nyren P (1998) A sequencing method based on real-time pyrophosphate. Science 281(363):365
Ronaghi M (2001) Pyrosequencing sheds light on DNA sequencing. Genome Res 11:3–11
Felsberg J, Wolter M, Seul H, Friedensdorf B, Goppert M, Sabel MC, Reifenberger G (2010) Rapid and sensitive assessment of the IDH1 and IDH2 mutation status in cerebral gliomas based on DNA pyrosequencing. Acta Neuropathol 119:501–507
Setty P, Hammes J, Rothamel T, Vladimirova V, Kramm CM, Pietsch T, Waha A (2010) A pyrosequencing-based assay for the rapid detection of IDH1 mutations in clinical samples. J Mol Diagn 12:750–756
Cykowski MD, Allen RA, Fung KM, Harmon MA, Dunn ST (2012) Pyrosequencing of IDH1 and IDH2 mutations in brain tumors and non-neoplastic conditions. Diagn Mol Pathol 21:214–220
Boisselier B, Marie Y, Labussiere M, Ciccarino P, Desestret V, Wang X, Capelle L, Delattre JY, Sanson M (2010) COLD PCR HRM: a highly sensitive detection method for IDH1 mutations. Hum Mutat 31:1360–1365
Pang B, Durso MB, Hamilton RL, Nikiforova MN (2013) A novel COLD-PCR/FMCA assay enhances the detection of low-abundance IDH1 mutations in gliomas. Diagn Mol Pathol 22:28–34
Horbinski C, Kelly L, Nikiforov YE, Durso MB, Nikiforova MN (2010) Detection of IDH1 and IDH2 mutations by fluorescence melting curve analysis as a diagnostic tool for brain biopsies. J Mol Diagn 12:487–492
Erali M, Voelkerding KV, Wittwer CT (2008) High resolution melting applications for clinical laboratory medicine. Exp Mol Pathol 85:50–58
Li J, Wang L, Mamon H, Kulke MH, Berbeco R, Makrigiorgos GM (2008) Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med 14:579–584
Andronesi OC, Kim GS, Gerstner E, Batchelor T, Tzika AA, Fantin VR, Vander Heiden MG, Sorensen AG (2012) Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med 4:116ra114
Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, Yang XL, Mashimo T, Raisanen JM, Marin-Valencia I, Pascual JM, Madden CJ, Mickey BE, Malloy CR, Bachoo RM, Maher EA (2012) 2-Hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med 18:624–629
Pope WB, Prins RM, Albert Thomas M, Nagarajan R, Yen KE, Bittinger MA, Salamon N, Chou AP, Yong WH, Soto H, Wilson N, Driggers E, Jang HG, Su SM, Schenkein DP, Lai A, Cloughesy TF, Kornblum HI, Wu H, Fantin VR, Liau LM (2012) Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neurooncol 107:197–205
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340:626–630
Acknowledgments
This work was supported by JSPS KAKENHI Grant Numbers 26861171 (H.A.), 25462283 (K.I.) and by the National Cancer Center Research and Development Fund 23-A-50 (K.I.).
