
Abstract
Adsorption of toxic hydrazine (N2H4) at the surface of pristine and Al-doped single-wall boron nitride nanotubes (BNNTs and Al-BNNTs) has been investigated using density functional theory (DFT). Single hydrazine molecule was allowed to interact with Lewis acid center (i.e., B atom) of BNNTs, where most stable gauche conformer of the guest is used. Both zigzag (8,0) and armchair (4,4) variants of BNNTs were considered and to estimate the effect of tube diameter on adsorption energies wider zigzag (9,0) and (10,0), and armchair (5,5) and (6,6), tubes were included in the host list. Estimated adsorption energies of 2–8 kcal/mol do not support strong binding between pristine BNNTs with hydrazine. However, Al doping makes BNNTs an attractive adsorbent of N2H4 with adsorption energy in the range of 32–35 kcal/mol. Stability of such complexes is found less sensitive to the chirality, but nominally decreases with tube diameter. Thus, a large quantity of hydrazine may be adsorbed by Al-BNNT samples. The dative N: → B/Al bond between hydrazine and host tubes is the source of stability of complexes. Molecular electrostatic potentials (MEPs) and frontier molecular orbitals of host and guest were calculated to explain interaction character and strength.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrazine (N2H4) is a highly toxic and colorless liquid with ammonia like odor [1]. Exposure to hydrazine may cause serious environmental health problems, such as damaging nervous system, liver, and brain [2,3,4]. On the positive side of properties, hydrazine exhibits diverse functionalities. It has been used in pharmaceutical, chemical, and agricultural industries [5,6,7]. Because of high hydrogen content (12.5 wt%), hydrazine found a suitable place in recent research on hydrogen energy [8, 9] and in direct hydrazine fuel cell (DHFC) [10, 11]. Hydrazine and derivatives are also utilized as fuel for missile and rocket propulsion systems [12].
Since its widespread usage in different industries, the release of traceable quantity of hydrazine from these sources into the environment is inevitable. Thus, detection and capture of such carcinogenic compound is highly demanding. Both theoretical and experimental research on adsorption of hydrazine at the surface of several materials, such as transition metals (for example Fe, Ni, Co, Cu, Ir, Rh), metal oxides (Co3O4), alloys (Ni-M, M = Fe, Pt, Pd, Ir), and nanostructures (gold nanocage/graphene), were reported [2, 5,6,7,8,9,10,11, 13,14,15,16,17,18,19,20,21,22,23,24]. Besides adsorption of hydrazine and interaction with hosts, several studies also explored the mechanism of decomposition of N2H4 to H2 and catalytic reduction to NH3 [9, 11] and hydrazine electrooxidation [11]. Magnitude of hydrazine adsorption energy depends on hosts and active sites (mostly at the surface of transition metal clusters and their oxides or alloys) and estimated value range between 10 and 70 kcal/mol [13,14,15,16].
Hydrazine is a weaker base than ammonia and the main source of interaction with the host is the lone pair of each nitrogen atom. Due to its basicity, N2H4 can form coordination complexes with metal, metal ions, and Lewis acids. Researchers interested in hydrazine adsorption mostly used metals or metal oxides or alloys as host. Boron nitride nanotube (BNNT) is a fascinating nanomaterial [25,26,27,28,29,30] that contains both Lewis acid and base centers, alternate to each other at the surface. Surface reactivity of single-wall (5,5) carbon nanotube [31, 32] and (5,5)-BN nanotube [33] has been extensively studied by Leszczynski and co-researchers. Based on Fukui indices and local hardness/softness, they showed boron atoms prefer nucleophilic attack, while electrophilic attack is possible on the N-centers of BNNT, where magnitude of both electrophilic and nucleophilic interactions depends on the locations of B and N atoms on the tube.
BNNT exhibits extraordinary mechanical properties, larger thermal conductivity, higher field emission property, and higher resistance towards oxidation and thermal stability than CNTs [34, 35]. A recent theoretical study [36] explored Lewis acid and Lewis base character of single-wall boron nitride nanotubes and the fundamentals of interactions with different guest acids and bases using density functional theory (DFT). Ammonia (NH3) was an example of base interacting with BNNTs where a dative N: → B covalent bond formation stabilizes the complex by about 4–6 kcal/mol.
Thus, it seems BNNT may be an alternative to metal-based materials for hydrazine adsorption. If so, then how stable are the resulting complexes? Samples of BNNTs are a mixture of a wide range of tubes with different chirality and diameter. How these two structural factors affect binding energies? In the case of weak interaction between tubes with N2H4 and selectivity towards one kind of tube over other, what alternative may reverse such situation to make tube as a good host for hydrazine? In the present investigation, we tried to answer all such question using density functional theory (DFT).
Computational details
The B3LYP variant of DFT [37, 38] was used to include correlation effects. Such DFT method was found reliable in predicting binding/interaction energies between BN-tubes and nitrogen containing (such as HBNH and NH3) guest molecule [36, 39]. A double-ζ quality 6-31G* basis set augmented with polarized spherical d-functions (5d) for all heavy atoms was initially used. (It may be noted that use of 5d than 6d functions reduces the number of overall basis function, and hence save computational cost significantly). Several previous studies [40,41,42,43,44] on BNNTs used a similar basis set. However, a basis function without a set of diffuse sp.-functions for electronegative atoms is inadequate, especially for adsorption energy [36]. So an additional set of diffuse sp.-functions was added to the 6-31G* basis function. Geometries of pristine and chemically modified BNNTs were fully optimized without any symmetry restriction using both 6-31G*(5d) and 6-31+G*(5d) basis sets, followed by vibrational analyses at B3LYP/6-31G*(5d) that insure the identification of true minima.
Adsorption energies (Eads) represent the electronic energy difference between the complex and constituents and are obtained using the following equation (Eq. 1), where E is the electronic energy.
For an attractive or favorable interaction, the adsorption energy (also termed as interaction/binding/complexation energies in the following discussion) is negative; otherwise, it is positive. All adsorption energies are corrected for basis set superposition error (BSSE, (where one unit uses basis sets of others in complex and vice-versa)) using the counter-poise (CP) method [45]. All calculations were performed using the Gaussian-09 [46] code.
Results and discussion
To estimate adsorption energies of hydrazine at the surface of BNNTs, the single guest molecule is allowed to interact with the Lewis-acid site (i.e., approaching from the top of the boron atom) of the BNNT tube. The gauche structure of N2H4 is more stable than anti and cis-conformers [13, 14, 16]; hence, the most stable gauche form is only considered in the present investigation. Based on the findings of previous investigation [36] that smaller diameter tubes most likely form stable complexes with guest bases and interaction strength diminishes as diameter increases, molecular models of smaller diameter tubes were considered in the present investigation. To verify the effect of chirality and diameter on adsorption of hydrazine at their surface, both armchair and zigzag tube models with varying diameter were included in the list. The specific tube models are armchair (n,n)-BNNT, n = 4, 5, 6 and zigzag (m,0)-BNNT where m = 8, 10, 12. Each of these tubes contains an equal number of boron and nitrogen atoms and edges were saturated with hydrogen atoms to avoid dangling bonds. Since the magnitudes of interaction of several Lewis acids and Lewis bases at the surface of BNNTs were found insensitive to the tube length [36], four layers of BN-ring are considered herein, where each layer contains 4n(2 m) number of atoms for armchair (n,n) (zigzag (m,0)) tubes. Thus, chemical compositions of these models are B32N32H16 for (4,4) and (8,0), B40N40H20 for (5,5) and (10,0), and B48N48H24 for (6,6) and (12,0). In these models, tube lengths are about 1.1 nm for armchair tubes and 0.9 nm for zigzag tubes.
BNNTs-hydrazine
Fully optimized geometries of BNNTs-hydrazine complexes obtained at the B3LYP/6-31+G* level are shown in Fig. 1 and adsorption energies (EAds) are summarized in Table 1. Energy data clearly reiterate the importance of diffuse sp.-functions in the basis set for BNNTs. For example, 6-31G* adsorption energies are overestimated by about 3–4 kcal/mol than 6-31+G* energies. Use of even larger basis sets would further lower adsorption energies, but such calculations are computationally highly demanding for such large systems. As an alternative, counter-poise (CP) correction to basis set superposition errors (BSSE) is most widely used technique for a reasonable estimated of adsorption energies. Such corrections further lower adsorption energies, given in the second and last column of Table 1, by about 5.5–6.4 kcal/mol at B3LYP/6-31G* and 3.1–3.6 kcal/mol at B3LYP/6-31+G* methods, respectively. As expected, BSSE correction energy is almost double for smaller basis set than larger basis set. Difference in BSSE corrected adsorption energies obtained from two basis sets indicates smaller basis set mostly overestimates adsorption energy by 0.2 to 1.0 kcal/mol. These results indicate non-CP corrected adsorption energies are overestimated by a meaningful factor that cannot be ignored in studying such interactions and BSSE correction is highly recommended while using similar basis functions. In the following discussions of adsorption energies, BSSE corrected B3LYP/6-31+G* energies are used.

Optimized structure of hydrazine-BNNT complexes: (4,4) (1a), (5,5) (1b), (6,6) (1c), (8,0) (1d), (10,0) (1e), and (12,0) (1f). BSSE corrected B3LYP/6-31+G* adsorption energies (in kcal/mol) of hydrazine at the surface of BNNTs are shown in bold. A negative value of adsorption energy indicates stability of the complex. B(BNNT)-N(hydrazine) and N-N (hydrazine) bond distances are given in angstrom (Å). Color scheme: boron—yellow, nitrogen—blue, and gray—hydrogen
Interaction of hydrazine at the surface of (4,4)BNNT nanotube is found favorable and resulting complex 1a is stabilize by − 7.9 kcal/mol. Such interaction is found stronger by 3.4 kcal/mol than the interaction of the same tube with NH3 (EAds = −4.5 kcal/mol [36]). In both cases, covalent dative N: → B(BNNT) bond formation is the source of attractive interaction. Ammonia-borane (H3N: → BH3) is the illustrative example of such dative covalent bond and exhibits binding energy of − 28.3 kcal/mol [36] at the same level of theory. Thus, the interaction between hydrazine and BNNT is about 3.5 times weaker than ammonia-borane. Although 1a complex is significantly weaker, N-B(BNNT) distance of 1.684 Å is closer to that of in ammonia-borane (R(B-N) = 1.682 Å) structure at the same level of theory. These results suggest that bond distance between BN-tube and guest molecule may not be a good indicator (shorter the distance, stronger the interaction) of interaction strength at the surface of BNNTs.
As the diameter of armchair BNNT increases from 0.59 nm (1a) to 0.72 nm (1b), EAds value decreases to − 2.6 kcal/mol and a further increase in diameter to about 0.87 nm again lower the adsorption energy of 1c to − 2.1 kcal/mol. Complexation resulted in slight elongation (about 0.03 Å) of the NN bond of hydrazine. With the increase in diameter of the tube, the distance between host and guest increases from 1.684 to 1.716 Å. These results suggest that hydrazine may form a weak complex with smaller diameter armchair tubes, but certainly not with wider tubes.
Zigzag BNNTs-hydrazine complexes follow almost similar trends in structure and interaction energy as their armchair cousins. Interestingly, the adsorption energy of (8,0)BNNT-N2H4 complex 1d is almost same (EAds = −7.8 kcal/mol) as its isoelectronic armchair counterpart (1a). With the increase in diameter, interaction gets weaker; EAds reduces to − 4.1 kcal/mol in (10,0)BNNT (1e) and then to − 1.7 kcal/mol in (12,0)BNNT (1f) complex. The weakening of interaction between hydrazine and zigzag tubes is also reflected in the host-guest distance that progressively increases from 1.687 to 1.714 Å.
Based on the interaction energy and host-guest distance, it may be concluded that hydrazine will not form stable complexes with both kinds of BNNT. However, smaller diameter tubes (< 0.6 nm), due to their high structural strain, may form weaker complexes. These findings limited the use of BNNT as an adsorbent of hydrazine.
Al-BNNTs-hydrazine
Substitutional doping is one possibility to improve the adsorption ability of BNNTs. Previous studies [47] showed that Al-doping enhances adsorption energy of BNNT (also single-wall carbon nanotube [48]) when guest molecule is ammonia. Since interaction with hydrazine is similar to NH3, Al-doped BNNT seems a reasonable choice to explore. One boron atom at the middle of the tube is substituted by an Al atom, and structures of all six Al-BNNTs and their complexes with hydrazine were fully optimized.
Adsorption energies calculated using different basis sets are summarized in Table 2, and fully optimized structures obtained at the B3LYP/6-31+G* level are shown in Fig. 2. Effect of additional diffuse sp.-functions to 6-31G* and correction for basis set superposition error to both basis sets are found similar to the corresponding energies of pristine BN-tubes. The overall lowering of EAds of Al-BNNT-N2H4 complexes is in the range of 6–7 kcal/mol. In the following discussion, we used results given in the third column of Table 2.

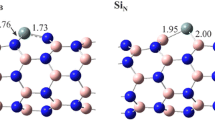
Optimized structures of Al-BNNT-hydrazine complexes: (4,4) (2a), (5,5) (2b), (6,6) (2c), (8,0) (2d), (10,0) (2e), and (12,0) (2f). BSSE corrected B3LYP/6-31+G* adsorption energies (in kcal/mol) of hydrazine at the surface of Al-BNNTs are shown in bold. A negative value of adsorption energy indicates stability of the complex. Al(Al-BNNT)-N(hydrazine) and N-N (hydrazine) bond distances are given in angstrom (Å). Color scheme: boron—yellow, nitrogen—blue, gray—hydrogen, and aluminum—pink
Al-doping increases the adsorption energy by a wide margin than the pristine tube. For example, both (4,4)Al-BNNT (2a) and (8,0)Al-BNNT (2d) tubes are stabilized by about 35–36 kcal/mol, which is more than four times the corresponding values of 1a and 1d. Unlike pristine tubes, enlargement in diameter of the tube has an insignificant effect on the adsorption energy. The EAds decreases from − 35.8 kcal/mol in 2a to − 33.3 kcal/mol in 2c, with an intermediate value of − 34.5 kcal/mol in 2b. Similarly, the stability of zigzag tube complex reduces from − 35.2 kcal/mol (2d) to − 33.3 kcal/mol (2e) and then to − 33.2 kcal/mol (2f). These results indicate a strong interaction between Al-doped BNNTs and hydrazine is possible even for much wider tubes than considered herein. This range of binding energy is even lower by 7–9 kcal/mol than the simplest ammonia-alane (H3Al-NH3) complex [36] (EAds = −26 kcal/mol), where N: → Al dative covalent bond is the source of interaction.
The Al-N bond distance in all 2a-2f complexes, shown in Fig. 2, is in the range of 2.01 to 2.03 Å and in excellent agreement with the reference distance of 2.10 Å in ammonia-alane complex [36]. Upon complexation with Al-BNNTs, the NN distance of hydrazine is stretched by about 0.04 Å in complexes and this change is independent of chirality and diameter of doped tubes.
Molecular electrostatic potentials
To understand what makes Al-doped BNNTs an attractive adsorbent of hydrazine over pristine tubes, we calculated molecular electrostatic potentials (MEP) at the B3LYP/6-31+G*(5d) level, describing charge distribution in a molecule in three dimensions, of pristine and Al-doped (4,4) and (8,0) tubes. According to Mulliken [49, 50] and natural population analyses [51, 52] boron atoms of the BN-tubes are positively charged (around + 1.0) and nitrogen atoms are negatively charged (around − 1.0), while Al of doped tube is less positively charged (by about 0.2 to 0.4, depending on the diameter of the tube) than B atoms.
In Al-doped tube, Fig. 3, MEP of pristine and doped (4,4) and (8,0) BNNTs are shown, where red and blue regions represent negative and positive potentials. Intermediate potentials from positive to negative ends are shown in light blue to green and then yellow/orange. The intense red regions in the MEP of hydrazine are located at the nitrogen atoms; one of them is approaching to the B/Al atoms of tubes to form a complex. The (4,4)BNNT (3a) exhibits alternate red (around N atoms) and light-blue/green regions (around B-atoms) spread across the tube surface, while blue regions are mostly located at the edges. Similar to the armchair tube, MEP of the zigzag tube (3b) also possess red, light blue/green regions, but not continuous as in armchair counterpart. So approaching N of N2H4 encounters repulsion from the negative potential of nitrogen atoms surrounded to the active B-site. Thus, two opposing forces are involved in dative bond formation between host and guest: attractive force between the red region of hydrazine and weaker positive potential at the active site and repulsive forces associated with surrounding negative regions of the active site and hydrazine red region. These opposing factors make hydrazine-BNNTs complexes less stable.

B3LYP/6-31+G*(5d) molecular electrostatic potential of hydrazine, (4,4)BNNT (3a), (8,0)BNNT (3b) (4,4)Al-BNNT (3c), and (8,0)Al-BNNT (3d)
Substitution of B by Al changes MEPs of doped BNNTs significantly at the active sites. An intense blue region around Al atom (3c and 3d, shown by arrows), which is also extended well above the surface, may responsible for strong interaction with hydrazine. Since Al is located above the surface in doped-BNNTs, negative potentials of neighboring N atoms are far away to approaching negative N atom of hydrazine, minimizes repulsive factor that hindered pristine tube for complex formation. Stronger bonding in Al-doped BNNTs than their pristine form may be explained by the character of the lowest unoccupied molecular orbital (LUMO) obtained at the B3LYP/6-31+G*(5d) level. The LUMO of the doped tube (4a and 4b in Fig. 4) is almost pure Al p-orbital available to accept electrons from lone-pair of N atoms (HOMO of hydrazine). The LUMO of pristine tubes represents p-orbitals of B atoms located near the edge(s) (4c and 4d) of the tube. Thus, adsorption of hydrazine at the surface of pristine and Al-doped BNNTs may be considered as chemisorption, where in former cases very week interaction while in doped tubes moderately strong interaction.

Highest occupied molecular orbital (HOMO) of hydrazine and lowest unoccupied molecular orbital (LUMO) of (4,4)Al-BNNT (4a), (8,0)Al-BNNT (4b), (4,4)BNNT (4c), and (8,0)BNNT (4d). MOs were obtained at the B3LYP/6-31+G*(5d) level
Conclusions
Interactions between toxic hydrazine and single-wall boron nitride tubes (pristine and Al-doped) have been investigated using B3LYP variant of DFT method. Due to the examination of electron-rich N atoms, the inclusion of diffuse sp. functions (such as 6-31+G* basis set) seems appropriate for the description of such interactions. Results from this study recommend considering counterpoise correction (due to basis set superposition error) to adsorption energies.
Adsorption energy (EAds) of pure BNNT strongly depends on the tube diameter; higher for smaller diameter tubes due to structural strain and sharply decreases with the enlargement of the tube diameter. For example, EAds value decreases from about 8.0 kcal/mol in isoelectronic pair of (4,4)BNNT-N2H4 and (8,0)BNNT-N2H4 to about 2.0 kcal/mol in (6,6) BNNT-N2H4 and (12,0) BNNT-N2H4. Such weak interaction limits the use of pure BNNTs as an adsorbent of hydrazine.
Replacement of a B-atom of the tube by an Al atom drastically increases the adsorption energy to the range of 33–35 kcal/mol which is almost independent on the chirality of the Al-doped tubes. The nominal diminishing value of EAds (about 2 kcal/mol for about 0.3 nm increment of the diameter) suggest that adsorption energy may be around 20 kcal/mol for Al-BNNT tubes as wide as 3 nm diameter. This range (20–35 kcal/mol) of binding energy is strong enough to form stable Al-BNNTs-hydrazine complexes, but interaction is not strong enough to easily release hydrazine. Thus, a significant quantity of hydrazine, as both types of tube with a wide range of diameter, may be adsorbed and stored on Al-doped BNNTs and may release hydrazine by applying milder condition (such as heating the sample) for a wide range of applications.
A dative covalent bond formation is the origin of the interaction between pure and doped BNNTs with hydrazine, where lone pair of one nitrogen of N2H4 donates electrons to vacant p-orbital of B/Al atom of the tube (hydrazine)N:→B (BNNTs). In the case of doped tube, LUMO represents pure p-orbital of Al, whereas in pure tube that is delocalized over several B atoms near the edge(s). Molecular electrostatic potential (MEP) maps provide a clear explanation in the differences between pristine and doped tubes. The negative potential of the approaching nitrogen atom encounters repulsion from the negative potentials of adjacent three nitrogen atoms of the link boron atom, due to their close proximity with hydrazine. In the case of doped tubes, Al atom, due to larger size than boron and longer Al-N distances, is well above the surface of the tube that reduces such repulsion, making tube more attractive adsorbent of hydrazine.
References
Agency UEP (1999) Integrated Risk Information System (IRIS) on hydrazine/hydrazine sulfate. National Center for Environmental Assessment, Office of Research and Development, Washington, DC
Reilly CA, Aust S (1997) Peroxidase substrates stimulate the oxidation of hydralazine to metabolites which cause single-strand breaks in DNA. Chem Res Toxicol 10(3):328–334
Zelnick SD, Mattie DR, Stepanial PC (2003) Occupational exposure to hydrazines: treatment of acute central nervous system toxicity. Abviat Space Environ Med 74(12):1285–1291
Garrod S, Bollard ME, Nicholls AW, Connor SC, Connelly J, Nicholson JK, Holmes E (2005) Integrated metabonomic analysis of the multiorgan effects of hydrazine toxicity in the rat. Chem Res Toxicol 18(2):115–122
Ragnarsson U (2001) Synthetic methodology for alkyl substituted hydrazines. Chem Soc Rev 30:205–213
Narayanan SS, Scholz F (1999) A comparative study of the electrocatalytic activities of some metal hexacyanoferrates for the oxidation of hydrazine. Electroanalysis 11:465–469
Yamada K, Yasuda K, Fujiwara N, Siroma Z, Tanaka H, Miyazaki Y, Kobayashi T (2003) Potential application of anion-exchange membrane for hydrazine fuel cell electrolyte. Electrochem Commun 5(10):892–896. https://doi.org/10.1016/j.elecom.2003.08.015
Singh SK, Xu Q (2013) Nanocatalysts for hydrogen generation from hydrazine. Catal Sci Technol 3:1889–1900
Pelegrini M, Parreira RLT, Ferrão LFA, Caramori GF, Ortolan AO, da Silva EH, Roberto-Neto O, Rocco JAFF, Machado FBC (2016) Hydrazine decomposition on a small platinum cluster: the role of N2H5 intermediate. Theo Chem Acc 135(3):58
Asazawa K, Sakamoto T, Yamaguchi S, Yamada K, Fujikawa H, Tanaka H, Oguro K (2009) Study of anode catalysts and fuel concentration on direct hydrazine alkaline anion-exchange membrane fuel cells. J Electrochem Soc 156(4):B509–B512
Yang H, AZhong X, Dong Z, Wang J, Jin J, Ma J (2012) A highly active hydrazine fuel cell catalyst consisting of a Ni–Fe nanoparticle alloy plated on carbon materials by pulse reversal. RSC Adv 2:5038–5040
Sutton GP, Biblarz O (2010) Rocket propulsion elements, 8th edn. Wiley, New York,
Agusta MK, Kasai H (2012) First principles investigations of hydrazine adsorption conformations on Ni(111) surface. Surf Sci 606:766–771
He YB, Jia JF, Wu HS (2015) The interaction of hydrazine with an Rh(1 1 1) surface as a model for adsorption to rhodium nanoparticles: a dispersion-corrected DFT study. Appl Sutf Sci 327:462–469
He Y-B, Jia J-F, Wu H-S (2015) Selectivity of Ni-based surface alloys toward hydrazine adsorption: a DFT study with van der Waals interactions. Appl Surf Sci 339:36–45
Fathurrahman F, Kasai H (2015) Density functional study of hydrazine adsorption and its N=N bond cleaving on Fe(110) surface. Surf Sci 639:25–31
Daemi S, Ashkarran AA, Bahari A, Ghasemi S (2017) Fabrication of a gold nanocage/graphene nanoscale platform for electrocatalytic detection of hydrazine. Sensors Actuators B Chem 245:55–65
Hao Y, Zhang Y, Ruan K, Chen W, Zhou B, Tan X, Wang Y, Zhao L, Zhang G, Qu P, Xu M (2017) A naphthalimide-based chemodosimetric probe for ratiometric detection of hydrazine. Sensors Actuators B Chem 244:417–424
Zhang J, Gao W, Dou M, Wang F, Liu J, Li Z, Ji J (2015) Nanorod-constructed porous Co3O4 nanowires: highly sensitive sensors for the detection of hydrazine. Analyst 140:1686–1692
Deng K, Li C, Qiu X, Zhou J, Hou Z (2015) Synthesis of cobalt hexacyanoferrate decorated graphene oxide/carbon nanotubes-COOH hybrid and their application for sensitive detection of hydrazine. Electrochim Acta 174:1096–1103
Tafreshi SS, Roldan A, Dzade NY, de Leeuw NH (2014) Adsorption of hydrazine on the perfect and defective copper (111) surface: a dispersion-corrected DFT study. Surf Sci 622:1–8
Esrafili MD, Teymurian VM, Nurazar R (2015) Catalytic dehydrogenation of hydrazine on silicon-carbide nanotubes: a DFT study on the kinetic issue. Surf Sci 632:118–125
Zhang P-X, Wang Y-G, Huang Y-Q, Zhang T, Wu G-S, Li J (2011) Density functional theory investigations on the catalytic mechanisms of hydrazine decompositions on Ir(1 1 1). Catal Today 165:80–88
McKay HL, Jenkins SJ, Wales DJ (2011) Dissociative chemisorption of hydrazine on an Fe{2 1 1} surface. J Phys Chem C 115:17812–17828
Terrones M, Hsu WK, Terrones H, Zhang JP, Ramos S, Hare JP, Castillo R, Prasside K, Cheetham AK, Kroto HW, Walton DRM (1996) Metal particle catalyzed production of nanoscale BN structures. Chem Phys Lett 259:568–573
Loiseau A, Willaime F, Demoncy N, Hug G, Pascard H (1996) Boron nitride nanotubes with reduced numbers of layers synthesized by arc discharge. Phys Rev Lett 76(25):4737–4740
Terauchi M, Tanaka M, Takehisa M, Saito Y (1998) Electron energy-loss spectroscopy study of the electronic structure of boron nitride nanotubes. J Electron Microsc 47(4):319–324
Golberg D, Bando Y, Eremets M, Takemura K, Kurashima K, Tamiya K, Yusa H (1997) Boron nitride nanotube growth defects and their annealing-out under electron irradiation. Chem Phys Lett 279(3–4):191–196
Laude T, Matsui Y, Marraud A, Jouffrey B (2000) Long ropes of boron nitride nanotubes grown by a continuous laser heating. App Phys Lett 76(22):3239–3241
Golberg D, Bando Y, Sato T, Grobert N, Reyes-Reyes M, Terrones H, Terrones M (2002) Nanocages of layered BN: super-high pressure nanocells for formation of solid nitrogen. J Chem Phys 116(9):8523–8532
Saha S, Dinadayalane TC, Leszczynska D, Leszczynski J (2012) Open and capped (5,5) armchair SWCNTs: a comparative study of DFT-based reactivity descriptors. Chem Phys Lett 541:85–91
Saha S, Dinadayalane TC, Murray JS, Leszczynska D, Leszczynski J (2012) Surface reactivity for chlorination on chlorinated (5,5) armchair SWCNT: a computational approach. J Phys Chem C 116:22399–22410
Saha S, Dinadayalane TC, Leszczynska D, Leszczynski J (2013) DFT-based reactivity study of (5,5) armchair boron nitride nanotube (BNNT). Chem Phys Lett 565:69–73
Garel J, Leven I, Zhi C, Nagapriya KS, Popovitz-Biro R, Golberg D, Bando Y, Hod O, Joselevich E (2012) Ultrahigh torsional stiffness and strength of boron nitride nanotubes. Nano Lett 12:6347–6352
Huang Y, Lin J, Zou J, Wang M-S, Faerstein K, Tang C, Bando Y, Golberg D (2013) Thin boron nitride nanotubes with exceptionally high strength and toughness. Nano 5:4840–4846
Sundaram R, Scheiner S, Roy AK, Kar T (2015) Site and chirality selective chemical modifications of boron nitride nanotubes (BNNTs) via Lewis acid/base interactions. Phys Chem Chem Phys 17:3850–3866
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Sundaram R, Scheiner S, Roy AK, Kar T (2015) B═N bond cleavage and BN ring expansion at the surface of boron nitride nanotubes by iminoborane. J Phys Chem C 119:3253–3259
Wu X, An W, Zeng XC (2006) Chemical functionalization of boron-nitride nanotubes with NH3 and amino functional groups. J Am Chem Soc 128:12001–12006
Li Y, Zhou Z, Zhao J (2007) Transformation from chemisorption to physisorption with tube diameter and gas concentration: computational studies on NH3 adsorption in BN nanotubes. J Chem Phys 127:184705–184706
Ahmadi A, Beheshtian J, Hadipour NL (2011) Chemisorption of NH3 at the open ends of boron nitride nanotubes: a DFT study. Struct Chem 22:183–188
Zahedi E (2012) Effect of tube radius on the exohedral chemical functionalization of boron-nitride zigzag nanotubes with NH3. Physica B 407:3841–3848
Rimola A, Sodupe M (2013) Physisorption vs. chemisorption of probe molecules on boron nitride nanomaterials: the effect of surface curvature. Phys Chem Chem Phys 15:13190–13198
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Jaramillo J, Stratmann RE, Yazyev O, Austin A, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian09. Gaussian, Inc., Wallingford,
Soltani A, Raz SG, Rezaei VJ, Dehno Khalaji A, Savar M (2012) Ab initio investigation of Al- and Ga-doped single-walled boron nitride nanotubes as ammonia sensor. Appl Surf Sci 263:619–625
Azizi K, Karimpanah M (2013) Computational study of Al- or P-doped single-walled carbon nanotube as NH3 and NO2 sensors. Appl Surf Sci 285P:102–109
Mulliken RS (1955) Electronic population analysis on LCAO-MO molecular wave functions. I. J Chem Phys 23(10):1833–1840
Mulliken RS (1955) Electron population analysis on LCAO-MO molecular wave functions. II. Overlap populations, bond orders, and bond energies. J Chem Phys 23(10):1841–1846
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Reed AE, Weinhold F, Curtiss LA, Pochatko DJ (1986) Natural bond orbital analysis of molecular interactions: theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO, and CO2 with HF, H2O, and NH3. J Chem Phys 84:5687–5705
Acknowledgements
Rajashabala Sundaram thanks to the University Grants Commission (UGC) of India for providing financial support to carry out research in the USA under Indo-US Raman Post-Doctoral Fellowship Program and the Department of Science and Technology (India, DST-PURSE Program). HPC center at Utah State University is acknowledged for providing computational facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Muniyandi, S., Sundaram, R. & Kar, T. Aluminum doping makes boron nitride nanotubes (BNNTs) an attractive adsorbent of hydrazine (N2H4). Struct Chem 29, 375–382 (2018). https://doi.org/10.1007/s11224-017-1034-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-1034-8