
Abstract
An alternative way of calculating the Fukui function and the partial derivative of second order of the electronic density with respect to the number of electrons N is presented, the new formulas agree with the usual ones but only in cases without degeneracy. The new operative formulas are more general than the previous ones and are the right ones for those problematic cases where one or both of the frontier molecular orbitals are degenerate. Finally, we present a new way of applying the finite difference approximation that leads to more realistic results than the usual formulas.

A new way of calculating the Fukui function is presented that results in a new operative formula of the function. It has also been obtained the partial derivative of second order of the electronic density with respect to the number of electrons N, and it agree with the usual formula of the dual descriptor function but only in cases without degeneration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Local reactivity parameters are necessary to differentiate the reagent behavior of atoms forming a molecule. The Fukui function [1,2,3,4] [f(r)] and local softness [5, 6] [s(r)] are two of the most commonly used local reactivity parameters (Eq. 1).
The Fukui function is associated primarily with the response of the density function of a system to a change in the number of electrons (N) under the constraint of a constant external potential [v(r)]. The mathematical definitions of the Fukui function and local softness (Eq. 1) come from the so-called ensembles of the conceptual density functional theory (C-DFT) [7] where all global, local and non local reactivity descriptors are hierarchically organized. The Fukui function arises from the canonical ensemble where the number of electrons and the external potential are the essential variables. Meanwhile, the number of electrons and the electronic chemical potential are the essential variables for the local softness.
Due to the discontinuity of the electron density with regard to N, finite difference approximation leads to three types of Fukui function: f+(r), f−(r) and f0(r). They are defined as follows:
Theoretical development
The energy change [8] (ΔE) due to the electron transfer (ΔN) satisfies the parabolic approximation:
where μ and η are the electronic chemical potential and global hardness. Perdew et al. [9] show how the Hohenberg-Kohn theorem is extended to a fractional electron number (N), and the implications of derivative discontinuity for conceptual DFT are explored by Ayers and colleagues [10, 11]. Taking into account Eq. (5), and that the total energy is a functional of the density, it is reasonable to think that the second order expansion Eq. (6) can be an appropriate approximation.
if we substitute the values ΔN= 1 and ΔN= −1 in Eq. (6), and also calculate \( \varDelta {\rho}_N^{-}\left(\mathbf{r}\right) \) (corresponding to ΔN= −1) and \( \varDelta {\rho}_N^{+}\left(\mathbf{r}\right) \) (corresponding to ΔN= 1). And finally, substituting in Eq. (6), we obtain:
and we find a very simple system of equations with two unknowns; by solving them, we obtain Eq. (8). Expressions of this type (Eq. 6) have been used in previous works [12,13,14]. Also, implicit in the two articles the introduction of the concept of the dual descriptor [15, 16] because the expression used for the second derivative of the density with respect to the number of electrons corresponds to this kind of quadratic interpolation. Figure 1 graphically represents the physical meaning of the main parameters of Eqs. (6–8). It can be seen that the new formula of f(r) (see Eq. 8) is the same as that of f0(r) [see Eq. (4), neutral attack], this is logical since the quadratic expansion does not imply an electrophilic or nucleophilic attack.

On the other hand, the original operational formula proposed by Morell et al. [16] for the dual descriptor [15, 17, 18] is:
As can be seen, the formula obtained by Morell et al. in Eq. (9) is the same as the formula of Eq. (8).
What changes when there is degeneracy of frontier molecular orbitals?
The use of these operational formulae, Eq. (9), can result in failure when applying them to molecular systems that present degeneracy in their frontier molecular orbitals [19]. The issue of degeneracy in conceptual DFT is not restricted to degenerate frontier orbitals because (in rare cases) you can have degenerate ground states in DFT without degenerate frontier orbitals [19,20,21,22].
To overcome this limitation of the dual descriptor (and Fukui functions), a more general operational formula was proposed by Martínez-Araya [23, 24]:
where p and q stand for the degree of degeneracy of LUMO and HOMO, respectively.
When we apply these ideas proposed by Martínez-Araya to the system of Eq. (7), we obtain a new operative formula of f(2)(r) for degenerate cases:
which is slightly different from the formula obtained by Martínez-Araya Eq. (10), but they are proportional. On the other hand, Eq. (12) is the operative formula for f0(r) in cases with degeneracy (applying the ideas of Martínez-Araya),
on the other hand, starting from the system of Eq. (7), we obtain:
In this case, it can be seen that f0(r)Martínez−Araya and f(r)Quadratic expansion are not proportional. Appendix I in supplementary material includes a simple example that complements this conclusion and shows that the new operative formulas f (r) and f (2)(r) are different from the old ones. Finally, we propose a parabolic expansion as an alternative methodology to the finite difference approximation, and we rationalize this affirmation with the very simple example shown in Appendix II.
The new operational formulae for Fukui function and dual descriptor taking into account degrees of degeneracy in HOMO and LUMO are different to those ones based on finite difference. That makes sense in the case of a fractional value of ΔN. But when there is a degree of degeneracy greater than 1, meaning ΔN > 1, in such case there is no certainty that the Taylor expansion (Eq. 6) converges. The,n from the mathematical point of view, finite difference is a suitable approximation because it does not depend on the means of truncation of the Taylor expansion. That is why Eqs. (10) and (12) can be used as reference expressions, so that:
and for the Fukui function (the expression is not so simple):
Computational details
All the structures included in this study were optimized at B3LYP/6-31G(d) [25, 26] theory level by using the Gaussian09 package. [27] The densities used in the new methodology were calculated at the same level of calculation for the neutral molecule, the cation and anion, through Gaussian09 software.
The new indices included in this study were calculated with a modified version of UCA-FUKUI v.2.1 software (http://www2.uca.es/dept/quimica_fisica/software/UCA-FUKUI_v2.exe) [28]. Figure 2 includes two screenshots of the main menu showing the calculation modules that have been added to obtain the new indexes. Figures S1–S3 in the supplementary material show some screenshots of the UCA-FUKUI software displaying the correspondence between the program interface and the equations of the text.

Upper panel UCA-FUKUI main menu: atomic Fukui indices based on a quadratic expansion. Lower panel UCA-FUKUI main menu: Fukui function based on a quadratic expansion
Results and discussion
Obtaining atomic indices (condensed-to-atom)
Starting from Eq. (7) and by taking into account the response-of-molecular-fragment approach [29], (which is equivalent to the fragment-of-molecular-response approach for Hirshfeld partitioning [30,31,32]), the next condensed-to-atom system can be obtained:
where r and p are the global net charges of the ions. Solving the system:
where \( {q}_k^N \), \( {q}_k^{N+p} \) and \( {q}_k^{N-s} \) are the net atomic charges calculated with some population analysis (Hirshfeld, Mulliken, ...) for the neutral molecule and the corresponding ions. As an example, Table 1 shows the condensed \( {f}_k^{\mathrm{Quadratic}\kern0.34em \mathrm{expansion}} \) and \( {f}_k^0 \)(finite difference approximation [33]) indices obtained for SF6, which has triply degenerate HOMO, using the three different population analysis: Hirshfeld [34,35,36], Mulliken [37] and natural population analysis (NPA) [38,39,40]. As can be seen in Table 1, the \( {f}_k^{\mathrm{Quadratic}\kern0.34em \mathrm{expansion}} \) and \( {f}_k^0 \) indices are different.
Advantages of this method: Generalization of the finite difference approximation
The quadratic expansion Eq. (6) provides an important advantage, allowing interpolation Δρ(r) for fractional values of ΔN (−1 <ΔN<1). Thanks to this, we can use the finite difference approximation more generally. Suppose that ΔN is the fractional value ΔN∗, and, that, by substituting this value in Eq. (6), we are led to Δρ∗(r):
Now, the function Δρ(r)∗ allows the finite difference approximation to be applied to calculate the Fukui indices in a more general way:
Note that Fukui functions f(r)+ and f(r)− are particular cases of Eq. (19), where ΔN∗ takes the non-fractional values +1 and − 1. The recent work from the Gazquez group [41, 42] is the closest approach that we have been able to find in relation to this idea.
On the other hand, the amount of charge transfer ΔNA : B associated to the formation of A:B complex from acid A and base B, may be written as [43]:
then, combining Eqs. (18), (19), and (20), we obtain the formula:
that allows to estimate an approximate Fukui function corresponding to a molecule (for example an acid A) when it is attacked by another concrete molecule (for example a base B). It is important to note that f(r)A : B (Eq. 21) is being calculated with a charge variation ΔNA : B with physical meaning, and the same can be said for variations Δρ(r)A : B. The idea of using a model based on chemical potentials μA and μB, instead of a finite-difference approximation, can be traced back to Parr and Bartolotti [44] (the value of such an approach and also its limitations was recently stressed by Heidar-Zadeh et al. [45, 46] and the Gazquez group recently showed that the parabolic model is especially relevant [47]).
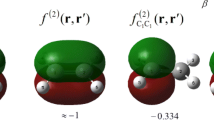
As an example, Fig. 3 (left) shows function f(r)A : B for the dienophile CH2CHCHO when it is attacked by a H2 molecule. In Fig. 3 (right) the function of Fukui f(r)− (Eq. 3) has been included to facilitate comparison. The images are similar because the two functions represent the nucleophile character of the CH2CHCHO molecule but they show some differences since function f(r)A : B takes into account the characteristics of the attacker (H2 molecule). All the images of Fig. 3 have been performed with Gaussview [48] and the “.cub” files used as a starting point were obtained from UCA-FUKUI software.

Left Function f(r)A : B of the CH2CHCHO when it is attacked by a H2 molecule. Right Fukui function f(r)− of the CH2CHCHO. The four images were obtained with isovalue: 0.0002
Starting from the previous idea and Eq. (17) the condensed-to-atom value \( \varDelta {q}_k^{\ast } \) of Eq. (22) can be obtained:
The values \( \varDelta {q}_k^{\ast } \) and ΔN∗ lead to the operative formula:
Note that the Fukui indices \( {f}_k^{+} \) and \( {f}_k^{-} \) are particular cases of Eq. (21). Finally, combining Eqs. (22), (23), and (20) we obtain the formula:
that is the condensed-to-atom version of Eq. (21).
Figure 4 shows the parameters \( {f}_k^{A:B} \) calculated for the dienophile CH2CHCl when it is attacked by different reagents (Hirshfeld population analysis was used to obtain atomic populations). The attackers A–C correspond to a set of dienes (see Fig. S4 in the Supplementary material), in addition the attackers H3O+ and OH− have been included. The condensed-to-atom indices \( {f}_k^{+} \), \( {f}_k^{-} \) and \( {f}_k^0 \) (Eqs. 2–4), nucleophilic, electrophilic and neutral attacks) have also been added for comparison. When the attacker is very electrophilic (for example see H3O+) the \( {f}_k^{A:B} \) values are close to the curve \( {f}_k^{-} \) (electrophilic attack); on the contrary, when the attacker is very nucleophilic (for example see OH−) the \( {f}_k^{A:B} \) values are close to the curve \( {f}_k^{+} \) (nucleophilic attack). Where the attacker possesses an electronic chemical potential similar to CH2CHCl (for example the dienes A-C) the \( {f}_k^{A:B} \) values tend to curve \( {f}_k^0 \) (neutral or radical attack). Figure S2 in the supplementary material shows an enlargement of Fig. 4 where D–H dienes, H2, HCl and Cl2 attackers have also been included (the data of Figs. 4 and S5 are shown in Table S1 in the supplementary material). Figures S6 to S10 of the Supplementary Material include some equivalent graphics for the CH2CHCHO, CH2CHNO2, CH2CHCN, CH2CHCH3 and CH2CHOCH3 molecules (see Fig. S11) and the results are equivalent to those shown in Fig. 4. In addition, Figure S12 in the Supplementary Material includes an additional graphic achieved from “atoms in molecules” (AIM) [49,50,51] theory through the AIMAll software (http://aim.tkgristmill.com/index.html), Figure S13 includes a graphic achieved from natural population analysis and Fig. S14 a graphic achieved from Mulliken approximation with a minimal basis set [52, 53] for the CH2CHCl reagent. The results, shown on these graphs, are qualitatively the same as those shown in Fig. 4. The comparison of all these methodologies led to the conclusion that the calculation method used to obtain the atomic populations does not change qualitatively the conclusions obtained.

Parameter \( {f}_k^{A:B} \) calculated with Eq. (21) and Hirshfeld population analysis for the CH2CHCl molecule taking into account different attackers. The parameters \( {f}_k^{+} \), \( {f}_k^{-} \) and \( {f}_k^0 \) have been added for comparison
Future perspectives
Our intention is to modify the UCA-FUKUI [28] software, based on “conceptual DFT” and specialized in the calculation of local reactivity indices, to introduce the new definition of Fukui’s function in some methodologies (bond-reactivity-indices calculation and improved-frontier-molecular-orbital approximation) implemented in the program [54,55,56]. Once the software works properly with the new definitions, we will make representative calculations in order to compare the results with those obtained from previous definitions.
Conclusions
A new way of calculating f(r) and f(2)(r) has been developed, resulting in new operative formulas. The Fukui function has been obtained for those cases where one or both of the frontier orbitals are degenerate, and a more general operative formula was obtained. The new formulae are in agreement with the usual formulae but only in cases without degeneracy. The new f(2)(r) function is identical to the previous formula of dual descriptor in all cases where the frontier molecular orbitals are not degenerate, and in those cases with degeneracy it has been found that they are proportional functions. Finally, a new way of applying the finite difference approximation has been developed that leads to more realistic results (with more physical meaning) than the usual formulas.
References
Yang WT, Parr RG, Pucci R (1984) Electron density, Kohn-sham frontier orbitals, and Fukui functions. J Chem Phys 81:2862–2863
Par RG, Yang W (1984) Density functional approach to the frontier-electron theory of chemical reactivity. J Am Chem Soc 106:4049
Ayers PW, Levy M (2000) Density functional approach to the frontierelectron theory of chemical reactivity. Theor Chem Accounts 103:353
Ayers PW, Yang WT, Bartolotti LJ (2009) The Fukui function. In: Chattaraj PK (ed) Chemical reactivity theory: a density functional view. CRC, Boca Raton, p 255
Yang W, Parr RG (1985) Hardness, softness, and the Fukui function in the electronic theory of metals and catalysis. Proc Natl Acad Sci USA 82:6723
Chandra AK, Nguyen MT (2008) Fukui function and local softness. In: Chattaraj PK (ed) Chemical reactivity theory: a density-functional view. Taylor and Francis, New York, pp 163–178
Geerlings P, Proft FD, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874
Parr R, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, Oxford
Perdew JP, Parr RG, Levy M, Balduz JL (1982) Density-functional theory for fractional particle number: derivative discontinuities of the energy. Phys Rev Lett 49:1691–1694
Yang WT, Zhang YK, Ayers PW (2000) Degenerate ground states and fractional number of electrons in density and reduced density matrix functional theory. Phys Rev Lett 84:5172–5175
Ayers PW (2008) The continuity of the energy and other molecular properties with respect to the number of electrons. J Math Chem 43:285–303
Gázquez JL (2009) Chemical reactivity concepts in density functional theory. In: Chattaraj PK (ed) Chemical reactivity theory: A density functional view. CRC, Boca Raton, p 7
Morell C, Gázquez JL, Vela A, Guegan F, Chermette H (2014) Revisiting electroaccepting and electrodonating powers: proposals for local electrophilicity and local nucleophilicity descriptors. Phys Chem Chem Phys 16:26832
Robles A, Franco-Pérez M, Gázquez JL, Cárdenas C, Fuentealba P (2018) Local electrophilicity. J Mol Model 24:245
Morell C, Grand A, Toro-Labbe A (2005) New dual descriptor for chemical reactivity. J Phys Chem A 109:205–212
Morell C, Grand A, Toro-Labbe A (2006) Theoretical support for using the Δf(r) descriptor. Chem Phys Lett 425:342–346
De Proft F, Ayers PW, Fias S, Geerlings P (2006) Woodward-Hoffmann rules in density functional theory: initial hardness response. J Chem Phys 125:214101–214109
Ayers PW, Morell C, De Proft F, Geerlings P (2007) Understanding the Woodward–Hoffmann rules by using changes in Electron density. Chem Eur J 13:8240–8247
Cárdenas AC, Ayers PW, Cedillo A (2011) Reactivity indicators for degenerate states in the density-functional theoretic chemical reactivity theory. J Chem Phys 134:174103–13
Bultinck P, Cardenas C, Fuentealba P, Johnson PA, Ayers PW (2013) Atomic charges and the electrostatic potential are ill-defined in degenerate ground states. J Chem Theory Comp 9:4779–4788
Bultinck P, Cardenas C, Fuentealba P, Johnson PA, Ayers PW (2014) How to compute the Fukui matrix and function for systems with (quasi-)degenerate states. J Chem Theory Comp 10:202–210
Bultinck P, Jayatilaka D, Cardenas C (2015) A problematic issue for atoms in molecules: impact of (quasi-)degenerate states on quantum theory atoms in molecules and Hirshfeld-I properties. Comput Theor Chem 1053:106–111
Martínez-Araya JI (2016) A generalized operational formula based on Total electronic densities to obtain 3D pictures of the dual descriptor to reveal nucleophilic and electrophilic sites accurately on closed-Shell molecules. J Comput Chem 37:2279–2303
Martínez-Araya JI (2015) Why is the dual descriptor a more accurate local reactivity descriptor than Fukui functions? J Math Chem 53:451–465
Becke AD (1993) Density-functional thermochemistry. III the role of exact exchange. J Chem Phys 98:5648–5652
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods. 25. Supplementary functions for gaussian basis sets. J Chem Phys 80:3265–3269
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford CT
Sánchez-Márquez J, Zorrilla D, Sánchez-Coronilla AM, de los Santos D, Navas J, Fernández-Lorenzo C, Alcántara R, Martín-Calleja J (2014) Introducing UCA-FUKUI software: reactivity-index calculations. J Mol Model 20:2492
Bultinck P et al (2007) Critical thoughts on computing atom condensed Fukui functions. J Chem Phys 127:034102–034111
Nalewajski RF, Parr RG (2000) Information theory, atoms in molecules, and molecular similarity. Proc Natl Acad Sci USA 97:8879–8882
Heidar-Zadeh F, Ayers PW, Verstraelen T, Vinogradov I, Vöhringer-Martinez E, Bultinck P (2018) Information-theoretic approaches to atoms-in-molecules: Hirshfeld family of partitioning schemes. J Phys Chem A 122:4219–4245
Roy RK, Pal S, Hirao K (1999) On non-negativity of Fukui function indices. J Chem Phys 110:8236–8245
Yang WT, Mortier WJ (1986) The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J Am Chem Soc 108:5708–5711
Hirshfeld FL (1977) Bonded-atom fragments for describing molecular charge densities. Theor Chem Accounts 44:129–138
Ritchie JP (1985) Electron density distribution analysis for nitromethane, nitromethide, and nitramide. J Am Chem Soc 107:1829–1837
Ritchie JP, Bachrach SM (1987) Some methods and applications of electron density distribution analysis. J Comp Chem 8:499–509
Mulliken RS (1955) Electronic population analysis on LCAO–MO molecular wave functions I. J Chem Phys 23:1833
Reed AE, Weinhold F (1983) Natural bond orbital analysis of near-Hartree-Fock water dimer. J Chem Phys 78:4066
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735
Reed AE, Weinhold F (1985) Natural localized molecular orbitals. J Chem Phys 83:1736
Orozco-Valencia U, Gazquez JL, Vela A (2018) Global and local charge transfer in electron donor-acceptor complexes. J Mol Model 24:250
Orozco-Valencia U, Gazquez JL, Vela A (2018) Role of reaction conditions in the global and local two parabolas charge transfer model. J Phys Chem A 122:1796–1806
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516
Parr RG, Bartolotti LJ (1982) On the geometric mean principle for electronegativity equalization. J Am Chem Soc 104:3801–3803
Heidar-Zadeh F (2016) When is the Fukui function not normalized? The danger of inconsistent energy interpolation models in density functional theory. J Chem Theory Comp. 12:5777–5787
Heidar-Zadeh F, Richer M, Fias S, Miranda-Quintana RA, Chan M, Franco-Pérez M, González-Espinoza CE, Kim TD, Lanssens C, Patel AHG, Yang XD, Vöhringer-Martinez E, Cárdenas C, Verstraelen T, Ayers PW (2016) Chem Phys Lett 660:307–312
Franco-Perez M, Gá́zquez JL, Ayers PW, Vela A (2018) Thermodynamic justification for the parabolic model for reactivity indicators with respect to Electron number and a rigorous definition for the Electrophilicity: the essential role played by the electronic entropy. J Chem Theory Comp 14:597–606
Dennington R, Keith T, Millam J (2009) Gauss View 5.0. Semichem Inc, Shawnee Mission, KS 7
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford University Press, Oxford
Matta CF, Boyd RJ (2007) The quantum theory of atoms in molecules: from solid state to DNA and drug design. WILEY-VCH, Weinham
Bader RFW (2005) The quantum mechanical basis for conceptual chemistry. Monatsh Chem 136:819–854
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (1999) A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J Chem Phys 110:2822
Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (2000) A complete basis set model chemistry. VII. Use of the minimum population localization method. J Chem Phys 112:6532
Sánchez-Márquez J (2016) Introducing new reactivity descriptors: “bond reactivity indices.” comparison of the new definitions and atomic reactivity indices. J Chem Phys 145:194105–194112
Sánchez-Márquez J, Zorrilla D, García V, Fernández M (2018) Introducing a new bond reactivity index: Philicities for natural bond orbitals. J Mol Model 24:25
Sánchez-Márquez J, Zorrilla D, García V, Fernández M (2018) Introducing a new methodology for the calculation of local philicity and multiphilic descriptor: an alternative to the finite difference approximation. Mol Phys 116:1737–1748
Acknowledgment
Calculations were performed through CICA (Centro Informático Científico de Andalucía).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 749 kb)
Rights and permissions
About this article

Cite this article
Sánchez-Márquez, J. New advances in conceptual-DFT: an alternative way to calculate the Fukui function and dual descriptor. J Mol Model 25, 123 (2019). https://doi.org/10.1007/s00894-019-4000-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4000-0