
Abstract
Nowadays, scientists are trying to develop low-cost fullerene free acceptors for small organic photovoltaic cells in order to overcome the limitations of fullerene derivatives. Current research work deals with theoretical study on three non-fullerene acceptors based on indaceno, dithiophene core, and thiophene bridge units linked with dissimilar end non-fullerene groups which act as strong acceptor moieties. Different optoelectronic characteristics of the designed molecules were calculated and compared with the reference compound R (indaceno dithiophene–based fused ring acceptor) which is recently reported. Results shows that C2 and C3 exhibit broad absorption spectrum and lower band gap whereas C2 and C1 exhibit highest open-circuit voltages VOC value with B3LYP and MPW1PW91 functionals respectively as compared with the R. All designed molecules have high dipole moment values, lower value of hole reorganization energy λh than electron reorganization energy λe which reflects that our designed acceptor molecules are good candidates for organic photovoltaics.

Absorption spectra of R and three designed non-fullerene acceptors with strong absorption band in the visible region of solar cells spectrum.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nowadays, organic solar cells (OSCs) gain much more attentions due to their different characteristics such as environment friendly, low cost, good mechanical strength, and light weight. Solar energy is most appealing, sustainable, safe, and clean energy [1]. At first, silicon-based inorganic materials were used for manufacturing solar cells. Semiconductor organic polymers have tremendous influence on the efficiencies of photovoltaic cells [2]. Previously, organic solar cells achieved maximum power conversion efficiencies (PCEs) by optimizing semiconducting polymers [3]. Organic bulk heterojunction solar cells have been considered next-generation renewable power source because of their benefits such as flexibility, low cost, and light weight devices [4] having long life span as compared with the perovskite photovoltaic devices. Power conversion efficiencies of OSCs exceed more than 14% nowadays. Organic polymers act as electron-donating compounds, while derivatives of fullerenes, i.e., 6,6-phenyl-C61-butyric acid methyl ester, act as electron acceptors which have near high electron mobility for efficient separation and transfer of charge. Although fullerene-based derivatives have many advantages, there are some limitations such as weak absorption in visible region [5]. Due to shortcomings of fullerene derivatives, small molecules with non-fullerene acceptors have obtained great attention because of having competitive orbital energies [6]. Non-fullerene acceptors exhibit the better performance and improved absorption coefficient when blend with polymers [5].
For large-scale production, semiconductors based on small molecules are suitable than polymers. Small molecules A-D-A type comprise of electron-accepting part as end-capped group, electron-donating part as central core unit, and thiophene as pi conjugated bridge. These types of construction can be helpful for tuning the HOMO-LUMO energy levels, minimized band gap and increased absorption strength. A-D-A type small molecules showed inspiring performance in polymer solar cells [3]. Many fused ring electron acceptors have been reported, i.e., diketopyrrolopyrrole, naphthalene diimide, perylene diimide, ITIC, and so on. Indaceno dithiophene–based fused ring acceptors IDIC have already been designed and synthesized [7]. Indaceno dithiophene (IDT) is promising organic semiconductor due to its efficient electron-donating abilities and charge mobility. IDT-based copolymers show high mobility of holes and wide absorption and maximum molar extinction coefficient which is helpful in attaining large short circuit current density [3].
In current research work, three acceptors C1, C2, and C3 based on IDT central core units with different strong electron withdrawing acceptor moieties attached via π conjugated methyl thiophene bridge for enhancing the optoelectronic properties of organic photovoltaics have been designed (Fig. 1).

Designed chemical structures with non-fullerene acceptors
Computational details
Initially, the structures of R and our designed molecules were drawn on 5.0 program [8, 9]. All the neutral molecules were optimized (Fig. 2) by applying the density functional theory (DFT) [10] with four different functionals such as CAM-B3LYP [11], B3LYP [12], WB97XD [8], and MPW1PW91 [13] at 6-31G (d,p) level of theory [14]. UV/Visible absorption spectra of theoretically studied molecules have been computed with chloroform solvent/IEFPCM model and in gas phase conditions. Absorption spectra obtained with four different functionals are compared with experimental reported data of R molecule. Density of states (DOS) has been calculated with PyMOlyze-1.1 software. Absorption spectra were plotted in Origin 6.0 software.

Optimized geometries of theoretically studied and designed molecules
Results and discussion
Electronic properties
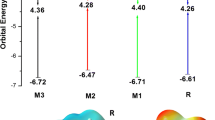
To understand the electronic behavior of designed and reference compound R, distribution of the electron density between HOMO and LUMO energy orbitals is very important. HOMO exhibit bonding character and LUMO exhibit anti-bonding character. The distribution patterns of frontier molecular orbitals (FMOs) with both functionals are shown in supporting information. Electronic transitions in excited states with B3LYP functional are 100% in gas phase and vary from 54 to 100% with chloroform solvent. Similarly in the case of MPW1PW91 functional, electronic excitations vary from 98 to 99% in both gas phase and with chloroform solvent. EHOMO-ELUMO of R is − 5.76 eV and − 3.51 eV respectively. HOMO values of C1, C3, and C2 are − 5.74 eV, − 5.72 eV, and − 5.41 eV. On the other hand, LUMO values are − 3.31 eV, − 3.11 eV, and − 3.02 eV for C3, C1, and C2 with B3LYP/6-31G (d,p) level of DFT in gas phase (Table 1). Eg values are 2.63 eV for C1, 2.39 eV for C2, and 2.41 eV for C3. Band gap decreases in order C1 > C3 > C2. Minimum Eg of C2 as compared with C3 shows effective intra-molecular charge transfer (ICT) that shifts the absorption spectra toward red shift. Eg of designed molecule C3 is smaller than C1 because of strong electron-accepting effect of end-capped units in C2 and C3. Among all compounds, C2 shows comparable Eg (2.39 eV) with respect to the reference compound R (2.25 eV). Eg value of the reference R is 2.21 eV with MPW1PW91/6-31G (d,p). Eg decreased in order of C1 > C2 > C3 with this functional as C1, C2, and C3 having 2.83 eV, 2.36 eV, and 2.22 eV respectively. Eg of C2 and C3 are comparable to reference R due to strong electron acceptor moieties. HOMO and LUMO values are − 5.99 eV and − 3.04 eV for C1, − 5.45 eV and − 3.11 eV for C2, and − 5.97 eV and − 3.27 eV for C3. Eg values are 2.95 eV, 2.68 eV, and 2.70 eV for C1, C2, and C3 respectively with chloroform solvent/IEFPCM model (Table 2). Eg decreases in the order of C1 > C3 > C2. Small band gap of C2 as compared with C3 and minimum band gap of C3 than C1 is due to the derivatives of thiazolidine acceptor group in C2 and malononitrile derivatives acceptor in C3. HOMO-LUMO values of R are − 6.01 eV and − 3.49 eV at the MPW1PW91/6-31G (d,p) level of theory. C2 has comparable Eg (2.68 eV) as compared with reference compound R (2.52 eV). The orbital energy comparisons of designed molecules with both functionals are shown in Fig. 3.

Orbital energy comparisons of R and C1 to C3 designed structures
Density of state (DOS) has also been computed with the both functionals B3LYP and MPW1PW91 at 6-31G (d,p) level of theory. DOS contributions reinforced the facts obtained by FMOs. HOMO arises from donor and spacer units with little overlapping effect of acceptor moieties in all compounds. LUMO mainly resides on acceptor with overlapping effect of donor moieties and spacer in all compounds. The graphs of DOS calculations are shown in supporting information.
Optical properties
For computing the electronic transitions of our designed molecules including reference compound R, UV/Visible absorption spectra were recorded with TD-B3LYP/6-31G (d,p) and TD-MPW1PW91/6-31G (d,p) level of theories in both phases gas as well as with chloroform solvent. UV/Visible spectra of absorption are shown in Fig. 4. π-π* transition decreases the absorption wavelength, and indaceno thiophene is responsible for red shift in absorption spectra among the donor and acceptor units which is confirmed during theoretical studies. UV/Visible spectra of designed molecules lie in the range of 499–561.2 nm in gas phase and 492.3–714.1 nm with solvent by B3LYP and by using MPW1PW91 functional absorption varies from 476.2 to 534.5 nm in gas phase and 504.5 to 567.4 nm with solvent/IEFPCM model (Table 3). Eg values have reciprocal trend with λmax. By using B3LYP functional, absorption spectra of C3 show red shift from C2 by 2.6 nm, C2 shows red shift from C1 by 59.6 nm, and absorption spectra of R is 604.4 nm in gas phase. C1 shows the blue shift from C2 and C3 by 59.6 nm and 62.2 nm respectively (Table 4). In the case of chloroform, absorption strength of R is 652.2 nm. C3 shows the red shift from R by 61.9 nm; it seems to be overestimated by B3LYP; C2 shows red shift from C1 by 104.5 nm. Red shift of C2 is due to strong electron withdrawing moieties present at the end of structure. In both phases, excitation energies decrease in order C1 > C2 > C3. Transitions which have larger oscillating strength (f) are most feasible transitions from HOMO to LUMO in both phases. With MPW1PW91 functional, C3 shows red shift from C2 by 2.6 nm, C2 shows red shift from C1 by 55.6 nm, and absorption strength of R is 577.4 nm in gas phase. In chloroform, C3 absorption spectra show red shift from C2 by 1.2 nm, C2 shows red shift from C1 by 61.7 nm, and absorption of R is 620.8 nm. Excitation energy is decreased in order C1 > C2 > C3. In both functionals, C3 is helpful to enhance photoelectric conversion efficiency of the photovoltaic cells because of C3 having more tendency to harvest the light at longer wavelength.

Absorption spectra of designed molecules with strong absorption bands
Dipole moment
Dipole moment of designed compounds and reference R is calculated with B3LYP/6-31G (d,p) and MPW1PW91/6-31G (d,p) level of theories (Table 5). All the compounds exhibit higher dipole moment than R. Higher value of dipole moment of designed acceptor molecules exhibits their good solubility in non-polar solvent for fabrication of OSCs. In self-assembly of molecules, dipole moment plays a key role which arranges the neighboring molecular dipoles in antiparallel fashion and enhances the crystallinity and self-assembly of thin film bulk heterojunction OSCs [15]. In the case of B3LYP, value of dipole moment in ground state of C1 to C3 is 4.23 D to 3.76 D and value of R is 0.0015 D whereas the value of dipole moment in excited states of C1 to C3 is 2.94 D, 5.51 D, and 8.86 D respectively and value of R is 0.0018 D (Table 5). In the case of MPW1PW91/6-31G (d,p) level of the theory, the value of dipole moment in ground states is 4.25 D, 3.75 D, and 0.0017 D for C1 to C3 respectively and value of R is 0.0014 D. The value of dipole moment in excited states is 4.83 D, 5.46 D, and 0.0019 D for C1 to C3 respectively and value of R is 0.0018 D as shown in Table 5. Molecules which have ability to self-assemble show supramolecular long chain which support the charge transfer by providing a straight way for hole and electron. Higher dipole moment increases the fill factor and lower the recombination of charge in donor acceptor moieties.
Reorganization energy
Reorganization energy plays a vital role in determination of charge mobilities of organic solar cells. The lower the energy of reorganization, the higher will be the charge mobilities. Hole reorganization energy λh and electron reorganization energy λe can be formulated [16] by using the following relationships:
where E+/E− shows the energy of cation/anion calculated by optimized structures. \( {E}_0^{+}/{E}_0^{-} \) represents the energy of cation and anion estimated by optimized neutral molecule. \( {E}_{+}^0/{E}_{-}^0 \) represents the energy of neutral molecule via optimized structure of cation and anion. E0 shows energy of neutral molecule at ground state. In the case of B3LYP, λe of C2 is 0.0100 eV lesser than C3 having 0.0102 eV which means that designed molecule C2 exhibits higher electron mobility as compared with C3. λe of C3 is lesser than C1 having 0.0144 eV which shows that electron transfer rate of C3 is greater than C1. λh of C3 is 0.0067 eV lesser than C2 having 0.0072 eV. λh of C2 is lesser than C1 having 0.0081 eV. λe of R is 0.0077 eV, and λh of R is 0.0057 eV which shows that hole transfer rate of R is greater than electron transfer rate (Table 6). In the case of MPW1PW91/6-31G (d,p) level of theory, λe of C2 is 0.0107 eV lesser than C3 having 0.0109 eV. λe of C3 is lesser than C1 having 0.0153 eV. λh of C3 is 0.0074 eV lesser than C2 having 0.0078 eV. λh of C2 is lesser than C1 having 0.0087 eV which shows that hole transfer rate of C3 is greater than C2 and hole transfer rate of C2 is greater than C1. λe of R is 0.0074 eV and λh of R is 0.0069 eV which shows that hole transfer rate of R is higher than electron transfer rate. λh is lesser than λe which inspire the material for transfer of holes (Table 6). Very small difference between λe and λh shows that molecules have equilibrium characteristics for electron and hole transfer.
Open-circuit voltages (V OC)
To permit the charge carriers toward the electrodes, VOC reaches the proficiency of excitation separation process. VOC is highest voltage quantity eradicated from solar cells. It is obtained at zero level of current which shows highest voltage attained from the solar cell devices. Larger VOC highest will be the PCEs. VOC can be measured by taking difference between LUMO of acceptor and HOMO of the donor moieties during DFT analysis. As illustrated in Fig. 5, differences of the HOMO of donor molecule, LUMO of designed acceptor molecules, and R with B3LYP and MPW1PW91 functional are clearly shown. In the case of B3LYP functional, VOC values of R, C1, C2, and C3 are 2.10 eV, 2.08 eV, 1.75 eV, and 2.06 eV, respectively. Result shows that the VOC of all compounds are greater than reference molecule R. C2 exhibits highest VOC among all compounds. These compounds can be utilized as acceptor molecules in solar cell fabrications because of their high VOC values. In MPW1PW91/6-31G (d, p) level of theory, the VOC of the R, C1, C2, and C3 are 2.34 eV, 2.33 eV, 1.79 eV, and 2.31 eV, respectively. C1 exhibits highest VOC value among all compounds in this functional.

Open-circuit voltage diagram of designed molecules with respect to the fullerene derivatives
Conclusions
In current research work three fused ring acceptor molecules namely C1, C2, and C3 containing pi-bridge with different acceptors at the ending parts by using DFT tools have been designed in order to examine the optoelectronic and photophysical characteristics of organic solar cells. Among our investigated compounds, C2 exhibit minimum band gap of energy as 2.39 eV in gas phase and C3 showed 2.22 eV in chloroform by B3LYP/6-31G (d, p) level of DFT. In MPW1PW91/6-31G (d,p), C2 exhibited minimum band gap energy as 2.68 eV in gaseous phase and in the chloroform solvent 2.65 eV. C2 and C3 showed broad absorption spectra with both functionals which are in the range of 558.6 and 561.2 nm, respectively. In gas phase and in chloroform, C2 exhibited broad absorption spectrum as giving 596.8-nm wavelength and C3 absorption wavelength was 714.1 nm during this simulation analysis which seems to be overestimated by B3LYP. C2 and C3 showed 531.8 nm and 534.5 nm respectively in gas phase and 566.2 nm and 567.4 nm in chloroform due to electron withdrawing groups present at the end of capped units. C2 showed highest VOC value 2.08 eV with B3LYP which confirms that it is suitable acceptor molecule for the construction of bulk heterojunction organic solar cell. With MPW1PW91/6-31G (d,p), C1 exhibited highest VOC as 2.33 eV. Dipole moment of designed molecules were calculated which are helpful in understanding their solubility in organic solvents. Reorganization energy calculation have shown that λh value is lower than λe which reflects that designed acceptor molecules could be ideal for transformation of holes. C1 showed highest VOC value at MPW1PW91/6-31G (d,p) level of theory. All these conclusions are helpful for the further construction of bulk heterojunction OSCs by using designed acceptor units.
References
Ku J, Lansac Y, Jang YH (2011). J Phys Chem C 115:21508–21516
Bagher AM (2014). Int J Renew Sustain Ener 3:53–58
Bai H, Wang Y, Cheng P, Li Y, Zhu D, Zhan X (2014). ACS Appl Mater Interfaces 6:8426–8433
Qiu B, Xue L, Yang Y, Bin H, Zhang Y, Zhang C, Xiao M, Park K, Morrison W, Zhang Z-G (2017). Chem Mater 29:7543–7553
Lin F, Huang W, Sun H, Xin J, Zeng H, Yang T, Li M, Zhang X, Ma W, Liang Y (2017). Chem Mater 29:5636–5645
Chang M, Wang Y, Qiu N, Yi YQQ, Wan X, Li C, Chen Y (2017). Chin J Chem 35:1687–1692
Q. Wang, Y. Li, P. Song, R. Su, F. Ma, Y. Yang, Polymers, 9 (2017) 692
Bari A, Irfan M, Zara Z, Eliasson B, Ayub K, Iqbal J (2017). J Mol Struct 1143:8–19
Bourass M, Amine A, Hamidi M, Bouachrine M (2017). New organic dyes based on phenylenevinylene for solar cells: DFT and TD-DFT investigation. Karbala International Journal of Modern Science, 3(2):75–82
Wang J-L, Xiao F, Yan J, Liu K-K, Chang Z-F, Zhang R-B, Wu H-B, Cao Y (2016). J Mater Chem A 4:2252–2262
Manzoor F, Iqbal J, Zara Z, Eliasson B, Mahr MS, Ayub K (2018). ChemistrySelect 3:1593–1601
Bourass M, Benjelloun AT, Benzakour M, Mcharfi M, Hamidi M, Bouzzine SM, Bouachrine M (2016). Chem Cent J 10:67
Zara Z, Iqbal J, Ayub K, Irfan M, Mahmood A, Khera RA, Eliasson B (2017). J Mol Struct 1149:282–298
Bourass M, Benjelloun AT, Benzakour M, Mcharfi M, Jhilal F, Serein-Spirau F, Sotiropoulos JM, Bouachrine M (2017). J Saudi Chem Soc 21:563–574
Turkoglu G, Cinar ME, Ozturk T (2017). Top Curr Chem 375:84
Ali U, Javed A, Tallat A, Iqbal J, Raza A (2019) Molecular designing of four high performance pyrazine-based non-fullerene acceptor materials with naphthalene diimide-based small organic solar cells. J Mol Model 25(2):50
Acknowledgments
The authors acknowledge the technical support provided by the Department of Chemistry, University of Agriculture, Faisalabad, 38040, Pakistan.
Funding
The study was financially supported by the Department of Chemistry, University of Agriculture, Faisalabad, 38040, Pakistan.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 551 kb)
Rights and permissions
About this article

Cite this article
Ajmal, M., Ali, U., Javed, A. et al. Designing indaceno thiophene–based three new molecules containing non-fullerene acceptors as strong electron withdrawing groups with DFT approaches. J Mol Model 25, 311 (2019). https://doi.org/10.1007/s00894-019-4198-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4198-x