
Abstract
Background
The presence of mutations in the isocitrate dehydrogenase 1 and 2 genes (IDH1/2) in glioma tumors is correlated with good prognosis upon standard-of-care treatment. Therefore, information on whether the glioma tumor has IDH1/2 mutations could be used in the correct diagnosis and management of glial tumors. The two most common techniques used to detect IDH1/2 mutations, immunohistochemistry (IHC) and Sanger sequencing, are prone to missing these mutations, especially if the tumor cells that carry the mutations constitute a small minority of the tumor itself.
Objectives
We developed and validated a rapid method (3-mismatch-amplification refractory mutation system [3m-ARMS]) that can be used for pre-, intra- and postoperative detection of the most common IDH1/2 mutations in glial tumors with high specificity and sensitivity. We also conducted a comprehensive IDH1/2 mutation analysis in 236 glial tumor samples comparing 3m-ARMS, IHC and Sanger sequencing.
Methods
3m-ARMS was optimized and validated for the specific and sensitive detection of the most common IDH1 and IDH2 mutations. We then analyzed 236 glial tumor samples for the presence of IDH1/2 mutations using 3m-ARMS, Sanger sequencing and IHC techniques. We then analyzed and compared the results, evaluating the diagnostic and screening potential of 3m-ARMS.
Results
Comparison of the three techniques used in the mutation analysis showed that 3m-ARMS-based IDH1/2 mutation detection was superior to IHC and Sanger sequencing-based IDH1/2 mutation detection in terms of accuracy, specificity and sensitivity, especially for tumor samples in which only a small minority of the cell population carried the mutation. 3m-ARMS could detect the presence of femtogram levels of IDH1/2 mutant DNA in DNA samples in which the mutant DNA-to-wild-type DNA ratio was as low as 1:100,000.
Conclusion
Sanger sequencing and IHC-based methods have shortcomings when detecting mutations in glial tumors so can miss IDH1/2 mutations in glial tumors when used alone without proper modifications. 3m-ARMS-based mutation detection is fast and simple with potential for use as a diagnostic test for the majority of hot spot mutations in IDH1/2 genes. It can detect IDH1/2 mutations within an hour so can be adapted for intraoperative diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Sanger sequencing and immunohistochemistry-based methods have shortcomings in the detection of isocitrate dehydrogenase 1 and 2 genes (IDH1/2) mutations in glial tumors if few of the tumor cells carry the mutation. |
We developed a rapid, sensitive and easy-to-use IDH1/2 mutation detection method. Technical validation of the new method was performed and compared with immunohistochemistry and Sanger sequencing. |
We analyzed 236 glioma tumor samples for the presence of IDH1/2 mutations and evaluated IDH1/2 mutation status within different tumor grades and histopathological subtypes. |
Use of co-amplification at lower denaturation temperature (COLD)-polymerase chain reaction (PCR) for the amplification of the DNA region to be sequenced by Sanger sequencing increased the sensitivity of IDH1/2 mutation detection in glial tumors. |
Two tumor samples had mutations in both IDH1 and IDH2 genes. The presence of mutations in both IDH1 and IDH2 genes is very rare in glioma tumors and could be associated with glioma tumor biology. We are following the progress of these patients. |
1 Introduction
The development of novel genetic markers in different glioma subtypes has led to a shift in the classification of glial tumors from a previously used and solely histology-based system into a more reliable, molecular marker-based system [1]. The current classification system offers improved diagnosis, prognosis prediction and response to therapy. Among the many parameters that play a critical role in the diagnosis and prognosis of gliomas, the mutation status of isocitrate dehydrogenase 1 and 2 (IDH1/2) genes is crucial, especially for low-grade gliomas [2]. IDH1/2 mutations disrupt enzymatic activity and cause the formation of 2-hydroxyglutarate, instead of α-ketoglutarate, which is necessary in the Krebs cycle [3] and other cellular mechanisms such as demethylation of cytosine. Many studies have shown that patients with gliomas have increased survival rates and better response to chemotherapy/radiotherapy if their glioma tumors have mutations in the IDH1/2 genes [2, 4].
Various mutation-detection methods have been used in brain tumors [5,6,7,8]. Conventionally, an immunohistochemistry (IHC)-based technique has been used to check for the presence of the most common mutation in the IDH1 gene, the R132H, for the post-surgical diagnosis of glioma tumors. However, IHC-based techniques provide limited information about IDH1 mutations, as the commercially available antibodies can only detect the R132H mutation in the IDH1 gene, and the R172K mutation in IDH2 gene. IHC is also prone to missing tumor cells with the R132H mutation if these cells are very few in the tumor analyzed. Alternatively, sequencing methods are widely used to detect mutations in the IDH1/2 genes. Sanger sequencing, especially, is considered the gold standard for IDH1/2 mutation detection. However, Sanger sequencing can also fail to detect a mutation in a tumor if the mutation is present only in the minority of the cells that constitute the tumor. In most cases, both IHC and sequencing-based techniques are also unsuitable for pre-and intraoperative detection of IDH1/2 mutations in glioma tumors because of the lack of biopsy samples prior to surgery and the lengthy time needed to conduct the analysis.
Herein, we introduce a new IDH1/2 mutation detection method called 3-mismatch-amplification refractory mutation system (3m-ARMS), which is based on a modified version of the amplification refractory mutation system (ARMS), to detect the most common IDH1/2 mutations in a short period of time with high sensitivity and specificity. Therefore, 3m-ARMS is a rapid and low-cost method of detecting the most common IDH1/2 mutations in glial tumors that can be adopted for pre- and intraoperative use.
Using 3m-ARMS in conjunction with Sanger sequencing and IHC, we analyzed and compared 236 glial tumors for the presence and type of IDH1/2 mutations. In our mutation analyses, 3m-ARMS detected IDH1/2 mutations that were originally missed by both Sanger sequencing and IHC in some tumor samples, because the cells that carried the mutation constituted the minority of the total cell population in the tumor.
2 Materials and Methods
2.1 Tumor Samples and DNA Isolation
This study is based on a mixed (retrospective and prospective) cohort of patient samples. Initially, 228 tumor samples from 228 patients were obtained from the “Bahcesehir University Brain Tumor Tissue Bank” and analyzed retrospectively. The pathology reports of glioma patients were reviewed following approval from our institutional research ethics committee (2017-14/01). The sample selection was only based on diagnosis, and only glioma tumors with a variety of grades were selected. Grade I tumors included pilocytic astrocytoma and ganglioglioma samples, and were retrieved as the IDH wild-type (WT) glial control group. Grade II included diffuse astrocytoma and oligodendroglioma and Grade III tumors included anaplastic astrocytoma and anaplastic oligodendroglioma. Grade IV tumors refer to glioblastoma samples. Histopathologic classification was based on the 2016 World Health Organization (WHO) classification [1]. An additional eight tumor samples were further obtained prospectively after we started our study and to test the potential for intraoperative analysis with this method. All patients provided informed written consent.
For DNA isolation from the tumor samples, a DNA isolation method based on alkaline lysis of the tissue allowed us to obtain DNA for polymerase chain reaction (PCR) in 8–10 min [9]. The DNA samples used in this study, except for those used in formalin-fixed paraffin-embedded (FFPE) tissues studies, were obtained from tumor samples that were flash frozen in liquid nitrogen and stored in a liquid nitrogen tank. Frozen tumor samples were dissected into small pieces, and 5–25 mg of tumor sample was used for DNA extraction. Each tumor sample was first incubated in an alkaline solution (1 mM Na2EDTA, 25 mM NaOH, pH 12) for 7–8 min at 95 °C. Then, an equal volume of a neutralization solution (40 mM Tris–HCl Buffer) was added at room temperature. After addition of the neutralization solution, DNA samples were stored at − 20 °C until analysis. Based on the Qubit fluorometric quantification method of some of the DNA samples, this method yielded about 19.06 ± 8.65 ng/µl DNA in a total volume of 400 µl (Supplementary Fig. 1). For the experiment in FFPE studies, FFPE tumor samples were used for DNA extraction. From each of the seven FFPE samples, five pieces of 4 µm paraffin sections were obtained and deparaffinized by heating. An alkaline lysis-based DNA extraction protocol was used on the deparaffinized FFPE sections using the same method.
2.2 3-Mismatch Amplification Refractory Mutation Detection (3m-ARMS) Polymerase Chain Reaction (PCR)
Following DNA isolation, ARMS-PCR was performed. For each 3m-ARMS reaction, we used 2 µl DNA, which on average corresponds to about 38 ng of total DNA. ARMS is based on the use of sequence-specific primers to amplify target DNA within a mixture of target and non-target DNAs [10]. ARMS is especially useful in detecting point mutations in tumors. Here, we modified the design of ARMS-PCR by incorporating additional mismatches to one of the PCR primers to increase the accuracy and specificity of mutant DNA amplification. In traditional ARMS, a 3′ terminal nucleotide of one of the PCR primers is designed to match mutant tumor DNA and mismatch to WT DNA. Penultimate nucleotide, next to the terminal mismatch nucleotide, is designed as a mismatch for both mutant and WT DNA. Selection of the penultimate nucleotide is based on the terminal mismatch nucleotide, and penultimate nucleotide confers DNA amplification of mutant DNA but not WT DNA as one mismatch for mutant DNA is tolerated but two mismatches for WT DNA are not. We noticed that having only two mismatched nucleotides did not always confer specific amplification of mutant DNA as sometimes WT DNA was also amplified. In this study, we designed our primers by introducing an additional mismatch nucleotide and called our method 3 mismatch-ARMS or 3m-ARMS. Basically, in 3m-ARMS, the three 3′ terminal nucleotides in one of the PCR primers show mismatch to the WT DNA, but only two of these, the penultimate and the nucleotide next to it located at its 5′ position, show mismatch to the mutant DNA. We determined the mismatch nucleotides empirically and adapted our primer design to detect the most common mutations in the IDH1 gene. Primer sequences are presented in Supplementary Table 1.
The presence of R132H (G395A), R132C (C394T) and R132G (C394G) mutations in the IDH1 gene and R172K (G515A), R172M (G515T) and R172W (A514T) were checked in tumor samples. Genomic DNA from the blood of two healthy volunteers was used as negative controls, and tumor samples proven to contain specific mutations in the IDH1 gene were used as positive controls. For real-time analysis of the 3m-ARMS assay, we used LightCycler-96 (Roche Applied Science) together with Taq 2X Master Mix (New England BioLabs) enzyme. Our real-time PCR analysis was performed with the addition of 0.35X EvaGreen (Biotium) and 1–2 µl of DNA to the optimized master mix solution.
To test the sensitivity of the 3m-ARMS PCR method, we cloned IDH1 and IDH2 genomic DNA sequences into pBlueScript-SK plasmid by Gibson cloning (NEB, NEBuilder HiFi DNA Assembly Master Mix) and then produced all clinically important mutations (G395A/T/C, G394A/T/G of the IDH1 gene and G515A/T/C, A514G/C/T of the IDH2 gene) using site-directed mutagenesis (Agilent, Pfu Ultravision II HS DNA polymerase). WT DNA and mutant DNA were purified (M&N, Nucleospin Plasmid), and DNA concentrations were measured with a Qubit double-stranded DNA BR assay kit. Different ratios of WT DNA and mutant DNAs were mixed, ranging between 1:1000 and 1:100,000 (mutant DNA:WT DNA ratio) at 1 ng, 1 picogram and 1 fg total DNA levels. Samples were tested with 3m-ARMS and results evaluated by comparing threshold cycle (Ct).
2.3 Sequencing
The mutation status of each specimen was checked via Sanger sequencing. IDH1 and IDH2 gene regions covering the mutation hot spots G395 and C394 for IDH1 and A514, G515 and G516 for IDH2 genes were amplified with specific primers and Q5® High-Fidelity DNA Polymerase (New England BioLabs).
Some samples (14/236) required additional sequencing because of either low-quality data or low tumor cellularity. Samples showing discordance between 3m-ARMS and Sanger sequencing were re-analyzed using the co-amplification at lower denaturation temperature (COLD)-PCR method to selectively increase the proportion of mutant DNA by amplifying the mutation-containing DNA irrespective of the mutation type and position [11]. The sequences of the COLD-PCR primers we designed and tested that were specific to mutant regions in IDH1 and IDH2 genes are shared in Supplementary Table 1. The COLD-PCR conditions were as follows: 95 °C for 30 s followed by 40 cycles of PCR with reduced denaturation temperature with a range of 75.7–75.9 °C as critical temperature for 30 s, annealing at 60 °C for 20 s and extension at 72 °C for 10 s. The final extension step was done at 72 °C for 5 min.
2.4 Immunohistochemistry (IHC)
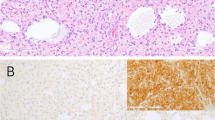
FFPE sections of all available tumor samples (n = 106) were evaluated for histopathological examination. Tissue sections were fixed in 10% buffered formalin and processed routinely, and 5-µm sections were cut and stained with H&E. Sections were stained with antibody IDH1 (dilution 1:20; Dianova, catalog no: DIA H09, Clone: H09) by immunohistochemical analysis using the classic avidin–biotin method. Tissue slides were counterstained with hematoxylin. Slides were processed in Autostainer (Dako EnVisionFLEX+) and PT Link. Mouse antibody was used as a secondary antibody, and the target retrieval solution was high PH as antigen retrieval.
2.5 Statistical Analysis
Since sequencing is considered the gold standard method in mutation detection, we compared the results of the 3m-ARMS and IHC methods and those received with Sanger sequencing. We calculated and compared the sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV) and accuracy of both techniques. We also evaluated the positive and negative likelihood ratios, which can represent the diagnostic utility of the test. Sensitivity and specificity were calculated via a 2 × 2 contingency table formed of true-positive (TP), true-negative (TN), false-positive (FP) and false-negative (FN) results. Graphs and statistical evaluations were prepared using GraphPad Prism-6®.
3 Results
3.1 3m-ARMS Enables Fast and Sensitive IDH1/2 Mutation Detection in Glial Tumors
We noticed that the techniques most commonly used to detect IDH1/2 mutations in glial tumors, IHC and Sanger sequencing, failed to detect these mutations in some glioma tumor samples. IHC provides information about the presence of just one type of IDH1/2 mutation, R132H in the IDH1 gene. Both IHC and Sanger sequencing also take too long to complete, precluding the adaptation of these techniques for use in preoperative and intraoperative diagnosis. Therefore, an alternative technique was needed that would allow us to detect the most common IDH1/2 mutations in glioma tumors with high sensitivity and specificity, and within an hour, for potential use as an intraoperative diagnostic tool. Among the different techniques we tried, the ARMS-based mutation detection with certain modifications provided us the best sensitivity and specificity for the detection of IDH1/2 mutations, using very little tumor tissue, and in the shortest time period compared with other alternatives. The most important modification we made was on the number of mismatches in the primers we used. We found that custom design 3 mismatches (3m-ARMS) gave the best results. We used alternative 3 mismatches for each mutation type and identified the primer sequence that yielded the most specific and sensitive amplification of the mutant allele (Fig. 1a, b and Sect. 2). We then adapted 3m-ARMS for quantitative real-time PCR, which enabled the real-time detection of mutations (Fig. 1c–f). The results of the 3m-ARMS analyses were always compared with, and confirmed by, Sanger sequencing (Fig. 1g–j).

a Depiction of the 3m-ARMS principle. IDH1 R132 WT DNA (black) has three mismatched nucleotides (red crosses) with the designed primer (blue) resulting in inhibition of primer extension and DNA amplification. IDH1 R132H MT DNA (purple) has one terminal match (green check) and one penultimate and one antepenultimate mismatch (red crosses) with the designed primer (blue) resulting in proper primer extension and DNA amplification (orange arrow). b Agarose gel analysis of the 3m-ARMS PCR for the detection of G395A (R132H) in tumor samples. The first well on the left was loaded with a DNA ladder. Note that each sample was loaded into the gel in duplicate wells adjacent to each other. A single well was kept empty in between different samples. Only the two tumor samples carrying the G395A mutation (marked as IDH1 R132H on the image) and the PC sample yielded a specific PCR band on this gel. Faint bands that are smaller in size are primer dimers. c–f Left: Tm curve analysis of WT samples (c, d) and MT samples (e, f) at the end of the qRT-PCR-based 3m-ARMS analysis. Only MT samples yield a distinctive peak with a unique Tm. g–j Sanger sequencing results of IDH1 gene for the WT (g, h) and MT samples (i, j) analyzed on the gel (a), showing nucleotide at position 395 and its periphery. WT samples yield only one peak for nucleotide G at position 395, whereas MT samples show two peaks for nucleotides G and A for the same position. IDH1/2 isocitrate dehydrogenase 1 and 2 genes, MT mutant, NC negative control DNA with no IDH mutation, NTC no tissue control, PC positive control that carries G395A mutation, PCR polymerase chain reaction, qRT-PCR quantitative real-time PCR, Tm melting temperature, WT wild type
Next, we measured the specificity and the sensitivity of 3m-ARMS in detecting IDH1/2 mutations using cloned DNA samples that contained WT and mutant IDH1 and IDH2 alleles at different ratios and concentrations. To evaluate the specificity of the tested primers, mutant DNA and WT DNA were mixed at ratios ranging from 1:1000 to 1:100,000 (mutant DNA:wt DNA ratio). To evaluate the sensitivity of the tested primers, hybrid DNA mixtures were used at quantities of 1 fg, 1 pg and 1 ng in 20 μl reaction volume. Mean amplification Ct values obtained for 1 fg, 1 pg and 1 ng levels per 20 μl reaction volume were 30.18, 23.26 and 13.46, respectively. The cut-off line for being positive was determined as a Ct value of ≥ 30. These results overall showed that the 3m-ARMS method enabled ultrasensitive and specific detection of IDH1/2 mutations, as specific mutation detection was possible even when femtogram levels of DNA were used and with a mutant DNA-to-WT DNA ratio as low as 1:100,000 in the samples (Fig. 2).

Sensitivity and specificity assessment of the 3m-ARMS primers. Amplification curves indicate the Ct (amplification threshold cycles) values of each DNA sample. a Arrows indicate varying degrees of DNA content, from 100 fg to 1 ng per reaction (R). In this analysis, WT and mutant DNAs were tested separately. b Arrows indicate varying ratios of mutant DNA/WT DNA mixtures ranging from 1:1000 to 1:100,000 with a total of 1 ng plasmid DNA per reaction. The higher concentration and higher relative ratio of the mutant DNA yields lower Ct values for the specific amplification of the mutant DNA. WT wild type
The fact that 3m-ARMS-based mutation detection can be conducted within 60 min allowed us to successfully conduct intraoperative checks for the presence of the most common IDH1/2 mutations in eight samples. The tumor samples were obtained from the operation room during surgeries, and IDH1/2 mutation analysis by 3m-ARMS was performed in less than 60 min. Results of the analyses confirmed the pathological evaluation of samples during and after surgery. Taken together, these results suggest that 3m-ARMS provides fast and accurate detection of the most common IDH1/2 mutations, even during surgery.
3.2 Comparative IDH1/2 Mutation Analysis in Glioma Tumors
In this study, we analyzed a total of 236 glial tumor samples from 236 patients for the presence of the most common mutations found in IDH1 and IDH2 genes in glioma tumors, using three different techniques. As mentioned, eight of the tumor samples were analyzed intraoperatively using fresh tumor tissues and the 3m-ARMS method initially and later by Sanger sequencing and IHC. Four of the eight samples tested intraoperatively were R132H (G395A) mutant, pathological classification of two cases confirmed anaplastic astrocytoma (AA) and two cases of oligodendroglioma (OD). Another four intraoperatively tested samples were IDH WT; three were diffuse astrocytoma (DA) and one was AA.
Our overall IDH1/2 mutation analysis relied on 3m-ARMS and Sanger sequencing results, which showed 100% concordance. Based on these results, among the 236 glial tumor samples analyzed, 94 had mutation(s) in IDH1 and/or IDH2 genes. Of those, 85 had G395A (R132H), two had C394T (R132C) and one had C394G (R132G) mutation only in the IDH1 gene. Two samples had a single mutation, G515A (R172K), only in the IDH2 gene. Two tumor samples carried both G395A and C394T mutations in the IDH1 gene. Interestingly, two tumor samples had G395A in the IDH1 gene and G515A in the IDH2 gene (Fig. 3a).

IDH1/2 mutation frequencies. aIDH1/2 mutation frequencies of all glioma samples analyzed in this study (n = 236). Each color indicates the number of samples with the corresponding mutation type. bIDH1/2 mutation frequencies of the 236 tumors categorized based on WHO grading (grades I, II, III, IV). c Mutation frequencies of the tumors based on histopathologic subtypes. Note that this information was available for 186 of the 236 tumor samples analyzed in this study. AA anaplastic astrocytoma, AO anaplastic oligodendroglioma, DA diffuse astrocytoma, GBM glioblastoma, IDH1/2 isocitrate dehydrogenase 1 and 2 genes, n number of tumor samples, OD oligodendroglioma, WHO World Health Organization
3.3 IDH1 and IDH2 Mutation Frequency in Different Glial Tumor Types
Corroborating previously published studies, the frequency of IDH1/2 mutations in our study also differed significantly among different types of glial tumors according to WHO grading and histopathological classification. Of the 236 tumors we analyzed, none of WHO grade I, 70% of WHO grade II, 64.42% of WHO grade III and 8.75% of WHO grade IV tumors had IDH1/2 mutations (Fig. 3b). Based on histopathological classification, IDH1/2 positivity in 186 samples (except pilocytic astrocytoma) was as follows: 42.9% of DA, 42.9% of AA, 100% of OD, 100% of anaplastic oligodendroglioma (AO) and 7.6% of glioblastoma samples carried one or two IDH1/2 mutation(s) (Fig. 3c).
3.4 Sanger Sequencing Without a Co-amplification at Lower Denaturation Temperature (COLD)-PCR Amplification Step Can Yield False-Negative Results
In our analysis, 90 tumor samples were initially found to be positive for an IDH1/2 mutation based on both Sanger sequencing and 3m-ARMS. 3m-ARMS but not Sanger sequencing detected an IDH1 mutation in four other tumor samples. This discordance in some results with 3m-ARMS and Sanger analyses prompted us to question the sensitivity of the Sanger sequencing as we detected some 3m-ARMS-positive/Sanger-negative but never 3m-ARMS-negative/Sanger-positive results. IDH mutations are known to be present only in tumor cells and not in normal or tumor-associated cells in patients with glioma tumors [12]. Therefore, if only a small percentage of the tumor cells in a tumor sample carry an IDH mutation, or if the tumor sample mostly comprises non-tumor cells such as vascular, perivascular, hematopoietic and normal neural and glial cells, it is possible that most cells in that tumor extract might be negative for an IDH mutation (even if the tumor has some cells that carry an IDH mutation). Therefore, we hypothesized that when the tumor sample has very few cells with an IDH mutation, Sanger sequencing based on PCR amplification of genomic DNA might not be sensitive enough to detect the mutation, as the PCR-based DNA amplification step required for Sanger sequencing would amplify both WT and mutant DNA without changing the mutant DNA-to-WT DNA ratio, thus rendering the mutant DNA undetectable by Sanger sequencing (Fig. 4a, d). This is because Sanger sequencing is known to fail to identify mutations or variations in DNA sequence in a DNA sample if only less than 20% of the DNA molecules of interest carry the variation or mutation [13]. To test our hypothesis, we repeated our Sanger sequencing analysis based on a regular PCR and a COLD-PCR amplification of the IDH1 genomic region for the four samples that originally showed 3m-ARMS-positive/Sanger-negative results. As opposed to regular PCR, COLD-PCR amplification increases the mutant DNA-to-WT DNA ratio if the mutation of interest is located in the amplified region [11] (Fig. 4b, e). As expected, the results based on COLD-PCR, but not regular PCR, followed by Sanger sequencing, showed the presence of the IDH1 mutations that 3m-ARMS results had already shown in the four samples (Fig. 4), suggesting that Sanger sequencing based on a regular PCR amplification of the target DNA region may fail to detect the mutation and therefore may give FN results.

Detection of IDH1 R132H mutations using different approaches. Agarose gel images of the IDH1 genomic region amplified by a regular PCR, b COLD-PCR and c 3m-ARMS. Sanger sequencing analyses for the same tumor sample failed to show the presence of the mutation if DNA amplification was conducted by regular PCR (d); however, G395A mutation as being heterozygous was clearly shown when COLD-PCR was conducted for DNA amplification prior to Sanger sequencing (e). f 3m-ARMS qRT-PCR results showing melting temperature peaks specific for the G395A mutation. The peaks shown by ** are visible if only mutant DNA is amplified. COLD-PCR co-amplification at lower denaturation temperature PCR, IDH1/2 isocitrate dehydrogenase 1 and 2 genes, PCR polymerase chain reaction, qRT-PCR quantitative real-time PCR
3.5 IDH1 Mutation Detection in Glial Tumors Based on IHC Has Multiple Shortcomings
Many pathology laboratories around the world have been using IHC to detect the most common IDH1 R132H (G395A) mutation in glioma tumors using a commercially available antibody against the IDH1 protein with R132H mutation. For tumor samples stored as FFPE blocks, we also conducted an IHC analysis using anti-IDH1 R132H antibody staining to check for the presence of R132H mutation (Supplementary Fig. 2). All samples analyzed by IHC were also analyzed with both Sanger sequencing and 3m-ARMS. In total, 91.51% (n = 97/106) of IHC results were concordant with Sanger sequencing results. For 7.55% (n = 8/106) of the samples analyzed, IHC analysis yielded FP results (IHC positive, Sanger negative and 3m-ARMS negative) (Supplementary Fig. 3) and for 0.94% (n = 1/106) of the samples analyzed IHC analysis gave FN results (IHC negative, Sanger positive and 3m-ARMS positive) (Fig. 5). For IHC FP samples, in order to confirm the mutation status, DNA was also extracted from FFPE tissues, and the 3m-ARMS PCR experiment was repeated. The results of this experiment show that tumor samples that were initially found to be IHC FP based on comparison of IHC, Sanger and 3m-ARMS analyses were indeed negative for the presence of the IDH1/2 mutations, even when 3m-ARMS PCR was conducted using DNA from FFPE tumor tissues (instead of fresh-frozen tumor tissues) (Supplementary Fig. 4).

Comparison of the 3m-ARMS and IHC mutation detection techniques with respect to Sanger sequencing. Coherency indicates matching results for the two techniques compared. Black columns show coherent results, grey columns show incoherent results. (+) indicates detection of the mutation(s) and (−) shows no mutation detection. COLD-PCR co-amplification at lower denaturation temperature polymerase chain reaction, IHC immunohistochemistry
These results show that IHC analysis as a single method to detect IDH mutations poses a risk because it yields a high FP rate. Furthermore, IHC analysis can only detect the R132H mutation in the IDH1 gene, which further limits its use as a sole method for IDH1 mutation detection.
4 Discussion
We analyzed the IDH1 and IDH2 mutations in 236 glioma tumors using three different methods. The 3m-ARMS technique that we introduce in this study is based on a modified ARMS PCR method and allowed us to detect mutations with a high sensitivity and specificity. Results obtained using 3m-ARMS showed high concordance with Sanger sequencing and IHC results. On the other hand, we detected FN results in some samples with both Sanger sequencing and IHC analysis, whereas 3m-ARMS showed accurate results in those samples. To overcome FN results by Sanger sequencing, we combined COLD-PCR with Sanger sequencing and showed the erroneous potential of Sanger sequencing without COLD-PCR in samples with low tumor cell frequency. Therefore, our study revealed that Sanger sequencing without COLD-PCR could be less accurate, but that 3m-ARMS was sensitive enough to be used in samples with few tumor cells carrying the mutation of interest.
Sanger sequencing and IHC are still the most commonly used techniques to perform IDH1/2 mutation analysis on glioma tumors. Sanger sequencing has the advantage of revealing the entire DNA sequence in regions where IDH1/2 mutations are mostly found. It is also relatively unbiased to the mutation type being analyzed, as the mutation type usually does not affect sequencing results. However, Sanger sequencing may not be sensitive enough to detect the presence of mutant DNA if the mutant DNA-to-WT DNA ratio is low (< 20%) [13]. Using Sanger sequencing to detect mutations in IDH1/2 is also time consuming and expensive [14]. To allow the relatively low levels of mutant DNA (with respect to WT DNA) in a genomic DNA extract to be detectable in Sanger sequencing, one must find a way to increase the mutant DNA-to-WT DNA ratio. COLD-PCR has been known as an effective method to increase this ratio as, in a COLD-PCR reaction, mutant DNA amplification is more efficient than WT DNA amplification [11]. In our IDH1/2 mutation analysis, Sanger sequencing based on regular PCR failed to detect IDH1 mutations in four tumor samples. We then amplified DNA from these four tumor samples using COLD-PCR conditions and reanalyzed them with Sanger sequencing. COLD-PCR-based Sanger sequencing indeed showed the presence of the IDH1 mutations, which were initially missed by regular PCR-based Sanger sequencing. These data suggest that Sanger sequencing-based mutation analysis of tumor samples with a low mutant DNA-to-WT DNA ratio can generate FN results. Therefore, COLD-PCR-based Sanger sequencing can be performed to reduce the risk of failure in detecting the mutations. As 3m-ARMS is based on selective amplification of the mutant DNA, it is compatible with tumor samples with a low mutant DNA-to-WT DNA ratio. Indeed, in our study, 3m-ARMS detected the IDH1 mutations in the four tumor samples that the Sanger sequencing without a COLD-PCR failed to detect. Furthermore, the most sensitive method in mutation detection is the next-generation sequencing (NGS) because of its extraordinary capacity for massive parallel sequencing of DNA fragments with high accuracy. Its superior sensitivity and specificity in IDH1 and other gene mutations has been previously shown in glial tumors [15]. Therefore, NGS should be the first choice in mutation detection. However, it is neither commonly available nor practical for intraoperative or postoperative use.
IHC-based mutation detection alone is insufficient to detect most of the mutation types in IDH1/2 genes. Currently, a limited number of antibodies have been validated to detect the most common mutations, including R132H in the IDH1 gene and a less common mutation, the R172K in the IDH2 gene. Although R132H mutation is the most common IDH1 mutation in glial tumors, for a certain diagnosis of glial tumors it is important to check other mutations in IDH1/2 genes. However, it is not practical to test all mutations with different antibodies by immunostaining. Previous studies have also shown varying degrees of concordance between IHC and Sanger sequencing. In Takano et al. [16], results of Sanger and IHC analyses matched in varying degrees ranging from 50 to 100% in gliomas with different grades. Another study showed that, for some samples with different forms of IDH1 mutations (R132L, R132C), IHC analysis might give FN results and for some WT specimens, IHC analysis gave FP rates for IDH WT tumor specimens in 14.3% of samples [13]. Multiple factors, ranging from antibody-related problems to operational errors could account for the high discordance between IHC and Sanger sequencing and for the high FP rate of IHC in the detection of IDH1 mutations. In our study, results for 8 of the 106 tumors analyzed with IHC were FP and one had FN results. Therefore, we do not recommend the use of IHC as the sole technique for mutation detection in IDH1/2 genes. If the use of IHC is desired, then combining IHC with Sanger sequencing and/or 3m-ARMS would be a safer approach than using IHC alone in the IDH1/2 status of glial tumors.
3m-ARMS is relatively simple compared with a few other existing techniques for intraoperative IDH mutation detection and does not require expensive reagents and equipment. Only 5–25 mg of tumor tissue is sufficient for 3m-ARMS-based mutation analysis.
Two other studies have reported alternative methodologies to detect IDH1/2 and other mutations intraoperatively. Kanamori et al. [6] showed the availability of real-time PCR with fluorescence melting curve analysis and COLD-PCR in intraoperative diagnosis of IDH1/2 mutations. Their results were satisfactory in terms of duration, sensitivity and ease of use, but they reported limitations in the specific and NPV of their test. Shankar et Al. [5] also developed PCR-based diagnostic test. They used peptide nucleic acid (PNA) to inhibit the WT amplification and locked nucleic acid (LNA) to specifically amplify the mutant allele, thereby increasing sensitivity and specificity. Although they obtained specificity of 100%, sensitivity was 96%. Our results appear to be superior to both of these methods in terms of sensitivity, specificity and accuracy. Despite the good levels of sensitivity and specificity reported in the previously published methods, utilization of PNA, LNA or specific fluorescence probes is neither more economical nor simpler than 3m-ARMS PCR. 3m-ARMS is also a good alternative and confirmatory technique to Sanger sequencing and IHC for routine postoperative analysis of IDH mutations, as it offers superior sensitivity compared with Sanger sequencing and IHC (Table 1).
Hereby, we offer a new diagnostic and screening test for common IDH1/2 mutations. The diagnostic potential of a novel test can be indicated by its specificity and PPV, whereas its screening potential can be indicated by sensitivity and NPV. The effectiveness of the test is determined by two factors—specificity and sensitivity—whereas its diagnostic utility is determined by the positive likelihood ratio (LR+) and negative likelihood ratio (LR−). We observed a relative superiority compared with IHC analysis in terms of specificity (97.40% for 3m-ARMS and 87.88% for IHC), PPV (95.35% for 3m-ARMS and 84.79% for IHC) and LR+ (38.50 for 3m-ARMS and 8.04 for IHC). Sensitivity was 100% for 3m-ARMS and 97.50% for IHC. Accuracy was 98.30% for 3m-ARMS and only 91.51% for IHC. Finally, the NPV was 100% for 3m-ARMS and 98.31 for IHC in the detection of IDH mutations (Table 1). Overall, in terms of the diagnostic and screening potential, the 3m-ARMS method was superior to IHC and Sanger sequencing with regular PCR.
Incorporation of molecular markers into diagnostic and prognostic classification in glial tumors enabled the preoperative and intraoperative diagnosis of brain tumors [5, 6]. Information regarding the IDH1/2 mutation status of glioma tumors before or during surgery can be critically important for surgeries in which the tumors to be resected are located in high-risk regions of the brain, as the extent of tumor resection will determine the risk of surgery-induced damage to the sensitive anatomical sites [17, 18]. Currently, two main obstacles impede the use of common histopathologic examination techniques to check for the presence of mutations in glioma tumor tissues obtained via either biopsy or surgery. First, depending on tumor location and size, the sample obtained for the test might not yield enough tumor cells for a definitive diagnosis. Second, the histology-based tests might take too long, prohibiting intraoperative diagnosis. Furthermore, frozen tissue sectioning-based methods are limited with only phenotype-based characterization rather than mutation analysis. Sequencing-based mutation-detection systems often provide accurate data but are incompatible with intraoperative use. Therefore, alternative methods are needed that are robust, rapid, accurate, and easily performed and that enable preoperative (sampling before surgery and detection of tumor mutations in the plasma sample of patients), intraoperative and postoperative detection of IDH1/2 mutations in glioma tumors. Knowing whether the tumor will be responsive to post-surgical therapies using molecular markers might be helpful for the surgeon when deciding how much resection to perform [18]. We recruited eight glioma tumors intraoperatively and analyzed prospectively. We informed neurosurgeons about the mutation status of the patients during the surgeries; however, none changed the extent of resection, as—first—this was a pilot study for mutation detection and—second—evidence showing a clear correlation between extensive resection and longer survival is lacking in the literature [19, 20]. Nevertheless, intraoperative detection of IDH mutations confirmed the frozen and sectioned tumor pathology results in < 1 h and surgeons were informed. Therefore, intraoperative detection can still be a significant tool that may affect surgical management in terms of changing intraoperative decisions regarding the extent of resection needed, especially for low-grade gliomas located at or nearby eloquent areas of the brain. This may increase patient quality of life by avoiding the possibility of major neurological deficits, especially knowing there is no survival benefit from a larger resection if IDH mutation is absent in the tumor [18, 21].
We developed 3m-ARMS as an alternative and complementary method to current methods and for both intraoperative and postoperative detection of the most common IDH mutations. However, 3m-ARMS also has the potential to be used for serum and cerebrospinal fluid-based liquid biopsy for the detection of various point mutations in many tumor types. Serum-based liquid biopsy is becoming more commonly utilized in detection of circulating tumor DNA to monitor mutations in tumors of various malignancies [22]. Currently, we are evaluating whether 3m-ARMS is sensitive enough to detect point mutations in circulating cancer-associated DNA obtained from serum samples. Preliminary data indicate that 3m-ARMS does have the potential for preoperative mutation detection. We are also in the process of optimizing 3m-ARMS analysis for the detection of other rare point mutations in the IDH1 and IDH2 genes and point mutations in other genes such as TERT and MGMT that have been implicated in the prognosis of glioma tumors.
4.1 Limitations of the Assay
The 3m-ARMS method proposed here detects G395A (R132H), C394T (R132C) and C394G (R132G) mutations in the IDH1 gene and G515A (R172K), A514T (R172W) mutations in the IDH2 gene. These mutations account for almost 98.3% of all IDH mutations in glioma tumors [23]. However, other IDH mutations remain very uncommon and can rarely be seen in glioma tumors. Although the 3m-ARMS method proposed here covers the majority of IDH mutations in glioma tumors, the possibility of these other rare IDH mutations that cannot currently be detected remains a limitation of our mutation-detection method. If results with the 3m-ARMS method are negative, Sanger sequencing of tumor samples is recommended if possible. We aim to design and add new primers that can also detect rare mutations. Nevertheless, the 236 glioma tumors studied in this manuscript did not show other rare mutation types with Sanger sequencing. The current antibody clone commonly used for IHC-based detection of IDH1 mutations is designed to detect only G395A (R132H) mutations. Several reports have indicated varying degrees of FN and/or FP results for IHC-based IDH mutation detection methods in glioma [15, 24, 25]. Therefore, we claimed 100% sensitivity of 3m-ARMS (Table 1) in our cohort in comparison with IHC.
5 Conclusions
Sanger sequencing and IHC-based methods of mutation detection have several shortcomings, especially when the relative ratio of cancer cells carrying the mutation of interest in tumor samples is low. 3m-ARMS offers a fast, specific and sensitive method for the detection of the most common IDH1/2 mutations in glial tumors. Compared with the conventional IHC technique, 3m-ARMS showed a greater diagnostic capability because of its favorable specificity, PPV and likelihood ratios. 3m-ARMS can be utilized simultaneously with Sanger sequencing or IHC for better diagnosis of challenging specimens. The design of our new molecular test may be applied in the future to detect several other mutations to facilitate and expedite the process of mutation detection.
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–20. https://doi.org/10.1007/s00401-016-1545-1.
Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75(17):1560–6. https://doi.org/10.1212/WNL.0b013e3181f96282.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465(7300):966. https://doi.org/10.1038/nature09132.
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372(26):2499–508. https://doi.org/10.1056/NEJMoa1407279.
Shankar GM, Francis JM, Rinne ML, Ramkissoon SH, Huang FW, Venteicher AS, et al. Rapid intraoperative molecular characterization of glioma. JAMA Oncol. 2015;1(5):662–7. https://doi.org/10.1001/jamaoncol.2015.0917.
Kanamori M, Kikuchi A, Watanabe M, Shibahara I, Saito R, Yamashita Y, et al. Rapid and sensitive intraoperative detection of mutations in the isocitrate dehydrogenase 1 and 2 genes during surgery for glioma. J Neurosurg. 2014;120(6):1288–97. https://doi.org/10.3171/2014.3.JNS131505.
Santagata S, Eberlin LS, Norton I, Calligaris D, Feldman DR, Ide JL, et al. Intraoperative mass spectrometry mapping of an onco-metabolite to guide brain tumor surgery. Proc Natl Acad Sci USA. 2014;111(30):11121–6. https://doi.org/10.1073/pnas.1404724111.
Boisselier B, Marie Y, Labussiere M, Ciccarino P, Desestret V, Wang X, et al. COLD PCR HRM: a highly sensitive detection method for IDH1 mutations. Hum Mutat. 2010;31(12):1360–5. https://doi.org/10.1002/humu.21365.
Shi S-R, Datar R, Liu C, Wu L, Zhang Z, Cote RJ, et al. DNA extraction from archival formalin-fixed, paraffin-embedded tissues: heat-induced retrieval in alkaline solution. Histochem Cell Biol. 2004;122(3):211–8.
Little S. Amplification-refractory mutation system (ARMS) analysis of point mutations. Curr Protoc Hum Genet. 2001. https://doi.org/10.1002/0471142905.hg0908s07.
Li J, Wang L, Mamon H, Kulke MH, Berbeco R, Makrigiorgos GM. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14(5):579–84. https://doi.org/10.1038/nm1708.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. https://doi.org/10.1056/NEJMoa0808710.
Agarwal S, Sharma MC, Jha P, Pathak P, Suri V, Sarkar C, et al. Comparative study of IDH1 mutations in gliomas by immunohistochemistry and DNA sequencing. Neuro Oncol. 2013;15(6):718–26. https://doi.org/10.1093/neuonc/not015.
Arita H, Narita Y, Matsushita Y, Fukushima S, Yoshida A, Takami H, et al. Development of a robust and sensitive pyrosequencing assay for the detection of IDH1/2 mutations in gliomas. Brain Tumor Pathol. 2015;32(1):22–30. https://doi.org/10.1007/s10014-014-0186-0.
Jabbar KJ, Luthra R, Patel KP, Singh RR, Goswami R, Aldape KD, et al. Comparison of next-generation sequencing mutation profiling with BRAF and IDH1 mutation-specific immunohistochemistry. Am J Surg Pathol. 2015;39(4):454–61.
Takano S, Tian W, Matsuda M, Yamamoto T, Ishikawa E, Kaneko MK, et al. Detection of IDH1 mutation in human gliomas: comparison of immunohistochemistry and sequencing. Brain Tumor Pathol. 2011;28(2):115–23. https://doi.org/10.1007/s10014-011-0023-7.
Poulen G, Goze C, Rigau V, Duffau H. Huge heterogeneity in survival in a subset of adult patients with resected, wild-type isocitrate dehydrogenase status, WHO grade II astrocytomas. J Neurosurg. 2018. https://doi.org/10.3171/2017.10.jns171825.
Delev D, Heiland DH, Franco P, Reinacher P, Mader I, Staszewski O, et al. Surgical management of lower-grade glioma in the spotlight of the 2016 WHO classification system. J Neurooncol. 2019;141(1):223–33.
Sanai N, Berger MS. Glioma extent of resection and its impact on patient outcome. Neurosurgery. 2008;62(4):753–66.
Clark VE, Cahill DP. Extent of resection versus molecular classification: what matters when? Neurosurg Clin. 2019;30(1):95–101.
Shankar GM, Kirtane AR, Miller JJ, Mazdiyasni H, Rogner J, Tai T, et al. Genotype-targeted local therapy of glioma. Proc Natl Acad Sci. 2018;115(36):E8388–94.
Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10(8):472–84. https://doi.org/10.1038/nrclinonc.2013.110.
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469–74. https://doi.org/10.1007/s00401-009-0561-9.
Preusser M, Wöhrer A, Stary S, Höftberger R, Streubel B, Hainfellner JA. Value and limitations of immunohistochemistry and gene sequencing for detection of the IDH1-R132H mutation in diffuse glioma biopsy specimens. J Neuropathol Exp Neurol. 2011;70(8):715–23.
Lee D, Suh YL, Kang SY, Park TI, Jeong JY, Kim SH. IDH1 mutations in oligodendroglial tumors: comparative analysis of direct sequencing, pyrosequencing, immunohistochemistry, nested PCR and PNA-mediated clamping PCR. Brain Pathol. 2013;23(3):285–93.
Author information
Authors and Affiliations
Contributions
TA and MA conceived and designed the study and wrote the manuscript. AS, GT, BNY, DA, KC, GD, AB, OY participated in data acquisition, analysis and interpretation of data. TK provided the tumor samples and patient information and participated in critical discussions.
Corresponding authors
Ethics declarations
Conflicts of interest
Timucin Avsar, Alihan Sursal, Gizem Turan, Berfu Nur Yigit, Deniz Altunsu, Kutay Cantasir, Gözde Duyu, Ahmed Bayoumi, Ozlem Yapicier, Melih Acar, and Turker Kilic have no conflicts of interest that are directly relevant to the content of this article.
Funding
This study was funded by Bahcesehir University, Scientific Research Projects Council, project no. BAP.2018-2.01, and The Scientific and Technological Research Council of Turkey (TUBITAK) Grant, project no. 118S539.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Avsar, T., Sursal, A., Turan, G. et al. Development of a Rapid and Sensitive IDH1/2 Mutation Detection Method for Glial Tumors and a Comparative Mutation Analysis of 236 Glial Tumor Samples. Mol Diagn Ther 24, 327–338 (2020). https://doi.org/10.1007/s40291-020-00461-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-020-00461-y