
Abstract
Three acceptor-donor-acceptor (A-D-A)-type small donor molecules (M1, M2 and M3) were evaluated for optoelectronic properties through density functional theory calculations. These designed molecules consist of a dithieno [2,3-b:3,2-d] thiophene (DTT) donor group linked with 2-(3-methyl-4-oxothiazolidin-2-ylidine)malononitrile acceptor through three different bridge groups. The effect of the donor and three different bridge spacer groups on the designed molecules for opto-electronic properties was evaluated in comparison with the reference molecule R. The reorganization energies of the designed donor molecules suggest very good charge mobility property. The lower value of hole mobility (λh), as compared to electron mobility (λe), revealed that the three designed molecules are best for hole mobility. Frontier molecular orbital (FMO) surfaces confirm the transfer of charge from donor to acceptor unit during excitation. The designed molecules show relatively low HOMO values (in the range of −2.19 to −2.36 eV), with strong absorption in UV-Visible region in the range of 459 nm to 500 nm in chloroform solvent. Electron-hole binding energy results indicate that the designed molecule M2 contains the highest amount of charge, which may dissociate into separate charge easily. Among all the studied molecules, the highest open circuit voltage (Voc) of 3.01 eV (with respect to HOMOdonor–LUMOPC61BM) was shown by M3. The open circuit voltages (Voc) of R, M1, M2 and M3 were 2.91 eV, 3.01 eV, 2.77 eV and 3.02 eV, respectively.

Three newly acceptor donor acceptor (A-D-A)-based donor small molecules (M1, M2 and M3) were designed by taking dithieno [2,3-b:3,2-d]thiophene (DTT) as a donor group linked with acceptor 2-(3-methyl-4-oxothiazolidin-2-ylidine)malononitrile through three different bridge groups. All the designed molecule were compared with the well-known reference compound R. Optical properties, electronic properties, photophysical and excited state energy were calculated and compared with the well-known, and recently published, reference molecule R . All the newly designed molecules shows good optoelectronic properties with respect to R.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The world now faces a serious energy crisis due to ever increasing energy demands. Primarily fossil fuels are used to meet current energy demands; however, they have certain limitations. For example, fossil fuels are non-renewable energy sources and cause environmental pollution [1]. These limitations have motivated scientists to explore alternate renewable energy sources that are environmentally friendly, such as, wind energy, hydro power energy, geothermal energy, solar energy, etc. [2]. Solar energy is of particular interest among the potential alternative to fossil fuels as a renewable and environmentally friendly energy source [3].
Different types of solar cells, including silicon-based solar cells, mono crystalline silicon solar cells, solution-processed solar cells and gallium-selenide-based solar cells, have been used previously [4]. Although these solar cells showed remarkable efficiency, inorganic solar cells have many disadvantages, such as rigid structure [5], high cost, non-tunable energy levels, etc. Organic solar cells have many advantages over silicon-based inorganic solar cells, such as low cost [6], light weight [7], flexibility, tunable structure and batch-to-batch reproducibility [8,9,10,11,12]. Over the past decade, organic photovoltaic cells have attained remarkable power conversion efficiency (PCE). For example, PCE values >10% can be achieved with polymer solar cells [13,14,15,16,17,18]. More recently, efforts have been exerted to design small molecules that can be used in solar cells. In this regard, a variety of donor molecules with diverse structures, e.g., star shaped, X-shaped, linear and others, have been reported in the literature in conjunction with PCBM acceptor ([6,6]-phenyl-C61-butyric acid methyl ester). Many fascinating characteristics of these small molecule organic solar cells reflect their potential as alternatives to solution-processed polymer solar cells (PSCs).
Although small molecule organic solar cells (SMOSCs) have emerged as an alternative to solution-processed PSCs, they still suffer from several limitations. For example, the frontier molecular orbitals (FMOs) of designed donor molecules do not align properly with the FMOs of PCBM, which significantly affects the open circuit voltage due to poor film morphology and low charge transport ability. To overcome the above cited problems, scientists have designed small molecule donor materials with special emphasis on solubility, absorption bands, suitable energy levels, and charge mobility.
The literature reveals several studies where computational tools have been used to design new molecules with improved efficiencies for solar cell applications. For example, computational studies were carried out for the design of dibenzosilol donor unit based A-D-A type molecules with different end-capped acceptor units for application in organic solar cells [19]. In the latter study, density functional theory (DFT) methods were used to evaluate different optoelectronic properties of organic solar cells [19]. In another DFT study, A-D-A type donor molecules based on the benzodithiophene donor core unit flanked with different acceptor moiety were evaluated for their optoelectronic properties. The study revealed that CAM-B3LYP/6-31G(d,p) level of theory is best for this class of compounds. Furthermore, the designed donor is blended with well-known acceptor PCBM [20].
To improve the performance of small molecules, we applied a dithieno (DTT) donor moiety to the organic solar cell due to its better optical properties [21]. Herein, we designed dithieno-based organic solar cells with donor and acceptor groups, linked together via a suitable bridge. Three molecules, M1, M2 and M3, were designed, where the donor dithieno(2,3-b:3,2-d)thiophene and acceptor 2-(3-methyl-5-methylene-4-oxothiazolidin-2-ylidene)malononitrile are linked through 3-methylthiophene (M1), 3-methlyfuran (M2) and 3-methylselenophen (M3) groups, respectively. The calculated properties are compared with well-known model molecule R, which is structurally comparable to the designed molecules.
Computational details
All calculations were performed using the Gaussian 09 program package [22]. Molecular structures were drawn with the GaussView 5.0 program [23]. Initially, for selection of basis set, the geometries and absorption spectra of the reference compound R [24] were calculated with different functionals, including B3LYP [25] CAM-B3LYP [26], and ωB97XD [27] at 6-31G (d,p) basic set [28], and the results compared with available experimental data. CAM-B3LYP gave closest agreement between theoretical and experimental results. The basis set 6-31G (d,p) gave the most reliable result for geometry optimization as well as for electronic properties [29, 30]. We evaluated different basis sets, such as 6–31 + G(d,p), 6–311 + G(d,p), 6–311++G(2d,2p), 6–311 + G(d,p) and 6-31G(d,p); however, the 6-31G(d,p) basis set proved best in term of results and computational cost (Table S1). Further, all calculations for the cationic, anionic and neutral species were performed at CAM-B3LYP/6-31G (d,p) level of theory.
For calculation of absorption spectra, time-dependent density functional theory (TD-DFT) with CAM-B3LYP at 6-31G (d,p) level of theory was used in gas as well as in solvent (chloroform) phase. The solvent effect was incorporated through the integral equation formalism polarizable continuum model (IEPCM) with chloroform solvent. UV-visible absorption spectra were drawn using the origin 6.0 program. Density of states (DOS) around HOMO–LUMO (highest occupied molecular orbital to lowest unoccupied molecular orbital gap) were calculated using PyMolyze software [31]. Reorganization energies of hole (λh) and electron (λe) were estimated with the selected functional. Reorganization energy was further divided into two parts: internal reorganization energy (λint) [32] and external reorganization energy (λext). The external environmental relaxation and effect of polarization on the external surrounding medium were explained by λext, while, on the other hand, λint revealed the fast changes in internal geometry. Herein, we are working on small donor molecules that have low dielectric constant value. Hence, we neglected the external surrounding interference on our study, i.e., only internal reorganization energy λint is discussed. Finally, the reorganization energy of hole (λh) and electron (λe) were calculated by the following Eqs. (1 and 2).
Where, \( {E}_0^{+},{E}_0^{-} \)shows the energy of cations and anions calculated at optimized structure of neutral molecules. The \( {E}_{-}^0,{E}_{+}^0 \) are the energies of neutral molecules calculated via optimized geometries of anion and cation, respectively. E0 is the single point energy of optimized structure of neutral molecule. Finally E+, E− are the energies of cation and anion at optimized geometry of cations and anions.
Results and discussion
Different DFT functionals including B3LYP, CAM-B3LYP, and ωB97XD with 6-31G (d,p) basis set were used for selecting suitable functionals. The maximum absorption values with B3LYP, CAM-B3LYP, and ɷB97XD are 626 nm, 450 nm, and 424 nm, respectively, while the experimental value is 532 nm (see Table S2). The UV-vis spectrum of reference R simulated with CAM-B3LYP provided the best match with the experimental spectrum. Hence, all further calculations were performed with this selected functional. We evaluated different basis sets: 6–31 + G(d,p), 6–311 + G(d,p), 6–311++G(2d,2p), 6–311 + G(d,p) and 6-31G(d,p). The λmax values at 6–311 + G(d,p), 6–311++G(2d,2p), 6–311 + G(d,p), and 6–31 + G(d,p) were 451.6 nm, 450.1 nm, 438.09 nm, and 448.7 nm, respectively. The 6-31G(d,p) basis set was chosen for this study by considering the efficiency and cost of the different basis sets tested here. The structure of reference molecule R, as well as the designed molecules M1, M2 and M3 are shown in Fig. 1. Optimized geometries at CAM-B3LYP/6-31G (d,p) level of theory are shown in Fig. 2. Donor and acceptor groups are arranged in one plane in optimized geometry.

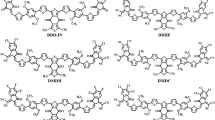
Molecular structures of reference R and designed molecules M1–M3

Optimized structures of reference R and designed molecules M1–M3 at the CAM-B3LYP/6-31G (d,p) level of theory
Frontier molecular orbitals
Charge transition character is strongly influenced by the distribution of FMOs [33]. Optical and electronic properties can be studied with the help of electronic distribution pattern of FMOs, i.e., HOMO and LUMO [34, 35]. The FMO diagram shows the relationship between conjugation and electron mobility. There is a direct relationship between conjugation and charge carrier mobilities. If conjugation/delocalization in the structure increases, the mobility of electrons also increases (Fig. 3). The calculated HOMO energy levels of designed molecules M1, M2 and M3 are −6.71 eV, −6.47 eV, and − 6.72 eV, respectively, while the LUMO energies levels are −2.32 eV, −2.19 eV, −2.36 eV, respectively (Table 1).

Frontier molecular orbitals (FMOs) and highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distribution pattern at the ground state of reference R molecule and molecules M1 to M3
The HOMO value of M2 (−6.47 eV) is higher than that of M1 (−6.71 eV), which can be attributed to the 3-methylfuran bridge group (linked with donor group) in M2 as compared to 3-methylthiophene in M1. The bridge 3-methylthiophene shows better conjugation with the donor group, due to the presence of the thio group in the ring. The LUMO values of M2 and M1 are −2.19 eV and − 2.32 eV, respectively. As a result, M2 has a low energy gap (4.28 eV) as compared to M1 (4.40 eV). The HOMO value of M3 (−6.72 eV) is even lower than that of M1 (−6.71 eV), which is due to the more conjugated 3-methylselenophene group (with donor) in M3 compared to 3-methylthiophene in M1. As a result, M3 has a low energy gap than M1. The HOMO and LUMO values of the reference molecule R are −6.61 eV and − 2.35 eV, respectively, which are higher than those of designed molecules M1 and M3.
The low HOMO value of M3 is useful in obtaining high open circuit voltage in organic solar cells. The designed molecule M2 has an orbital energy (eV) comparable to the corresponding value for the reference molecule R, as shown in Fig. 4. The density of states (DOS) calculations for all molecules were performed at CAM-B3LYP/6-31G (d,p) level of theory. DOS helps to find the density of electrons throughout the molecule. The presence of different bridge groups with donor, dithieno [2,3-b:3,2-d] thiophene, affects the distribution pattern of electron density over all the molecules. Density of states support the inferences obtained from FMO analysis, as shown in Fig. 5.

Evaluation of the calculated HOMO and LUMO energies for reference molecule R and designed donor molecules M1–M3 at CAM-B3LYP/6-31G (d,p) level of theory

Density of states around HOMO and LUMO for reference molecule R and designed donor molecules M1–M3
The FMO diagram shows the distribution of electron density around HOMO and LUMO. The HOMO of M1 is distributed all over the molecule including donor, bridge as well as acceptor sites, while the LUMOs are spread over the donor and 3-methylthipehene bridge groups only. The HOMO of M2 is distributed over the entire molecule except the spacer group, while the LUMO is distributed uniformly over the entire molecule. The HOMO of M3 shows a pattern similar to that of M2, i.e., it is distributed on donor and spacer groups while the LUMOs are distributed all over the molecule. The HOMO of the reference molecule R is distributed all over the molecule, except the acceptor groups, while the LUMOs are spread all over the molecules. DOS (Fig. 5) calculations were also performed with selected functionals to support the findings illustrated in the FMO diagram. The DOS further supports the idea of distribution of the electron pattern according to the donor moiety. DOS also shows the energies of HOMO–LUMO gap of reference R and designed molecules M1, M2 and M3.
Furthermore, the molecular electrostatic potential (MEP) shows the distribution of electron-rich and electron-deficient sites in a molecule [36]. The color MEP maps for R, M1, M2 and M3 are illustrated in Fig. 6. The same scale is used for all molecules. Three colors are present in MEP diagrams. The red region show the accumulation of positive charge (electron deficient) while blue illustrates negative charge (electron rich) and green the electrically neutral part. All designed molecules (M1–M3), including reference R, show similar MEP pattern. The end capped acceptor units contain positive charge (red color) whereas bridge units bear negative charge (blue color). The central donor core unit is neutral (green color). It can be seen that M1, M2 and R have greater positive and negative charges on acceptor and bridge units, respectively (with respect to M3). The lesser charge in the case of M3 may be due to the silicon atom on the bridge unit.

Molecular electrostatic potential (MEP) analysis of R, M1, M2 and M3
Optical properties
TD-DFT calculations at the CAM-B3LYP/6-31G (d,p) level were applied in gas phase as well as with solvent (chloroform) to obtain the absorption spectra (Tables 2 and 3). The calculated absorption value (λmax), excitation energy (Ex), dipole moment, and oscillator strength (f) in the gas phase are presented in Table 2.
The λmax of the designed molecules in gas phase ranged from 437 nm to 474 nm. The theoretical calculated λmax values for M1, M2 and M3 are 437 nm, 474 nm and 441 nm, respectively. The designed molecule M2 shows the highest λmax value as compared to the reference R, which is attributed to extended conjugation in the 3-methylfuran bridge group with donor (DTT) group. We can clearly see from the results that λmax values are strongly affected by extended conjugation with the donor unit, which causes a red shift in the absorption spectrum.
The simulated absorption spectra (Fig. 7) show two high intensity peaks in the case of M1. The peak around 310 nm is due to a 3-methylfuran moiety. M3 has a higher absorption wavelength than M1 due to bridge group 3-methylselonophene. The designed molecules M1 and M3 have similar absorption profiles with respect to the reference molecule R. The absorption value of M3 is 9 nm blue shifted as compared to R. The absorption value (λmax), excitation energy (Ex), dipole moment, and oscillator strength (f) with TD-CAM-B3LYP/6-31G (d,p) in chloroform solvent are shown in Table 3.

Simulated absorption spectra of reference R and designed donor molecules (M1–M3) in gas phase and chloroform solvent
The designed molecules M1, M2 and M3 show maximum absorption in chloroform solvent at 459 nm, 500 nm and 462 nm, respectively. Hence, the designed molecule M2 shows the highest λmax value (500 nm) among all designed and reference molecules. The reason for the high absorption of the M2 molecule is the extended conjugation between 3-methylfuran and the donor group (DTT). The simulated absorption spectrum in chloroform solvent shows two high intensity peaks due to the 3-methylfuran bridge group. M3 shows the largest red shift as compared to M1, due to the presence of the 3-methylselenophene group in conjugation with the donor group (DTT). The λmax value of the reference compound is 467 nm. The designed molecules M1 and M3 shows comparable λmax values with reference molecule R.
Reorganization energy
The performance of solar cells is evaluated by charge mobility values, which can be calculated with the assistance of reorganization energy values of electron mobility (λe) and hole mobility (λh). An inverse relation is found between reorganization energy and charge mobility. To obtain high charge mobility, there must be a low reorganization energy value of donor material [37, 38]. The reorganization energy value depends on different factors but is affected primarily by cation geometry and anion geometry (as defined by Eqs. 1 and 2). Reorganization energies of all molecules (M1–M3) including reference R are shown in Table 4.
In our study, we neglected external environmental relaxation and external reorganization and dealt only with internal reorganization energy. The results revealed that the designed molecules have better electron (λe) and hole (λh) motilities as compared to the reference molecule R. The lower value of hole reorganization energy, as compared to electron reorganization energy revealed that the designed molecules are better suited for hole mobility. The λe values of the reference R, and the designed molecules M1, M2 and M3 are 0.02407 eV, 0.02448 eV, 0.01916 eV, and 0.02564 eV, respectively. The designed molecule M2 has the lowest λe value; therefore, it shows the highest electron mobility among all designed and reference molecules. The λe value of M1 and M2 are almost comparable to the λe value of reference molecule R; hence, both M1 and R have same electron mobility. Therefore, the designed molecule M2 is recommended for use in organic solar cells due to its highest hole mobilities. The λe values of all studied molecules are in the order of M2 > M1 > M3.
On the other hand, hole mobility λh for the reference molecule is 0.02099 eV, while the λh of the designed molecules M1, M2 and M3 are 0.02090 eV, 0.01495 eV, and 0.02100 eV, respectively. The designed molecule M2 is the best for hole transport mobility due to having the lowest λh value (0.01495 eV) of all the designed and reference molecules. The designed molecules M1 and M3 have comparable hole transfer abilities to the reference molecule R. The λh values are in the order M2 > M1 > M3.
The above discussion shows that the designed molecules are good candidates for electron and hole mobilities. Furthermore, the designed molecule M2 shows the lowest λe as well as λh values among all the designed and reference molecules. All the designed molecules show good hole and electron mobility, but M2 is the lead molecule, with lowest λe and λh values.
Dipole moment
Another promising factor to evaluate the performance of OSCs is dipole moment. Dipole moments of all designed and reference molecules (M1, M2, M3 and R) were also calculated with CAM-B3LYP/6-31G(d,p) level of theory. The values of dipole moment have a great influence on the fabrication of organic solar cells, and a direct relationship with solubility in organic solvent. The higher the dipole moment, the greater the solubility in organic solvents. From Table 2, it is obvious that the designed molecule M2 has the highest dipole moment among of all designed molecules, as evidenced by its good solubility in organic solvent. The dipole moments are even higher in chloroform solvent (Table 3) than in gas phase (Table 2). The value of dipole moments of designed molecules M1, M2, and M3 in chloroform solvent are in order of M2 > M3 > M1. The dipole moments of M1, M2, and M3 are 3.14 D, 3.79 D, and 3.13 D in gas phase, and 3.66 D, 4.15 D, and 3.81 D, respectively, in chloroform solvent. The higher values of dipole moments facilitate the self-assembly of molecules and formation of long chains that provide a strong pathway for charge mobility if the designed molecules have the ability to pack. The dipole moment at ground as well as in excited state and their difference are shown in Table 5.
Open circuit voltage
Open circuit voltage (Voc), which shows the maximum amount of voltage that can be taken from any device, is an important parameter to check the performance of organic solar cells [39]. Voc shows the bias of junction of current taken from the solar cell. Both light-generated current and saturation current rely on recombination in the devices, and Voc relies on these both currents. Open circuit values depend on the difference between donor and acceptor molecules (corresponding to HOMO and LUMO energies, respectively) and are approximately proportional to Voc. Herein, our designed molecules are donors. Therefore, we compared our designed molecules with the well-known acceptor, PCBM [28, 21].
Figure 8 presents a molecular orbital diagram, where the energy differences between the HOMOs of designed molecules and LUMOs of the acceptor PCBM can be observed. The open circuit voltages of reference R and designed molecules M1, M2 and M3 with respect to PCBM are 2.91 eV, 3.01 eV, 2.77 eV, and 3.02 eV, respectively. The calculated open circuit voltages are in the order of M3 > M1 > R > M2. The comparison between of HOMOs–LUMOs of the reference R and designed molecules M1, M2 and M3 with respect to PCBM are shown in Fig. 9. The above result shows that M1 and M3 have higher Voc values than M2.

Molecular orbital energy level diagrams of reference R and designed donors M1–M3 and [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) acceptors

Expected open circuit voltage (Voc) of references R and designed donor molecules (M1–M3) with respect to PCBM
Transition density matrix and exciton binding energy
The transition density matrix (TDM) of dithieno [2,3-b:3,2-d] thiophene (DTT)-based small molecules donor were computed to estimate the nature of the transition. CAM-B3LYP/6-31G (d,p) level of theory was used to compute absorption and emission of the S1 state in a vacuum, and the results are illustrated in Fig. 10. TDM explains the interaction between donor and acceptor groups in the excited state, in addition to explaining hole-electron localization and electronic excitation [40, 41]. Due to the small contribution of hydrogen atoms in transition, the effect of hydrogen atoms was ignored by default in the present study.

Transition density matrix (TDM) of reference R and designed molecules (M1–M3)
We divided our designed molecules into three types to evaluate the TDM results: A, B and D (A for end capped acceptor, B for bridge unit, and D for central donor unit). From the TDM diagram, it is obvious that the electron coherence of reference R and designed molecules (M1–M3) is partially available on the diagonal of donor and the bridge segment, while there is very little on the end-capped acceptor moieties. Additionally, the coefficient of interaction between donor and acceptor group of designed molecules are in the order of M1 > M3 > M2. The coupling of hole and electron of M2 may be lower with respect to other three molecules, but perhaps it shows higher and easier exciton dissociation in the excited state. Exciton binding energy (Eb) reveals that M2 has the highest number of charges, which may cause easy dissociation into separated charges. Therefore, as a result, it has higher charge dissociation energy with respect to M1 and M3.
Binding energy holds a promising key to evaluating the performance of organic solar cells. It helps to determine exciton dissociation potential, interaction of columbic force between hole and electron, and the optoelectronic properties of OSCs. Binding energy and columbic interactions have a direct relationship. The lower the resultant binding energy, the lower the columbic interaction between electron and hole. Lower binding energy leads to higher exciton dissociation in the excited state. Binding energy can be calculated by taking the difference between energy gap (Eg) and single point energy Eopt forms S0 to S1 by producing pair of electron and hole [42,43,44]. The binding energy (Eb) values of reference R and designed molecules M1–M3 are calculated using following Eq. 3.
The theoretical calculated binding energies of designed molecules (M1–M3) including reference R are shown in Table 6.
All newly designed molecules have binding energies comparable to that of the reference molecule R. Moreover, the order of binding energy of all molecules is M1 (1.774) > M3 (1.592) > M2 (1.488) > R (1.433), which is in excellent agreement with the TDM result.
Charge transfer analysis of our designed donor and acceptor PCBM
In order to estimate the charge transfer (CT) between designed donor molecules (M1) and PCBM, the complex was subjected to CT analysis, as shown in Fig. 11.

Charge transfer (CT) between the M1 and PCBM, the complex analyzed geometry for CT analysis at CAM-B3LYP/6-31G (d,p) level of theory
The PCBM acceptor interacts with the designed molecule M1 in such a way that the designed molecule is aligned parallel to the acceptor PCBM. The relative orientation of M1 and PCBM has significant effect on the electronic structure of the complex. The dipole from PCBM to M1 suggests efficient excitation at the M1/PCBM interface [45,46,47,48]. The dipole μ in the M1:PCBM complex is due to electrostatic interactions of permanent electric moment of PCBM with that of M1. The orientation of the dipole in our case is in accordance with this statement. The dipole moment vector originates from the polymer side, and points towards the end capped acceptor group of designed molecule M1.
The HOMO–LUMO distribution pattern and electronic structure of the complex are calculated at CAM-B3LYP/6-31G (d,p) level of theory (Fig. 12). The HOMO is spread mainly on the donor molecule, while the LUMO is present mainly on the acceptor PCBM. The fact shown in the orbital diagram shows the HOMO to LUMO excitation as CT from donor M1 to the acceptor PCBM molecule. Shifting of density from donor to acceptor is concrete evidence for CT between different groups.

Distribution patterns of FMOs (HOMO and LUMO) of M1 and PCBM at CAM-B3LYP/6-311G (d,p) level of theory
Conclusion
Three new molecules M1, M2 and M3 are designed to enhance the optical, photophysical and electronic properties of OSCs. These molecules comprising dithieno [2,3-b:3,2-d] thiophene (DTT) donor group linked with acceptor 2-(3-methyl-4-oxothiazolidin-2-ylidine)malononitrile through three different bridge groups, 3-methylthiophene (M1), 3-methylfuran (M2) and 3-methylselenophene (M3), respectively. Optical properties, electronic properties, photophysical and excited state energy were calculated and compared with the well-known reference molecule R, which was recently published. All designed molecules (M1–M3) have suitable FMO diagrams for CT. From their binding energy, M2 has the highest number of charges, which may cause easier dissociation into separated charges. Therefore, as a result, it has higher charge dissociation energy with respect to M1 and M3. All designed molecules show potential photovoltaic parameters with respect to the reference molecule R. The designed molecule M2 exhibits the lowest band gap (4.28 eV) and shows the highest λmax value at 474 nm in gas phase and 500 nm in chloroform solvent. The lower λe and λh of M2 (among all molecules) revealed its higher charge mobility (electron and hole) with respect to all designed and reference molecule R. The lower λh value compared to λe values revealed that all designed molecules have higher hole mobility than that of the electron mobility. Conclusively, all designed donor molecules (M1–M3), especially M2, hold promising optoelectronic properties, and, therefore, are suitable donor materials for their use in organic solar cells.
References
Koroneos C, Spachos T, Moussiopoulos N (2003) Exergy analysis of renewable energy sources. Renew Energy 28(2):295–310
Kyaw AKK, Wang DH, Gupta V, Zhang J, Chand S, Bazan GC, Heeger AJ (2013) Efficient solution-processed small-molecule solar cells with inverted structure. Adv Mater 25(17):2397–2402
Irfan A, Al-Sehemi AG (2012) Quantum chemical study in the direction to design efficient donor-bridge-acceptor triphenylamine sensitizers with improved electron injection. J Mol Model 18(11):4893–4900
Lee JW, Choi YS, Jo WH (2012) Diketopyrrolopyrrole-based small molecules with simple structure for high V OC organic photovoltaics. Org Electron 13(12):3060–3066
Bruton T (2002) General trends about photovoltaics based on crystalline silicon. Sol Energy Mater Sol Cells 72(1–4):3–10
Ni W, Li M, Kan B, Zuo Y, Zhang Q, Long G, Feng H, Wan X, Chen Y (2014) Open-circuit voltage up to 1.07 V for solution processed small molecule based organic solar cells. Org Electron 15(10):2285–2294
Kaltenbrunner M, White MS, Głowacki ED, Sekitani T, Someya T, Sariciftci NS, Bauer S (2012) Ultrathin and lightweight organic solar cells with high flexibility. Nat Commun 3:770
Lou SJ, Szarko JM, Xu T, Yu L, Marks TJ, Chen LX (2011) Effects of additives on the morphology of solution phase aggregates formed by active layer components of high-efficiency organic solar cells. J Am Chem Soc 133(51):20661–20663
Walker B, Kim C, Nguyen T-Q (2010) Small molecule solution-processed bulk heterojunction solar cells. Chem Mater 23(3):470–482
Günes S, Neugebauer H, Sariciftci NS (2007) Conjugated polymer-based organic solar cells. Chem Rev 107(4):1324–1338
Ans M, Ayub K, Muhammad S, Iqbal J (2019) Development of fullerene free acceptors molecules for organic solar cells: a step way forward toward efficient organic solar cells. Comput Theor Chem (in press) https://doi.org/10.1016/j.comptc.2019.06.003
Li G, Zhu R, Yang Y (2012) Polymer solar cells. Nat Photonics 6(3):153
You J, Dou L, Yoshimura K, Kato T, Ohya K, Moriarty T, Emery K, Chen C-C, Gao J, Li G (2013) A polymer tandem solar cell with 10.6% power conversion efficiency. Nat Commun 4:1446
Zhou H, Yang L, You W (2012) Rational design of high performance conjugated polymers for organic solar cells. Macromolecules 45(2):607–632
Li CZ, Chang CY, Zang Y, Ju HX, Chueh CC, Liang PW, Cho N, Ginger DS, Jen AKY (2014) Suppressed charge recombination in inverted organic photovoltaics via enhanced charge extraction by using a conductive fullerene electron transport layer. Adv Mater 26(36):6262–6267
Lu L, Yu L (2014) Understanding low bandgap polymer PTB7 and optimizing polymer solar cells based on it. Adv Mater 26(26):4413–4430
He Y, Chen H-Y, Hou J, Li Y (2010) Indene− C60 bisadduct: a new acceptor for high-performance polymer solar cells. J Am Chem Soc 132(4):1377–1382
Kang H, Kee S, Yu K, Lee J, Kim G, Kim J, Kim JR, Kong J, Lee K (2015) Simplified tandem polymer solar cells with an ideal self-organized recombination layer. Adv Mater 27(8):1408–1413
Zara Z, Iqbal J, Iftikhar S, Khan SU-D, Haider S, Eliasson B, Ayub K (2018) Designing dibenzosilole and methyl carbazole based donor materials with favourable photovoltaic parameters for bulk heterojunction organic solar cells. Comput Theor Chem 1142:45–56
Mikroyannidis JA, Kabanakis AN, Sharma S, Sharma GD (2011) A simple and effective modification of PCBM for use as an electron acceptor in efficient bulk heterojunction solar cells. Adv Funct Mater 21(4):746–755
Manzoor F, Iqbal J, Zara Z, Eliasson B, Mahr MS, Ayub K (2018) Theoretical calculations of the optical and electronic properties of dithienosilole-and dithiophene-based donor materials for organic solar cells. ChemistrySelect 3(5):1593–1601
Frisch M, Trucks G, Schlegel HB, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Mennucci B, Petersson G (2009) Gaussian 09, revision D. 01. Gaussian, Inc., Wallingford, CT
Dennington RD, Keith TA, Millam JM (2008) GaussView 5.0. 8. Gaussian Inc., Wallingford, CT
Kan B, Li M, Zhang Q, Liu F, Wan X, Wang Y, Ni W, Long G, Yang X, Feng H (2015) A series of simple oligomer-like small molecules based on oligothiophenes for solution-processed solar cells with high efficiency. J Am Chem Soc 137(11):3886–3893
Becke AD (1993) Becke’s three parameter hybrid method using the LYP correlation functional. J Chem Phys 98:5648–5652
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393(1–3):51–57
Chai J-D, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys 10(44):6615–6620
Irfan M, Iqbal J, Sadaf S, Eliasson B, Rana UA, Ud-din Khan S, Ayub K (2017) Design of donor–acceptor–donor (D–A–D) type small molecule donor materials with efficient photovoltaic parameters. Int J Quantum Chem 117(10):e25363
Ans M, Iqbal J, Ahmad Z, Muhammad S, Hussain R, Eliasson B, Ayub K (2018) Designing three-dimensional (3D) non-fullerene small molecule acceptors with efficient photovoltaic parameters. ChemistrySelect 3(45):12797–12804
Ans M, Iqbal J, Ayub K, Ali E, Eliasson B (2019) Spirobifluorene based small molecules as an alternative to traditional fullerene acceptors for organic solar cells. Mater Sci Semicond Process 94:97–106
O'boyle NM, Tenderholt AL, Langner KM (2008) Cclib: a library for package-independent computational chemistry algorithms. J Comput Chem 29(5):839–845
Hutchison GR, Ratner MA, Marks TJ (2005) Hopping transport in conductive heterocyclic oligomers: reorganization energies and substituent effects. J Am Chem Soc 127(7):2339–2350
Paramasivam M, Gupta A, Raynor AM, Bhosale SV, Bhanuprakash K, Rao VJ (2014) Small band gap D-π-A-π-D benzothiadiazole derivatives with low-lying HOMO levels as potential donors for applications in organic photovoltaics: a combined experimental and theoretical investigation. RSC Adv 4(67):35318–35331
Ans M, Iqbal J, Eliasson B, Saif MJ, Javed HMA, Ayub K (2019) Designing of non-fullerene 3D star-shaped acceptors for organic solar cells. J Mol Model 25(5):129. https://doi.org/10.1007/s00894-019-3992-9
Ans M, Iqbal J, Eliasson B, Ayub K (2019) Opto-electronic properties of non-fullerene fused-undecacyclic electron acceptors for organic solar cells. Comput Mater Sci 159:150–159
Goszczycki P, Stadnicka K, Brela MZ, Grolik J, Ostrowska K (2017) Synthesis, crystal structures, and optical properties of the π-π interacting pyrrolo [2, 3-b] quinoxaline derivatives containing 2-thienyl substituent. J Mol Struct 1146:337–346
Kong H, Cho S, Lee DH, Cho NS, Park MJ, Jung IH, Park JH, Park CE, Shim HK (2011) The influence of electron deficient unit and interdigitated packing shape of new polythiophene derivatives on organic thin-film transistors and photovoltaic cells. J Polym Sci A Polym Chem 49(13):2886–2898
Sun M, Niu B, Zhang J (2008) Computational study on optical and electronic properties of the “CH”/N substituted emitting materials based on spirosilabifluorene derivatives. J Mol Struct THEOCHEM 862(1):85–91
Irfan M, Iqbal J, Sadaf S, Eliasson B, Rana UA, Khan UD, Ayub K (2017) Design of donor–acceptor–donor (D–A–D) type small molecule donor materials with efficient photovoltaic parameters. Int J Quantum Chem 117(10)
Ans M, Ayub K, Bhatti IA, Iqbal J (2019) Designing indacenodithiophene based non-fullerene acceptors with a donor–acceptor combined bridge for organic solar cells. RSC Adv 9(7):3605–3617
Gangala SK, Paramasivam M, Dyaga B, Rao VJ (2019) Energy levels tuning of ‘Z’-shaped small molecular non-fullerene electron acceptors based on dipyrrolo [2, 3-b: 2', 3'-e] pyrazine-2, 6 (1H, 5H)-dione acceptor unit for organic photovoltaic applications: a joint experimental and DFT investigation on the effect of fluorination. New J Chem
Köse ME (2012) Evaluation of acceptor strength in thiophene coupled donor–acceptor chromophores for optimal design of organic photovoltaic materials. J Phys Chem A 116(51):12503–12509
Dkhissi A (2011) Excitons in organic semiconductors. Synth Met 161(13–14):1441–1443
Kim BG, Zhen CG, Jeong EJ, Kieffer J, Kim J (2012) Organic dye design tools for efficient photocurrent generation in dye-sensitized solar cells: exciton binding energy and electron acceptors. Adv Funct Mater 22(8):1606–1612
Arkhipov V, Heremans P, Bässler H (2003) Why is exciton dissociation so efficient at the interface between a conjugated polymer and an electron acceptor? Appl Phys Lett 82(25):4605–4607
Marchiori C, Koehler M (2010) Dipole assisted exciton dissociation at conjugated polymer/fullerene photovoltaic interfaces: a molecular study using density functional theory calculations. Synth Met 160(7–8):643–650
Koehler M, Santos M, Da Luz M (2006) Positional disorder enhancement of exciton dissociation at donor/acceptor interface. J Appl Phys 99(5):053702
Baranovskii S, Wiemer M, Nenashev A, Jansson F, Gebhard F (2012) Calculating the efficiency of exciton dissociation at the interface between a conjugated polymer and an electron acceptor. J Phys Chem Lett 3(9):1214–1221
Acknowledgments
The computations/simulations/SIMILAR were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at Umeå University, 901 87, Umeå, Sweden. The authors acknowledge the financial and technical support from Punjab Bio-energy Institute (PBI), University of Agriculture Faisalabad (UAF).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 22 kb)
Rights and permissions
About this article

Cite this article
Ans, M., Manzoor, F., Ayub, K. et al. Designing dithienothiophene (DTT)-based donor materials with efficient photovoltaic parameters for organic solar cells. J Mol Model 25, 222 (2019). https://doi.org/10.1007/s00894-019-4108-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4108-2