
Abstract
The present study reports the effect of oxygen addition on small size Nin (n = 1-6) clusters in different spin states within the framework of linear combination of atomic orbital (LCAO) density functional theory (DFT) under spin polarized generalized gradient approximation (GGA) functional. Relative stabilities of the optimized clusters are discussed on the basis of the calculated parameters, such as, binding energy (BE), embedding energy (EE) and fragmentation energy (FE). Other parameters, like ionization potential (IP), electron affinity (EA), etc. show that though the additions of oxygen can affect the chemical properties of Nin clusters with an additional stability to NinO. In most of the cases the magnetic moment of the stable isomers are geometry dependent for a particular size both in pure and oxidized clusters. Calculated magnetic moments of NinO (n = 1-6) clusters reveal that the magnetic moment of ground state Ni4O isomers in different geometries is same as in pure Ni4 isomers. Present study also explains the cause of stable magnetic moment in Ni4O cluster through the distribution of electrons in different orbitals.

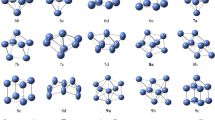
(b) Orbital and spin density distribution of ground state Nin and NinO clusters among different structural isomers and the different spin multiplicities (singlet to nonet) studied of different sizes. All clusters shown in figure, correspond to the minimal energy among the different structural isomers, and the different spin multiplicities studied in a particular size.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the last few decades transition metal (TM) oxide clusters are becoming interesting field of theoretical as well as experimental research because of their novel electronic, magnetic and optical properties as well as the prominent role in the catalytic activities, environmental process in addition to several other applications in nanoscience and nanotechnology [1–12]. Although there are many experimental studies by using mass spectrometry and photoelectron spectroscopy of TM oxides, but determining the relative stabilities of these systems remains problematic. Several experimental works on TM oxide cluster in the gas phase have contributed fundamental information about their chemical bonding, reactivity, magnetic, and electronic properties. Herman et al. [13] studied the gas phase reaction of different transition metal ions (Ti+, V+, Fe+, Co+, Ni+, Cu+, Zn+) with CO and CO2 followed by ab initio calculations for further understanding of reaction mechanism, reaction kinetics and thermochemistry of the nanoclusters. Xu et al. [14] studied the oxygen affinity of pure Ptn clusters (n = 1-10) using density functional theory (DFT). By using laser vaporization and electro-spray experimental techniques followed by photoelectron spectroscopy Zhai et al. [15] studied electronic and geometric structures of doubly and singly charged species M2O 2-7 , MM´O 2-7 , and M2O -7 (M, M´ = Cr, Mo, W) followed by DFT calculations to understand the experimental evolution of geometric and electronic structures as a function of charge state. Liu et al. [16] studied the effect of addition of oxygen on the small size Co clusters. They found that the addition of oxygen could not produce much effect on the average magnetic moment of the Co clusters. Li et al. [17] calculated total atomization energies and normalized clustering energies of (MO2)n (M = Ti, Zr, Hf) and (MO3)n (M = Cr, Mo, W) transition metal oxide clusters up to n = 4 at the coupled cluster [CCSD(T)] and density functional theory (DFT) levels. In another study Zhai et al. [18] reported a comparative study of reduced transition metal oxide clusters, M3O −8 (M = Cr, W) in neutral and anionic state via anion photoelectron spectroscopy, density functional theory, molecular orbital theory (CCSD(T)) calculations and predicted the redox reaction in thermochemistry of the system.
Among several other transition metal oxides, small size NinO clusters is one of the most important candidates because of its high magnetic moment and stability. Nickel oxide is a highly insoluble thermally stable nickel source suitable for glass, optics and ceramic applications. Oxide compounds are usually not electrical conductor; however certain nickel oxides are electronically conductive and therefore have useful applications in fuel cells and oxygen generation systems where they exhibit ionic conductivity. Moreover, nickel oxide is also an important element as a component in lightweight aerospace applications. Several other theoretical [19–24] and experimental [25–30] studies on nickel oxides in neutral and charged states are also reported. Most of the theoretical studies are focused on the geometrical, electronic and magnetic properties of pure and oxidized nickel clusters Nin (n ≤ 60) [19–23]. On the other hand, most of the experimental studies [25–30] on nickel oxide clusters are focused on their magnetic and chemical properties by using gas phase reaction, photoelectron spectroscopy and Mössbauer spectroscopy. Both theoretical and experimental research on NinO clusters are challenging because of its complicated electronic and magnetic properties due to the 3d sub-shells and in addition, the energy difference between different spin states is very small. Therefore, determination of the ground state in a particular size is a difficult work. In this report a detailed theoretical investigation of small size Nin and NinO (n = 1-6) clusters of different geometries and spin states (singlet to 11) is presented to understand their electronic as well as magnetic behavior and their variation with the geometry, size and spin states in a systematic manner. In the latter part, both Nin and NinO (n = 1-6) optimized ground state geometry in each size are then selected to study their binding energy, embedding energy, stability, chemical potential, ionization potential etc. Comparisons of the present calculations with the available experimental and theoretical results are also done.
Computational
In the present report geometry optimization of Nin and NinO (n = 1-6) clusters are performed by using density-functional theory (DFT) with the unrestricted B3PW91 exchange-correlation potential [31–34]. Self-consistent field (SCF) electronic structure calculations were carried out on all clusters within the framework of Kohn-Sham DFT [35]. Molecular orbital (MO) are expressed as linear combination of atom-centered basis functions for which the standard Gaussian LanL2DZ basis set and associated effective core potential (ECP) is used on all the atoms. Spin-polarized calculations are carried out using the Becke three-parameter exchange and the Perdew-Wang generalized gradient approximation (GGA) functional [36–39]. The standard LanL2DZ basis sets are employed to provide an effective way to reduce difficulties in calculations of two-electron integrals caused by transition metal Ni atom [40]. In order to obtain the lowest-energy NinO structures, we have chosen a considerable number of possible structures as initial geometries in each cluster size (n = 1-6). Based upon the available theoretically and experimentally verified geometries of Nin [23] different evolution patterns for determining the NinO clusters, including Ni-substituted and O-capped patterns, are first taken into account and then the equilibrium structure in a particular size is obtained by varying the geometry starting from high to low symmetric initial guess structures. For each stationary point of a cluster, the stability is reassured by calculating the frequency of harmonic vibration. If any imaginary frequency is found, a relaxation along that vibrational mode is carried out until the true local minimum is obtained. In all clusters, geometries were optimized with no symmetry constraints in each initial guess structure. Both in case of pure and oxygenated nickel clusters optimizations are done with the spin multiplicity varied from singlet to 11 and are independent of the size of the clusters. Optimized ground state geometries in each size with their spin multiplicity are shown in Fig. 1. In case, when the total optimization energy decreases with increasing spin, increasingly higher spin states is considered until the energy minimum with respect to spin is reached. To check the reliability of the present level of calculation, calculated parameters of Ni2 and NiO dimers, as example, the bond length, ionization potential, electron affinity and the lowest vibrational frequency are compared with the reported experimental and theoretical data available as shown in Tables 1 and 2. It shows that the present calculated values are comparable to the reported values and hence it is expected that the present level of calculation can be applied to a bigger size clusters in the same system. All theoretical calculations are carried out with the Gaussian 03 program package [41].

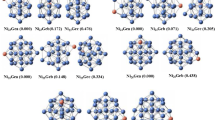
(a) Optimized structures of Nin and NinO clusters. In the figure, the last digit in name represents the spin multiplicity of the clusters, as: Triplet (3), Quintet (5), Septet (7) and Nonlet (9). All clusters shown in figure, correspond to the minimal energy among the different structural isomers, and the different spin multiplicities studied in a particular size. (b) Orbital and spin density distribution of ground state Nin and NinO clusters among different structural isomers and the different spin multiplicities (singlet to nonet) studied of different sizes. All clusters shown in figure, correspond to the minimal energy among the different structural isomers, and the different spin multiplicities studied in a particular size
Results and discussion
Optimized structures of different bare nickel and nickel oxide clusters calculated in the present study are shown in Fig. 1a-b along with their HOMO-LUMO orbital distributions and total spin densities. Different physical and chemical parameters of the pure and doped clusters such as bond length, frequency, ionization potential, point group symmetry, electronic states, relative energies etc. are shown in Tables 1 2 and 3. In the following section geometries, stabilities, electronic and magnetic properties of the nanoclusters will be discussed on the basis of the calculated parameters and their variation with the cluster size.
Geometries and stabilities
Ni2 and NiO clusters
As the first members in the series, different physical and chemical parameters of Ni2 (Dαh) and NiO dimers are calculated and presented in Tables 1 and 2 with the reported theoretical and experimental data. Variation of the optimized energies of NiO dimer in different spin states are presented in Table 3 with respect to the optimized ground state dimer with C∞ν point group symmetry and 3Σ+ electronic state. In the present calculation the bond length of the pure nickel dimer varies with different spin multiplicities from singlet to 11 in the range of 2.32 Å to 2.45 Å with a value of 2.36 Å in triplet ground state, which is comparable to the experimental values presented in Table 1. Similarly, the bond length of NiO cluster is found as 1.70 Å. This is close to the experimental value 1.63 Å. Other calculated parameters, as like, IP and EA are in good agreement with the reported theoretical and experimental data shown in Tables 1 and 2. From the parameters presented in Tables 1 and 2, it is clear that present level of calculation is appropriate for both pure nickel and nickel oxide clusters. In both the clusters calculated natural vibrational frequencies are also in good agreement with the previously reported values. The ionization potential calculated for Ni2 cluster is 7.06 eV and this is within the range of reported experimental and theoretical values that varies from 5.4 eV to 8.3 eV as shown in Table 2. Electron affinities of Ni2 and NiO clusters are also quite close to the reported values shown in Table 1. Calculated HOMO and LUMO orbital distributions of the optimized ground state of Nin (n = 1-7) and NinO (n = 1-6) clusters along with the total spin magnetic moment distributions are shown in Fig. 1b. In Ni2 dimer structure, the HOMO distribution is like a double quadruple charge distribution (π-bonding) whereas the LUMO orbital is like a perfect dumbbell structure (σ-bonding). In NiO dimer, most of the orbital charge density distributed around the nickel atom and this is even more in LUMO orbital. The total spin density distribution is symmetrical in Ni dimer but it is more around the nickel atom in NiO cluster as shown in Fig. 1b.
Ni3 and Ni2O cluster
In bare Ni3 cluster two different geometries are obtained. The ground state geometry is a triangle with internal angle close to 56.7o in triplet spin state with C s point group symmetry. This structure is lower in energy than the linear chain structure by an amount 0.06 eV. In linear chain geometry the ground state is a quintet with D ∞h point group symmetry. At this size the spin state in different ground state geometries is geometry dependent. The same behavior is also found in other systems and will be discussed in afterward section. By replacing the end and the middle Ni atoms by oxygen in the ground state Ni3_A and in Ni3_B (Fig. 1a), four different optimized structures at different spin states are obtained. The first triangular geometry (Ni2O_A) is the ground state structure in triplet spin state. Out of the other three remaining structures, two are linear chain like (Ni-O-Ni or Ni-Ni-O) and the third one is a bend structure (C S symmetry) where the Ni-Ni-O angle is 1090 as shown in Fig. 1a. The first linear chain structure Ni-Ni-O has a C∞v point group symmetry, whereas the 2nd linear chain structure with an oxygen at the middle position of the chain (Ni-O-Ni) has Dαh symmetry. Here the first and third structures of Ni2O are in triplet state, whereas, the second and fourth structures are in quintet and singlet states respectively. It is to be noted that the ionized Ni2O+ cluster is a linear chain structure with the oxygen atom at the middle (Ni-O-Ni). In Ni2O+ the Ni atoms contribute charge in ionization. Calculated ionization potential of Ni3 cluster is in good agreement with the reported value. The reported value of ionization potential varies from 3.9 eV to 6.8 eV (Table 2) whereas the present value is 5.54 eV.
Ni4 and Ni3O clusters
Between two different isomers of Ni4, the triangular based pyramid geometry is the optimized ground state in quintet spin state with D2d point group symmetry and 5A2 molecular orbital electronic state represented as irreducible representation (IR) in Table 3. The other structure is a bend rhombus. The ionization potential of the ground state structure is 5.46 eV is close to reported values as given in Table 2. Replacing one-nickel atom in bend rhombus isomer of Ni4 structure, Ni3O optimized structure with C3v point group symmetry is obtained as shown in Fig. 1a. In the present calculation the linear chain structures are not considered because of the presence of an imaginary frequency. Relative energies of the structures in different spin state with respect to the ground state structure are presented in Table 3.
Ni5 and Ni4O clusters
Two different optimized structures are obtained in Ni5 size. The distorted Ni capped rhombus based structure is the ground state geometry with Cs point group symmetry in septet spin state. The other structure is a bi-capped triangular Ni3 structure (not shown). A number of stable geometries are obtained in Ni4O cluster as shown in Fig. 1a. The ground state geometry is obtained by replacing a capped nickel atom from the bi-capped Ni3 triangular structure by oxygen. The ground state of Ni4O is in a quintet spin state as like ground state Ni4 isomers. The binding energy of this structure is higher than Ni4 or Ni5 structures.
Ni7, Ni6, Ni5O and Ni6O clusters
The most stable structure in Ni6 is bi-capped quadrilateral with Cs point group symmetry. By replacing one nickel with oxygen, three different kinds of optimized structures are obtained. Among them, bi-capped Ni rhombus structure in septet spin state is the ground state geometry with C s point group symmetry (Fig. 1b). The ground state geometry of Ni5O structure is in septet. Three other ground state geometries are also obtained in Ni5O series in triplet; quintet and septet spin states respectively. Energetically these three structures are very close to the overall ground state structure within the energy difference of 0.05 eV. Addition of a Ni or an oxygen atom on the surface of Ni6 pure cluster gives optimized slightly distorted Ni7 and Ni6O structures respectively. Optimized Ni7 cluster is a nonet and Ni6O is in septet spin state. Different orbital (HOMO and LUMO) and spin distributions of Nin and NinO ground state structures are shown in Fig. 1b.
It is clear from the above observation that for the clusters from n = 2 to 6, except for n = 4, the spin state is geometry dependent in a particular size as shown in Table 3. It is commonly observed that in these geometries of different sizes, the structures where O atom is bonded with three-nickel atoms are the most stable. In these structures oxygen atom prefers to sit as surface capped atom on the Nin clusters.
The relative stabilities, electronic and magnetic properties
In order to get the idea about the relative stabilities, electronic and magnetic properties of NinO (n = 1-6) clusters, different parameters like, binding energy (BE), HOMO-LUMO gap, embedding energy (EE), fragmentation energy (FE or Δ(n,n-1)), stability or second order change in energy (Δ2(n)), ionization potential (IP), etc. are calculated. Variation of these parameters with the size of NinO and Nin are then plotted and compared. On the basis of these calculated parameters and their variations with the size, stabilities and other properties of the clusters are discussed.
In the present work the following relation defines binding energy per atom of a cluster:
for NinO and Nin respectively. Here, E(NinO) and E(Nin) is the energy of the NinO and Nin clusters respectively, E(O) is the energy of the isolated oxygen atom; E(Ni) is the energy of a nickel atom. Variation of binding energy curve is shown in Fig. 2 for pure and oxidized nickel clusters. It is clear from Fig. 2 that with the increase of n during the growth process of the cluster, the binding energy increases initially and shows a relative maximum at n = 4 in NinO cluster. Beyond this point there is hardly any increase of BE per atom. However, the total optimized energy of the NinO cluster can continue to gain energy during the growth processes. On the other hand, the binding energy in pure clusters tend to increase even if it is a cluster with n = 7, which is basically indicating that clusters are gaining binding energy as well as the total optimized energy during their growth process. Comparing the binding energy curves it is clear that binding energy of the oxidized cluster is much higher than the nickel clusters of same size (n) within the range of the present calculation. This is an indication of improved stability due to the addition of oxygen in pure nickel structures.

Variation of binding energy per atom of Nin and NinO (n = 1–6) clusters with n
The other parameters, which can explain the thermodynamic stability of the clusters, are embedding energy (EE), fragmentation energy (FE) and the second order difference in energy (Δ2(n)). The last parameter Δ2(n) is also known as stability parameter. In the section calculated EE, FE and Δ2(n) of NinO and Nin clusters will be discussed. Variation of the embedding energy is shown in Fig. 4. Embedding energy is defined as,
It is actually the gain in energy to incorporate a foreign atom in a cluster during its growth process with an aim to produce a particular targeted product. In the present work since the growth process of NinO studied by adding oxygen with Nin cluster, therefore, here EE is the gain in energy by the product cluster due to addition of oxygen with Nin. Variation of embedding energy shown in Fig. 3 clearly shows that the cluster with n = 4 in quintet state has an affinity to absorb oxygen atom to increase its stability compared to its surrounding clusters of same or different sizes. Or in other words, Ni4 in quintet spin state is the most favorable size that can easily incorporate an oxygen atom in the cluster. A related but significantly related question is which is the most stable cluster as successive Ni atoms are added to increase the cluster size of NinO starting from NiO. This is given by Δ2(n) parameter or stability parameter of the clusters which is defined as,
With this definition, large positive values of Δ2(n) are indicative of enhanced stability as they correspond to a gain in energy during formation from the preceding size and lower gain in energy to the next size. In Fig. 4 calculated Δ2(n) as a function of cluster size n is plotted. It is to be noted that Δ2(n) exhibits a peak at n = 4 which is the stability parameter of Ni4O cluster in quintet state. Furthermore, the relative stability of Ni4O (Fig. 4) cluster in terms of the calculated second order change in energy is the biggest among all the NinO clusters in the present range of calculation showing that the cluster has stronger relative stability. Therefore, the structure with n = 4 is the most stable geometry among all the members of NinO (n = 1-6) clusters.

Variation of embedding energy of NinO (n = 1–6) clusters with n

Variation of stability of NinO (n = 1–6) clusters with n
Calculated fragmentation energies (Δ(n,n-1) or FE) of NinO clusters can give the information of relative stabilities of the clusters. FE of the cluster is defined as,
Variation of FE with the size of the cluster (Fig. 5) shows that there is a sharp increase of FE from n = 3 to n = 4 and then a drop from n = 4 to 5 indicate that during the growth process of NinO by adding Ni atom one by one the cluster Ni4O is found most stable with quintet spin state.

Variation of fragmentation energy of NinO (n = 1–6) clusters with n
While the above parameters indicate thermodynamic stability of a cluster, kinematic stability of the clusters in the chemical reaction is indicated by HOMO-LUMO gaps, ionization potential (IP), electron affinity (EA), chemical potential and hardness. Usually the larger the HOMO-LUMO gap, the less reactive the cluster is. HOMO-LUMO gaps of Nin and NinO are plotted in Fig. 6. Overall, there is a decrease of gap with the increase of the cluster size in both Nin and NinO clusters. However, there are some local oscillations over and above the decreasing trend. Although, there is no sharp global peak in HOMO-LUMO gap variation of NinO cluster as in other quantities, but there is a visible local peak at n = 4 in NinO series. This again points out the enhanced stability at n = 4 in NinO series as like other quantities discussed before. This is precisely seen in the variation of ionization potential and electron affinity with the cluster size shown in Figs. 7 and 8 respectively. Variation of the IP of NinO first decreases from n = 1 and the show a local maxima at n = 4. Therefore, the cluster at n = 4 has relatively higher chemical stability compare to the surrounding sizes. The sharp dip of electron affinity (EA) in Fig. 8 indicates the reaction by capturing electron from surrounding environment is relatively less in Ni4O cluster compared to the other oxidized and pure nickel clusters. This also supports the enhanced stability of Ni4O cluster. The common feature in all of these graphs at n = 4, is indicating that with the size-evolution of NinO clusters, the cluster Ni4O in quintet spin state has an enhanced local stability.

Variation of HOMO-LUMO gap of Nin and NinO (n = 1–6) clusters with n

Variation of ionization potential of NinO (n = 1–6) clusters with n

Variation of electron affinity of NinO (n = 1–6) clusters with n
Further, to verify the chemical stability of the clusters, chemical potential (μ) and chemical hardness (η) of the ground state isomers are calculated. In practice chemical potential and chemical hardness can be expressed in terms of electron affinity and ionization potential. In terms of total energy consideration, if E(N) is the energy of the N electron system, then energy of the system containing N+ΔN electrons where ΔN < <N can be expressed as:
Then, μ and η can be defined as:
Since,
By setting ∆N = 1, μ and η are related to IP and EA via the following relations:
Consider two systems with μ i and η i (i = 1,2) contracting each other, where some amount of electronic charge (ΔM) transfer from one to other. The quantity ΔM and the resultant energy change (ΔE) due to the charge transfer can be determined in the following way:
If E (N+ΔM) is the energy of the system after charge transfer then it can be expressed for the two different systems 1 and 2 in the following way:
and
Corresponding chemical potential becomes,
to first order in ΔM after the charge transfer. In chemical equilibrium, \( {\mu \prime_1} = {\mu \prime_2} \)which gives the following expressions:
In the expression, ΔE is the energy gain by the total system (1 and 2) due solely to the alignment of chemical potential of the two systems at the same value. It is evident from the above expressions of ΔM and ΔE that a guiding principle to predict the occurrence of an easier charge transfer is a large difference in μ together with low η1 and η2.
Keeping these aspects in mind, chemical potential (μ) and chemical hardness (η) of the clusters are calculated. The size variations of chemical potential and chemical hardness are shown in Figs. 9 and 10 respectively. Variations of these parameters also support the higher stability of Ni4O cluster in neutral state. Variation of chemical potential shows a relative minimum at n = 4. It means that the reaction affinity of this cluster is relatively low compare to its surrounding sizes. Again the variation of chemical hardness, shows a stable minimum nature of all the sizes for n > 2, which is another indication of non-reactive nature of the cluster. Therefore the nature of chemical potential and chemical hardness graphs support the highest stability of the neutral Ni4O cluster.

Variation of chemical potential of NinO (n = 1–6) clusters with n

Variation of chemical hardness of NinO (n = 1–6) clusters with n
To get an idea about the effect of the addition of oxygen in the pure nickel clusters HOMO-LUMO of the ground state structures at different sizes and geometries are shown by using contour mapping in Fig. 1b in addition to the resultant distribution of the total spin density. Comparing the maps of pure and doped clusters, it can be seen that due to addition of oxygen there is a drift of charge and spin densities toward the nickel atoms. Therefore, in the contour maps the wavefunction representing the HOMO orbital or in other words, electron density in the neighboring space of oxygen atoms are comparatively less. The change of electron density from HOMO-1 to HOMO and LUMO to LUMO+ 1 are also studied (not shown in figure) in ground state stable structures. It shows that the oxygen atom is able to attract only a weak orbital charge density in its surrounding space. Specially, in the most stable ground state structure, Ni4O, the electron clouds mainly localizes around Ni atoms while the distribution of electron density around O atom is relatively low. This is indicating that the orbital hybridization between Ni and O atoms is relatively less than orbital hybridization between the Ni and Ni atoms in the cluster. Therefore, the shapes of the HOMO and bonding properties between bare Nin clusters and NinO clusters are obviously different. Furthermore, the Ni-Ni bonding in the bare Nin clusters is the localized σ -type bonds or partly delocalized σ-type bonds while the bonding among Ni-Ni atoms in the NinO clusters is completely delocalized σ-type bonds. Consequently, the hybridization between Ni and O atoms are responsible for the change of electronic properties of Nin clusters after adsorbing O atom and it helps in improving the stability of NinO clusters because of high ionization potential, relatively high HOMO-LUMO gap, less electron affinity and chemical potential.
Variations of calculated magnetic moments of pure and doped clusters of the most stable structures are shown in Fig. 11. It is important to mention here that the average magnetic moment of the clusters changes discontinuously with the size of clusters. This behavior of the magnetic moments of the low energy small sized NinO clusters is clearly indicating that the magnetic moments for the NinO clusters are not stable in a particular size; it is very much geometry dependent and this finding is analogous to those of the reported Zrn clusters [55] as well as in transition metal doped Na clusters [56]. Furthermore, the magnetic moment is stable or in other words it is independent of geometry of the stable isomers in Ni4O. Therefore, the incoming oxygen does not have much effect on the magnetic moment of the ground state of Ni4O clusters. But the question is how the ten (eight from four nickel and two from one oxygen atoms) electrons are accommodating in the Ni4O ground state cluster so that the magnetic moment of Ni4 cluster is not affected by addition of oxygen and at the same time increased the chemical stability? The answer can be given by Mulliken population charge and spin density analysis. Mulliken charge analysis clearly shows that out of ten electrons, six electrons take part in the bonding orbital’s px, py and pz. Out of four remaining electrons, two electrons go into the p *y orbital. One electron goes into the majority spin level p *z orbital, whereas the down spin level orbital p *z is empty. Since the nickel is strongly ferromagnetic element, so the remaining one electron goes into the lowest unoccupied spin minority level. Again from the spin density analysis it is found that the oxygen atom has positive magnetic moment in the Ni4O cluster in quintet state. This is because of the splitting of sub-bands originating from oxygen 2py and 2pz levels due to ferromagnetism of nickel clusters.

Variation of magnetic moment of NinO (n = 1–6) clusters with n
Conclusions
In the present study a systematic investigation of the Nin and NinO (n = 1-6) clusters in various spin states are performed at the unrestricted B3PW91/LanL2DZ level. Total optimized energies, equilibrium geometries, and stabilities of NinO (n = 1-6) clusters along with the binding energy, embedding energy, fragmentation energies, ionization potential, electron affinity etc. are presented and discussed. Conclusions drawn from the calculated results can be summarized as follows:
-
(1)
The ground state geometry in NinO (n = 1-6) clusters show that the O atom prefers the surface capped Nin pattern. The oxygen atom interacts with the three Ni atoms in the most stable Ni4O cluster in the NinO series in such a way that the stability of the cluster increases.
-
(2)
The relative stabilities of NinO (n = 1-6) clusters in terms of different calculated parameters show that Ni4O cluster is the most stable structure among all small size (n < 7) nickel and nickel oxide clusters.
-
(3)
Calculated magnetic moments of small-sized NinO clusters show that the magnetic moment is not stable and it depends upon the geometrical structure of the cluster in a particular size. Whereas, in the most stable cluster Ni4O in Cs symmetry structure has the magnetic moment of 5.0 μB, which is the same as the most stable pure Ni4 cluster. At this size the magnetic moment is independent of the geometry of the cluster and also negligible effect of addition of oxygen on it.
-
(4)
Calculated HOMO-LUMO gaps indicate that the most stable Ni4O cluster in quintet spin state has stronger chemical stability as compared to the neighboring sizes. The distribution of electron density of the HOMO states for the most stable Ni4O cluster mainly localizes around Ni atoms while the distribution around O atom is relatively low. The shapes of the HOMO and bonding properties between bare Nin clusters and NinO clusters are obviously different. Our calculated results on electron affinity, chemical potential etc. indicate that the incoming oxygen does not have much effect on the magnetic property of Nin clusters, but at the same time it helps in improving the stability of the cluster after forming NinO.
References
Veliah S, Xiang KH, Pandey R, Recio JM, Newsam JM (1998) J Phys Chem B 102:1126–1135
Cui XY, Morrison I, Han G (2002) J Chem Phys 117:1077–1084
Han JG, Ren ZY, Lu BZ (2004) J Phys Chem A 108:5100–5110
Molek KS, Jaeger TD, Duncan MA (2005) J Chem Phys 123:144313–144322
Andrews L, Chertihin GV, Ricca A, Bauschlicher CW Jr (1996) J Am Chem Soc 118:467–470
Wang LS, Wu H, Desai SR (1996) Phys Rev B 53:8028–8031
Tono K, Terasaki A, Ohta T (2006) J Chem Phys 124:184311–184316
Asmis KR, Sauer J (2007) Mass Spectrom Rev 26:542–562
Feyel S, Scharfenberg L, Daniel C, Hartl H, Schröder D, Schwarz H (2007) J Phys Chem A 111:3278–3286
Tsipis AC, Tsipis CA (2000) J Phys Chem A 104:859–865
Molek KS, Reed ZD, Ricks AM, Duncan MA (2007) J Phys Chem A 111:8080–8089
Li S, Dixon DA (2007) J Phys Chem A 111:11908–11921
Herman J, Foutch JD, Davico GE (2007) J Phys Chem A 111:2461–2468
Xu Y, Shelton WA, Schneider WF (2006) J Phys Chem A 110:5839–5846
Zhai HJ, Huang X, Waters T, Wang XB, O’Hair RAJ, Wedd AG, Wang JS (2005) J Phys Chem A 109(46):10512–10520
Liu L, Zhao RN, Han JG, Liu FY, Pan GQ, Sheng LS (2009) J Phys Chem A 113:360–366
Li S, Hennigan JM, Dixon DA, Peterson KA (2009) J Phys Chem A 113:7861–7877
Li S, Zhai HJ, Wang LS, Dixon DA (2009) J Phys Chem A 113:11273–11288
Rodríguez-Lopez JL, Aguilera-Granja F, Vega A, Alonso JA (1999) Eur Phys J D 6:235–241
Yao YH, Gu X, Ji M, Gong XG, Wang DS (2007) Phys Lett A 360:629–631
Grigoryan VG, Springborg M (2001) Phys Chem Chem Phys 3:5135–5139
Lathiotakis NN, Andriotis AN, Menon M, Connolly J (1996) J Chem Phys 104:992–1003
Reddy BV, Nayak SK, Khanna SN, Rao BK, Jena P (1998) J Phys Chem A 102:1748–1759
Czekaj I, Wambach J, Kröcher O (2009) Int J Mol Sci 10:4310–4329
Bahl CRH, Hansen MF, Pederse T, Saadi S, Nielsen KH, Lebech B, Mørup S (2006) J Phys Condens Matter 18:4161–4175
Khadar MA, Biju V, Inoue A (2003) Mater Res Bull 38:1341–1349
Ahmad T, Ramanujachary KV, Lofland SE, Ganguli AK (2006) Solid State Sci 8:425–430
Biesinger MC, Payne BP, Lau LWM, Gerson A, Smart RSC (2009) Surf Interface Anal 41:324–332
Ghosh M, Biswas K, Sundaresana A, Rao CNR (2006) J Mater Chem 16:106–111
Vann WD, Castleman AW Jr (1999) J Phys Chem A 103:847–857
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1992) Phys Rev B 46:6671–6678
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1993) Phys Rev B 48:4978
Perdew JP, Burke K, Wang Y (1996) Phys Rev B 54:16533–16539
Burke K, Perdew JP, Wang Y (1998) In: Dobson JF, Vignale G, Das MP (eds) Electronic density functional theory: recent progress and new directions. Plenum
Kohn W, Sham LJ (1965) Phys Rev 140A1:135–138
Langreth DC, Perdew JP (1980) Phys Rev B 21:5469–5493
Langreth DC, Mehl MJ (1983) Phys Rev B 28:1809–1834
Perdew JP, Wang Y (1986) Phys Rev B 33:8800–8802
Perdew JP (1986) Phys Rev B 33:8824
Bandyopadhyay D (2008) J Appl Phys 104:084308–084314
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu B, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA (2004) Gaussian 03, Revision E.01. Gaussian Inc, Wallingford
Ho J, Polak ML, Ervin KM, Lineberger WC (1993) J Chem Phys 99:8542–8551
Wu H, Wang LS (1997) J Chem Phys 107:9606–9611
Sadanov VI, Harris DO (1988) J Chem Phys 89:2748–2753
Noell JO, Newton MD, Hay PJ, Martin RL, Bobrowicz FW (1980) J Chem Phys 73:2360–2371
Gutsev GL, Lutatskaya VD, Klyagina AP, Levin AA (1986) Chem Mater Sci 28:829–833
Doll K, Dolg M, Fulde P, Stoll H (1997) Phys Rev B 55:10282–10288
Bauschlicher CW, Maitre P (1995) Theor Chim Acta 90:189–203
Mitchell KAR (1985) Surf Secrets 149:93–104
Basch H, Newton MD, Moskowitz JW (1980) J Chem Phys 73:4492–4510
Nygren MA, Siegbahn PEM, Wahlgren U, Akeby H (1992) J Phys Chem 96:3633–3640
Tomonari M, Tatewaki H, Nakamura T (1986) J Chem Phys 85:2875–2884
Pastor GM, Dorantes-Davila J, Bennemann KH (1988) Chem Phys Lett 148:459–464
Blyholder G (1974) Surf Sci 42:249–260
Wang CC, Zhao RN, Han JG (2006) J Chem Phys 124:194301–194308
Pradhan K, Sen P, Reveles JU, Khanna SN (2008) Phys Rev B 77:045408–045414
Acknowledgments
Gaussian 03 calculations were performed on the cluster computing facility at HRI (Harish-Chandra Research Institute), http://cluster.hri.res.in.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bandyopadhyay, D. Chemisorptions effect of oxygen on the geometries, electronic and magnetic properties of small size Nin (n = 1-6) clusters. J Mol Model 18, 737–749 (2012). https://doi.org/10.1007/s00894-011-1090-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1090-8