
Abstract
The reliability of ONIOM approach have been examined in calculations of adsorption energies, transition structures, change of HOMO-LUMO energy gaps and equilibrium geometries of the interaction between NH3 and N-enriched (A) or B-enriched (B) open ended boron nitride nanotubes. To these ends, four models of the A or B, with different inner and outer layers have been studied. In addition, various low-levels including, AM1, PM3, MNDO and UFF have been examined, applying B3LYP/6-31 G* in all high-levels. It was shown, that in the case of A, (choosing two atom layers of the tube open-end as inner layer) the results of ONIOM approach are in best agreement with those of the pure density functional theory (DFT) calculations, while their results significantly differ from those of DFT in the case of B in same conditions. All above and population analysis demonstrate that the ONIOM may be a reliable scheme in the study of weak interactions while it is a controversial approach and should be applied cautiously in the case of strong interactions. We also probed the effect of tube length and diameter on the consistency between ONIOM and DFT results, showing that this consistency is independent of the mentioned parameters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The applications of computational chemistry span predicting the structure, spectra, transition states and reactivity of complicated molecules. To serve as a predictive tool, however, the methods should be applicable to a large enough portion of the system, reflecting the features of the real system. Among the all quantum mechanical methods, a few of them can be easily applied to the study of thermodynamics and reaction mechanisms in large systems such as proteins, nanotubes, etc. In fact, the calculation time in accurate ab initio methods grows much faster than the number of atoms. This growth is roughly relative to the third power of the number of atomic basis functions used to solve the Schrödinger equation, at least within the density functional theory (DFT) context.
Developed by Morokuma et al., ONIOM is a method to study the large molecules by dividing them into two or three layers, where a high-level calculation is performed on the smallest layer (inner layer) and the rest layer (outer layer) effects are included at a low-level of theory [1, 2].
In spite of several theoretical studies on single-walled carbon nanotubes, applying ONIOM method, [3, 4] only few works have been published on the case of boron nitride nanotubes (BNNTs) [5]. BNNTs as inorganic nanomaterials, have received considerable research interests [6, 7] because of their remarkable electronic, mechanical, and thermal properties.
The ONIOM method is still a controversial method, it has been recently reported that the often-used ONIOM (B3LYP:AM1) approach is not appropriate for some nanotube systems [8, 9]. In the present work, we are interested in ONIOM study of the N-H bond cleavage of NH3 at the open ends of BNNTs, evaluating its reliability by comparing its results with our previous reported full DFT ones [10]. To this aim, several combinations of different levels of theory as well as different partitions of the inner and outer layers were considered.
The N-H bond cleavage is a challenging problem not only toward the transformation of NH3 into a useful amino compound but also toward the starting of many catalytic reactions [11–13]. Since only methodological aspects of the ONIOM method have been studied in the present work, no discussion was addressed with respect to either experimental data or absolute accuracy of the chosen levels of calculations.
Computational details
All calculations were carried out with the Gaussian 98 suite of programs [14]. A zigzag (4, 0) BNNT, B20N20H4 was chosen with open ends, in which only one end was saturated with four H atoms. Existing two different terminals for zigzag BNNTs, two forms of open-ended types were used in order to model the NH3 dissociation at the tube ends, including N-enriched (A) and B-enriched (B) types (Fig. 1).

(a) The model of A (N-enriched open-ended BNNTs), (b) The model of B (B-enriched open ended BNNTs)
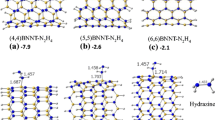
Firstly, four models of the A tube were selected in which the inner layers consist of N4B4 (A1), N8B4 (A2), N8B8 (A3) and N5B5 (A4), Fig. 2. In models of A1, A2 and A3, two, three and four rows of atoms were placed respectively in the inner layer, and in the A4 two hexagonal rings were selected as inner layer. All of models with and without NH3 were optimized using ONIOM (B3LYP/6-31 G*:AM1) and adsorption energy (Ead ) were computed (Table 1). The Ead is defined here as follows:
where Etot is the total energy of a given system.

Four optimized models of the A tube at ONIOM(B3LYP/6-31 G*:AM1)
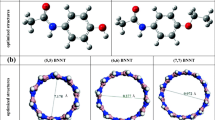
In addition, the other low-levels were applied in the ONIOM study of A3 model including the semiempirical MNDO and PM3 methods and also the molecular mechanics universal force field (UFF). All calculations performed on A model were repeated for B model, as well. Finally, to explore the effect of tube length and diameter on the consistency of ONIOM and DFT, the A3 model of three other BNNTs were studied, including: (5,0), (6,0) and (7,0) zigzag types.
Results and discussion
At first, we probed reliability of the ONIOM(B3LYP/6-31 G*:AM1) level of theory in calculation of geometrical parameters and Ead of NH3 dissociation at open ends of A1, A2, A3, and A4 models. B3LYP, has the most generality and predictive capacity providing a sufficiently accurate description of finite-size nanotubes [15–19]. As we have recently showed, during the NH3 adsorption process, a single NH3 molecule dissociates into an -H atom and an -NH2 group at the open ends of BNNTs [10].
In the present work, we compare the results of ONIOM method with those of full DFT in ref. [10]. The calculated Ead amounts for A1, A2, A3 and A4 models are −80.8, -70.5, -64.8 and −74.7 kcal mol-1, respectively (Table 1). The results indicate that only the Ead of NH3 on A3 model partly agrees with B3LYP/6-31 G* result (−63.1 kcal mol-1) and the energy difference is about 1.7 kcal mol-1 (with 3% error). As shown in Table 1, the other models show significant errors. The calculated values for three representative bond lengths including, N1-NH2, N2-H and N1-N2 (Table 1, Fig. 3), indicate that geometrical parameters have no dependency upon the type of tube models.

The structural geometry of A3/NH3 complex with different ONIOM methods
However, adopting the A3 as the appropriate model, we subsequently compared the reliability of four different low-levels (AM1, PM3, UFF and MNDO) in Ead, activation energy (Eact), HOMO-LUMO energy gap (Eg), charge transfer (QT), dipole moment (μ) and structure geometry calculations. The Eg, μ and QT were computed using full B3LYP/6-31 G* level of theory (performing a single point (SP) calculation on the optimized structures of ONIOM), due to inability of the ONIOM method in their calculations. Frequency calculations verified the obtained transition structures (with one imaginary frequency). Ugliengo et al. showed that the results of frequency calculations using ONIOM method are comparable with full DFT ones [20–22].
All calculated parameters are shown in Table 2, indicating that the results of Ead are near to that of ref. [10], except those of UFF, showing an error of 7.4%. However, among all low-levels, Ead of MNDO is the nearest to that of B3LYP. We once more performed a SP energy calculations on the A3 and H-A3-NH2 complexes, using B3LYP/6-31 G*. It is observed that the difference of Ead between ONIOM and full DFT is relatively reduced in the all cases (Table 2). It seems that performing a high-level SP calculation on the ONIOM-based optimized structures improve the initial results of Ead.
The UFF has the most deviation in Eg calculations. Either PM3 or MNDO overestimates Eg about 0.11 eV, while AM1 underestimates it about 0.13 eV. Generally, all methods give eligible results, except the UFF. In Eact calculations, we observe a good consistency between ONIOM and DFT as the result of PM3 is in the best agreement with that of full DFT.
In the cases of structure geometry and QT calculations, the results of the different ONIOM methods show no significant difference in comparison to those of DFT, indicating that these properties are not dependent upon low-levels. Finally, the largest and lowest deviations belong to the UFF and PM3, respectively, in the case of μ calculation.
However, PM3 is the most reliable among all low-level methods and the results of UFF are the most misleading, especially in the calculations of Ead, Eact and μ. The results of MNDO are somewhat similar to those of PM3 by experience, the PM3 usage is difficult in comparison to that of MNDO, due to many convergence failures in the Gaussian program.
Subsequently, we assessed the reliability of the ONIOM method in Ead calculation of NH3 dissociation at the open end of B model, using the same three semiempirical low-levels. The data of Table 3 show that the results of ONIOM method for all B models are not in agreement with DFT. The best agreement belongs to the B3 model with 25% error. It is noteworthy to mention that the designation of B models is similar to those of the A models. In the B3 case, the calculated Ead values for AM1, PM3, and MNDO are −156.5, -202.5 and −182.6 kcal mol-1 while that of full DFT is −131.1 kcal mol-1 [10].
In contrast to the case of A, the results of ONIOM method significantly differ with that of DFT in all models of B. For example in B3 model, the calculated errors are 26%, 63% and 47% for AM1, PM3, and MNDO, respectively. This induces very cautiously usage of ONIOM method in computational studies.
Here, we interpret in detail why the ONIOM method is appropriate for A3 model, while it is not for B3 (with the same atoms in low-level). To this end, we performed Mulliken population analysis on the A3 and B3 tubes and their NH3 adsorbed complexes. It is noteworthy to say that our main objective is a comparative study and, the exact values of Mulliken charges (MCs) is not the purpose of the present manuscript. However, during the NH3 adsorption process, the MCs of atoms change within the tubes, whereas the amounts of changes reduce going away from their ends.
The percentage of MC changes were computed during the adsorption process for five layers at the open end of both A3 and B3. We have designated the name of “layer” for a row of N atoms with a row of B atoms. The results are depicted in Fig. 4 and collected in Table 4. In the case of A3, the changes are 34.7% and 10.1% for layers 1 and 2 (region 1) while those are only 2.6%, 0.3% and 0.1% for layers 3, 4 and 5 (region 2), respectively. It demonstrates that the MCs of atoms of region 1 significantly change during the adsorption process, while their changes in region 2 are not significant.

The percentage change of Mulliken charges in layers 3, 4 and 5 of both A3 and B3 models
In other words, region 1 is a chemically active site and it is necessary to locate in high-level of ONIOM scheme while region 2 is not a sufficiently active area and can be placed in low-level. As discussed above, this strategy has been applied in our ONIOM calculations for the case of A3, justifying the reliable results. In the models of A1, A2 and A4 some atoms of region 1 are located in low-level of theory, justifying the large errors in the Ead results.
In the case B3, we observed different results so as the changes of MCs in region 2 are not negligible. These changes are 31.3%, 27.3% and 6.2% in layers 3, 4 and 5, respectively. This suggests that region 2 may be a part of chemically active site that should be studied in high-level of theory. As shown before, this region has been placed in low-level of theory and it may be the origin of the large errors in Ead of B3 model. As a result, we conclude that the ONIOM may be a reliable scheme in the study of weak interactions, while in the case of strong interactions it is a controversial approach and should be applied cautiously. In other words, in the case of ONIOM-based strong interaction studies, more atoms should be located in high-level in comparison to that of weak interaction studies.
Subsequently, we probed the effect of tube length and diameter on reliability of ONIOM results. Here we only considered case A, because the ONIOM results of B models are very different from those of DFT. To this end, we calculated Ead values of NH3 dissociation at open ends of A3 models of (5,0), (6,0) and (7,0) tubes with ONIOM(B3LYP/6-31 G*:AM1) and full B3LYP/6-31 G*. The data of Table 5 indicate that the results of ONIOM and full DFT are in best agreement for different diameter tubes. In addition, to investigate the effect of length, we considered the (5,0)-A3 model with various length including: 9, 8, 7, 6 and 5 layers. The results (Table 6) show that Ead values for all tubes with various lengths do not differ significantly for both ONIOM and DFT and there is good consistency between these approaches. Generally, we conclude that the agreement between the results of ONIOM and DFT approaches are independent of tube length and diameter.
We mention that the absolute values of Ead are increased as the tube diameter is elongated. This phenomenon is justified as the weakening of tube edge bonds resulting from the bond length elongation because of diameter enlargement.
Conclusions
We used the ONIOM method to calculate the adsorption energies (Ead), transition structures, the change of HOMO-LUMO energy gaps (Eg) and structure geometries of the NH3 adsorption on the A (N-enriched) and B (B-enriched) models of open ended BNNTs. Different low-levels including, AM1, PM3, MNDO and UFF have been investigated, applying B3LYP/6-31 G* in all high-levels of ONIOM calculations. PM3 method is the most reliable among all low-levels used here especially in calculation of Ead, Eact and dipole moment and UFF is the most misleading. Either PM3 or MNDO overestimates the Eg, while AM1 underestimates it. We showed that in the case of A, by selecting two atom layers of the open end of the tube as inner layer, the results of ONIOM approach is in best agreement with those of the pure DFT calculations while in the case of B with the same condition, the results of ONIOM significantly differ with those of DFT. Finally, the results demonstrated that the ONIOM might be a reliable method in the study of weak interactions while in the case of strong interactions it is a controversial approach and should be applied cautiously. In addition, we showed that the agreement between the results of ONIOM and DFT approaches are independent of tube length and diameter.
References
Froese R, Humbel S, Svensson M, Morokuma K (1997) IMOMO(G2MS): a new high-level G2-like method for large molecules and its applications to Diels−Alder reactions. J Phys Chem A 101:227–233
Svensson M, Humbel S, Froese R, Matsubara T, Sieber S, Morokuma K (1996) ONIOM: a multilayered integrated MO + MM method for geometry optimizations and single point energy predictions a test for Diels−Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J Phys Chem 100:19357–19363
Wang L, Yi C, Zou H, Gan H, Xu J, Xu W (2011) Initial reactions of methyl-nitramine confined inside armchair (5,5) single-walled carbon nanotube. J Mol Model. doi:10.1007/s00894-011-0967-x
Lu X, Yuan Q, Zhang Q (2003) Sidewall epoxidation of single-walled carbon nanotubes: a theoretical prediction. Org Lett 5:3527–3530
Li F et al. (2006) Theoretical study of hydrogen atom adsorbed on carbon-doped BNnanotubes. Phys Lett A 357:369–373
Peralta-Inga Z et al. (2002) Characterization of surface electrostatic potentials of some (5,5) and (n,1) carbon and boron/nitrogen model nanotubes. Nano Lett 3:21–28
Wu HS, Cui XY, Qin XF, Strout D, Jiao H (2006) Boron nitride cages from B12N12 to B36N36: square–hexagon alternants vs boron nitride tubes. J Mol Model 12:537–542
Chu Y-Y, Su M-D (2004) Theoretical study of addition reactions of carbene, silylene, and germylene to carbon nanotubes. Chem Phys Lett 394:231–237
Bettinger HF (2004) Effects of finite carbon nanotube length on sidewall addition of fluorine atom and methylene. Org Lett 6:731–734
Ahmadi A, Beheshtian J, Hadipour N (2011) Chemisorption of NH3 at the open ends of boron nitride nanotubes: a DFT study. Struct Chem 22:183–188
Blum O, Milstein D (2002) Oxidative addition of water and aliphatic alcohols by IrCl(trialkylphosphine)3. J Am Chem Soc 124:11456–11467
Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G (2007) Facile splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center. Science 316:439–441
Zhao J, Goldman AS, Hartwig JF (2005) Oxidative addition of ammonia to form a stable monomeric amido hydride complex. Science 307:1080–1082
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski J, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, AlLaham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PM, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (1998) Gaussian Inc. Pittsburgh, PA
Kilina S, Badaeva E, Piryatinski A, Tretiak S, Saxena A, Bishop AR (2009) Bright and dark excitons in semiconductor carbon nanotubes: insights from electronic structure calculations. Phys Chem Chem Phys 11:4113–4123
Tabtimsai C, Keawwangchai S, Wanno B, Ruangpornvisuti V (2011) Gas adsorption on the Zn–, Pd– and Os–doped armchair (5,5) single–walled carbon nanotubes. J Mol Model. doi:10.1007/s00894-011-1047-y
Ruangpornvisuti V (2010) Molecular modeling of dissociative and non-dissociative chemisorption of nitrosamine on close-ended and open-ended pristine and Stone-Wales defective (5,5) armchair single-walled carbon nanotubes. J Mol Model 16:1127–1138
Ahmadi A, Beheshtian J, Hadipour NL (2011) Interaction of NH3 with aluminum nitride nanotube: electrostatic vs. covalent. Physica E 43:1717–1719
Ahmadi A, Kamfiroozi M, Beheshtian J, Hadipour N (2011) The effect of surface curvature of aluminum nitride nanotubes on the adsorption of NH3. Struct Chem. doi:10.1007/s11224-011-9820-1
Rimola A, Tosoni S, Sodupe M, Ugliengo P (2006) Does silica surface catalyse peptide bond formation? New insights from first-principles calculations. ChemPhysChem 7:157–163
Rimola A, Zicovich-Wilson CM, Dovesi R, Ugliengo P (2010) Search and characterization of transition state structures in crystalline systems using valence coordinates. J Chem Theor Comput 6:1341–1350
Roggero I, Civalleri B, Ugliengo P (2001) Modeling physisorption with the ONIOM method: the case of NH3 at the isolated hydroxyl group of the silica surface. Chem Phys Lett 341:625–632
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ahmadi, A., Beheshtian, J. & Kamfiroozi, M. Benchmarking of ONIOM method for the study of NH3 dissociation at open ends of BNNTs. J Mol Model 18, 1729–1734 (2012). https://doi.org/10.1007/s00894-011-1202-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1202-5