
Abstract
A theoretical study of structural, electronic, topological and vibrational parameters of the ternary hydrogen-bonded complexes C2H4O···2HF, C2H5N···2HF and C2H4S···2HF is presented here. Different from binary systems with a single proton donor, the tricomplexes have the property of forming multiple hydrogen bonds, which are analyzed from a structural and vibrational point of view, but verified only by means of the quantum theory of atoms in molecules (QTAIM). As traditionally done in the hydrogen bond theory, the charge transfer between proton donors and acceptors was computed using the CHELPG calculations, which also revealed agreement with dipole moment variation and a cooperative effect on the tricomplexes. Furthermore, redshift events on proton donor bonds were satisfactorily identified, although, in this case, an absence of experimental data led to the use of a theoretical argument to interpret these spectroscopic shifts. It was therefore the use of the QTAIM parameters that enabled all intermolecular vibrational modes to be validated. The most stable tricomplex in terms of energy was identified via the strength of the hydrogen bonds, which were modeled as directional and bifurcated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Oxirane (C2H4O), aziridine (C2H5N) and thiirane (C2H4S) are some of the most important heterocyclic compounds found in nature [1, 2]. The chemistry of these compounds is immensely rich and they have been the subject of a vast quantity of scientific research [3–6], ranging from spectroscopic characterization as products derived from organic synthesis mechanisms [7] to theoretical analysis of transition states using keywords in computational methods [8], for example. It is now known that these heterocyclic compounds, and oxirane and thiirane in particular, are structures capable of forming intermolecular systems through interactions with either haloacids [9] or nucleophilic species [10, 11]. The aforementioned interaction is therefore intermolecular, or a hydrogen bond [12] of the form (Y···HX), where X is an element with a higher electronegativity than hydrogen, whereas Y = n lone pairs, π electrons [13, 14], or in some cases hydrides derived from earth alkaline elements, which occasionally generate dihydrogen bonds [15]. On the basis of this insight, and work done by Kuczkowski et al. [16], Legon et al. [17], Alonso et al. [18], Goswami and Arunan [19] and many other important researchers [20, 21], dimers or bimolecular systems have been studied carefully under experimental conditions and from a theoretical perspective through the application of Fourier transform microwave spectroscopy (FTMS) [22–24], which allows rotational parameters to be measured [25], and through the computation and subsequent interpretation of electronic properties [26]. Experimental procedures and theoretical computations have thus been successfully applied to the study of bimolecular structures [27], although some research highlights the importance of trimolecular or ternary systems as foci of investigation [28].
Nevertheless, the kinetic treatment that led to the theory of trimolecular reactions formulated by Gershinowitz and Eyring [29] is a crucial factor in determining that competition between bimolecular and trimolecular systems must, under abnormal conditions, govern the chemical process. However, over the years, the question of which is the correct mechanism—bimolecular or trimolecula—has remained unanswered [30]. Some time ago, however, Jursic showed that trimolecular reactions are more favorable and energetically stable [31]. This result suggests that trimolecular hydrogen complexes are reliable structures. Taking this reasoning into account, we have developed a theoretical study of bimolecular and trimolecular heterocyclic hydrogen-bonded complexes [32], the main conclusions of which are that (i) there are nonlinear deviations in the hydrogen bonds of bimolecular complexes [33, 34], and (ii) multiple hydrogen bonds define the preferential configuration of trimolecular complexes [35, 36]. Although the bimolecular hydrogen complex C2H4O···HF is the only structure that has been evaluated using FTMS [37], the theoretical results show good correlation with the available experimental data [38]. On the other hand, the trimolecular complex C2H4O···2HF has only been studied theoretically [32], using density functional theory (DFT) [39] and topological parameters computed in light of the quantum theory of atoms in molecules (QTAIM) [40], which was likewise applied to the bimolecular complexes C2H5N···HF and C2H4S···HF [9, 13]. In a direct comparison, the actions of two haloacids yield more stable structures, but one additional and important aspect must be mentioned: ternary complexes have shorter and stronger hydrogen bonds [41]. In terms of hydrogen bond strength, it is worth assuming that more stable complexes are the consequence of stronger hydrogen bonds, or of multiple hydrogen bonds being formed, such as intramolecular hydrogen bonds besides the intermolecular ones. So far as computational approaches are concerned, it is extremely important to choose theoretical methods that can describe the aforementioned conditions, i.e., describe the hydrogen bond strength or multiple hydrogen bonds.
Very recently, Xu et al. [42] performed a large computational study in order to evaluate the efficiencies of modern density functionals. They concluded that Beck, Lee, Yang and Parr’s B3LYP hybrid [43, 44] is the most appropriate for measuring hydrogen bond strength. Furthermore, our main goal was to search in the literature for stable structures in order to characterize the formation of hydrogen bonds [45, 46], for which the B3LYP approach always yields excellent results. From a theoretical point of view, it is possible to apply a methodology that is capable of validating the formation of nonbonded interactions (such as hydrogen bonds) or bonded interactions (i.e., covalent bonds). It is through the identification and quantization of the charge density on the molecular surface within the framework of Bader’s QTAIM calculations [47] that the concentration and depletion of charge density are computed, thus indicating the existence of hydrogen bonds and covalent bonds, respectively [48]. This was our immediate objective, since, if hydrogen bonds can be identified [49–51], they subsequently constitute an important criterion for identifying the preferential structures of the trimolecular hydrogen complexes C2H4O···2HF, C2H5N···2HF and C2H4S···2HF, as has already been reported for other bimolecular [52, 53] and trimolecular [54, 55] complexes. The historical background of hydrogen bond theory [56] has highlighted the importance of electrostatic potential and charge transfer, in addition to polarizability, dispersion forces, and exchange repulsion. In the case of hydrogen bonds, electrostatic potential clearly has a significant influence on molecular stability. However, it is worth noting the importance of charge transfer, which makes it possible to confirm the existence of intermolecular contacts between the HOMO and LUMO frontier orbitals [57]. In fact, the electronic distribution profile and the vibrational stretch frequencies are quite clearly explained by charge transfer analysis [58]. For this reason and many others, charge transfer should be included in investigations of hydrogen bonds. However, owing to the existence of many atomic charge methods, which are nevertheless not observable from the viewpoint of quantum mechanics [59], atomic charge calculations have been successfully applied to studies of hydrogen complexes. Therefore, although Mulliken's population analysis [60] and generalized atomic polar tensors (GAPT) [61] are very popular methods, the present study adopts the widely used charges from electrostatic potential using a grid-based (CHELPG) method to examine hydrogen bonds [62], owing to its low computational cost and the excellent interpretation it provides of intermolecular charge transfer and vibrational events, such as red- and blueshift effects on proton donor bonds [63].
Geometric criteria
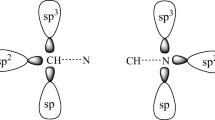
Taking the molecular structures of the C2H4O (a), C2H5N (b) and C2H4S (c) monomers as starting points, it is essential to know the stereochemistry of these heterocyclics, in particular the orientations of their n lone electron pairs. Figure 1 shows how different the features of the n lone electron pairs are in a, b and c [64]. In terms of trimolecular hydrogen complexes, it can be clearly seen that c has only one n-electron pair, whereas a and b each have two n-electron pairs. Henceforth, in contrast to f, the formation of trimolecular hydrogen complexes of d and e will be aided by double electrophilic attack from the hydrogen fluoride on the n-electron pairs of oxygen and sulfur, as can also be seen in Fig. 1. However, it is hoped that this “limitation” of c will not compromise the characterization of the most stable trimolecular hydrogen complex, since trimolecular structures can be obtained from f if the interaction of the HF···HF dimer is taken to be the proton donor [65].

Diagram of the lone pairs on the heterorings oxirane, thiirane and aziridine, as well as the preferential structures for forming trimolecular complexes with hydrogen fluoride
Computational scheme
The full optimized geometries of the C2H4O···2HF, C2H4S···2HF and C2H5N···2HF trimolecular hydrogen-bonded complexes in their possible configurations were obtained at the B3LYP/6-311++G(d,p) level of theory (no imaginary frequencies were obtained), and all calculations were performed by the GAUSSIAN 98 W quantum chemistry software package [66]. The charge transfer computations were calculated using CHELPG in GAUSSIAN 98 W with isolated analysis of the atomic point charges, while the fluxes were determined by the simple differences between complexes and monomers. The QTAIM topography was established via topological calculations executed in GAUSSIAN 98 W [67–69] and the AIM 2000 1.0 program [70].
Results
Structural parameters
The geometries of the C2H4O···2HF (g, h and i), C2H4S···2HF (j, l and m) and C2H5N···2HF (n and o) trimolecular complexes are presented in Figs. 2, 3 and 4, respectively. Before commencing analysis, it is important to mention that, in contrast to Fig. 1, several trimolecular hydrogen-bonded complexes were obtained from the oxirane, thiirane and aziridine heterorings. It was decided that all structural possibilities should be explored in an effort to establish the most stable complex. Some time ago, Gilli et al. [71] reported a hydrogen bond structure in which very strong hydrogen bonds have distance values that are shorter than 2.50 Å. Given this, Table 1 clearly shows that very few of the hydrogen bonds were very strong; their strengths are overestimated. However, if medium-strength and weak hydrogen bonds are characterized by lengths of 2.65–2.80 Å and >2.80 Å, respectively, the values shown in Table 1 indicate medium strength if not weak bonds. More recently, Grabowski et al. [72] presented very strongly bonded hydrogen complexes with intermolecular distances of between 1.094 and 1.946 Å. Taking into account the criteria explained above, accurate predictions of the strengths of the structural hydrogen bonds of the trimolecular heterocyclic complexes under study here would seem to be possible. However, a systematic analysis can be constructed to elucidate the true consequences of the formation of the hydrogen bonds. A review of the literature [73–75] shows that it is well known that the hydrogen bond distances of the bimolecular hydrogen complexes formed by oxirane, thiirane and aziridine exhibit the following trend: C2H5N···HX > C2H4O···HX > C2H4S···HX, where HX represents a monoprotic acid such as hydrogen fluoride or hydrogen cyanide. The same behavior is found in a direct comparison of the primary hydrogen bond (Y···Hλ), where the mean values of the hydrogen bond distances R λ(Y···H ) for the trimolecular complexes of aziridine (n and o), oxirane (g, h and i) and thiirane (j, l and m) are 1.4, 2.0 and 1.6 Å, respectively. Based on relatively recent research, very much shorter hydrogen bond distances such as those reported here present some covalent character [76]. In light of this, the results in the range 1.444–1.468 Å reported here could also be considered covalent hydrogen bonds. However, it would not be fair to claim that hydrogen bonds are covalent only across shorter distances, since an examination of charge density [77] and energetic parameters [78] would provide better evidence of this.

Optimized geometries of the three structures (g, h and i) of the tricomplex C2H4O···2HF obtained through B3LYP/6-311++G(d,p) calculations

Optimized geometries of the three structures (j, l and m) of the tricomplex C2H4S···2HF obtained through B3LYP/6-311++G(d,p) calculations

Optimized geometries of the three structures (n and o) of the tricomplex C2H5N···2HF obtained through B3LYP/6-311++G(d,p) calculations
The only interaction in g and j involves the primary hydrogen bonds (Y···Hλ) = (Y···Hλ′). For the aziridinic systems n and o, in agreement with their bimolecular relatives (the 2HF dimer in particular), the (Fφ···Hλ′) values of 1.633 Å and 1.606 Å are the shortest. On the other hand, a slight difference can be observed between the (Fφ···Hλ′) results for the oxirane (h and i) and thiirane (l and m) tricomplexes, which contradicts the (Fφ···Hλ′) values computed for the aziridinic tricomplexes. One explanation for this phenomenon concerns the deviations of the hydrogen bonds from linearity (θ) [13]; the values of this parameter (listed in Table 1) indicate that the most extensive deformation of the hydrogen bonds can be found in tricomplexes j, l and m. It is for this reason that there is a similarity between the (Fφ···Hλ′) values of the oxirane and thiirane tricomplexes. It should be noted that shortening of the hydrogen bond distances R φ(F λ′···H ) occurs due to the orientation of the n lone pairs of the sulfur atom—see Fig. 1 b—in which a tertiary interaction between the fluorine (Fφ′) of the 2HF dimer and the axial hydrogen atoms (Hα and Hβ) of the thiirane is facilitated [79]—see Fig. 1 e. In other words, if we assume a value of 1.831 Å for the length of the (Fφ···Hλ′) hydrogen bond in the isolated hydrogen fluoride dimer, it can be assumed that a slight shortening of this interaction will be observed upon the formation of the j, l and m systems. However, it should be emphasized that, although slight reductions in the (Fφ···Hλ′) hydrogen bonds of the thiirane tricomplexes have been computed, these corroborate the observations that large deviations from linearity of 18, 10.1 and 9.6° for θ1 as well as 18, 3.7 and 20.0° for θ2 are a consequence of the stereochemistry of the thiirane ring, not the strength of the hydrogen bond. Were this the case, aziridinic tricomplexes would have the largest deviations from linearity, which they do not. It is through this identification of linearity that the tertiary interaction emerges as a decisive parameter in the formation of the heterocyclic complexes. In the structures i, m and o, the R(Fφ′···Hα) and R(Fφ′···Hβ) values are in concordance with the tabulated van der Waals radii for fluorine and hydrogen [80], 1.47 Å and 1.20 Å, which add up to 2.67 Å. The structure of the tertiary interactions in i and m should therefore be analyzed carefully. The values of 2.755 Å and 2.677 Å depicted in Table 1 thus suggest that only tricomplex m shows the tertiary interactions (Fφ′···Hα) and (Fφ′···Hβ), which are in essence dual and bifurcated. In exceptional cases, the tricomplex o shows distinct tertiary interactions, where the value of 2.503 Å indicates the existence of the (Fφ′···Hα) hydrogen bond, whereas the result of 3.551 Å for (Fφ′···Hβ) reveals that there is no interaction.
The most important factor to be analyzed in hydrogen bonding is the alterations that occur to the molecular structures of individual monomers when they undergo complexation [81]. Part of this study will therefore also examine hydrofluoric acid bond lengths (H–F), as well as heteroring structures (C–H and C–C), results for which are reported in Table 2. In short, (Hλ–Fφ) and (Hλ′–Fφ′) exhibit elongation, as expected from the proton donors in intermolecular systems [82–85]. In the remaining (C–C) bonds, very slight but systematic variations were calculated, which also fully concords with the historical background of small heterocyclic compounds [86]. Although the (C–Hα), (C–Hβ), (C–Hα′) and (C–Hβ′) bonds behave in their own specific ways, showing slight reductions, no changes and or lengthenings, various kinds of reductions were also found in the (C–Hα) and (C–Hβ) bond lengths of the tricomplex o. It is likely that these different variations are caused by the tertiary interaction (Fφ′···Hα). Although the structure of this is well known [29], it has been reconfirmed by this study.
Charge transfer distribution and dipole enhancement
The charge transfer mechanism is undoubtedly one of the most important criteria for describing the formation of hydrogen bonds. Table 3 groups together all charge transfers for hydrogen (Hλ and Hλ′), fluorine (Fφ and Fφ′), and oxygen, sulfur and nitrogen atoms. It is important to note that these elements were chosen by considering the charge transfer phenomenon in hydrogen complexes, where the electronic flux flows from the HOMO to the LUMO frontier orbitals of the proton acceptor and donor, respectively. Therefore, as complexes g and j possess a peculiar structure compared to the other systems, this analysis can be better explained in two steps. First, the g and j complexes exhibit higher Δq(Y) values, since oxygen and sulfur are simultaneously donating charge. This means that, under these conditions, the two haloacids (Hλ–Fφ and Hλ–Fφ′) become heavily loaded when complexes g and j are formed. The charge transfer Δq (Y) is smaller for trimolecular complexes formed via the hydrogen fluoride dimer. Figure 5 explains the charge transfer mechanism by illustrating the charge distribution profiles of the tricomplexes g, h, i, j, l, m, n and o. In concordance with the structural analysis regarding the hydrogen bond distance R λ(Y···H ), here expressed in terms of electronic parameters, this also proves that the aziridine complexes n and o are more strongly bonded, with their charge transfers increasing the electropositive character of nitrogen by +0.375 e.u. and +0.335 e.u., respectively. By contrast, the charge transfer calculated for complexes g and j indicates an increase in charge density concentration in both of the acids Hλ–Fφ and Hλ–Fφ′, as discussed above, but this is also found in tricomplexes n and o. However, moderate charge transfer was obtained on the hydrofluoric dimers in h, i, l, and m, which is a natural consequence of the nonadditive electron distribution [87]. Both the atomic charge transfers and the dipole moment supply satisfactory evidence that the nonadditive phenomenon can be debated and explained. A special note should be made of the electronic consequences represented by the high and medium-sized dipole enhancements in complexes h, l and n).

Charge transfer profiles for the proton donors and acceptors of the tricomplexes
The definition of the nonadditive or cooperative effect is related to the equivalent charge distribution along the electronic structure formed by three or four bodies [88–90]. This distribution is uniform and no significant variations are detected, especially on the central components of an oligomer [91] for instance. The systems studied here exhibited nonuniform charge distributions on complexes h, l and n. Table 3 shows that the Δq(Hλ) charge transfer values of −0.061, −0.095 and −0.210 e.u., as well as the Δq(Hλ′) charge transfer values of −0.034, −0.014 and −0.043 e.u., are indicative of additive electron distribution. It should be noted that these are the strongest charge transfers within the hydrogen fluoride dimer upon the formation of the tricomplexes h, i, l, m, n and o. Investigation focused on charge fluxes in proton donors, because these form the central points in hydrogen bonding [92]. Moreover, as the relationship between charge transfer and dipole variations is well known [93, 94], it is possible that the increase in the dipole moment is a consequence of the Δq(Hλ′) and Δq(Hλ) charge transfers. This is clearly not a general conclusion, but it can be assumed that larger dipole variations are closely related to enhanced charge transfer mechanisms [95]. Finally, it should also be noted that the slight variations in atomic charges on axial hydrogen atoms (Hα, Hβ, Hα′ and Hβ′) indicate the absence of charge transfer in tertiary interactions. From an electronic viewpoint, this information at the very least suggests that no hydrogen bond is formed or that the interaction will be only very weak, although this requires more careful investigation.
Vibrational spectrum analysis
Analysis of the vibrational infrared spectrum is crucial in studies of molecular systems, especially hydrogen complexes [84, 85]. It is highly doubtful that a hydrogen complex would form at intermolecular sites. In other words, can we observe the harmonic oscillator activities of the stretch frequencies and absorption intensities for weak interactions? This study provides an affirmative answer to this question. In this context, the values of the stretch frequencies and absorption intensities for all the hydrogen bonds in the tricomplexes are given in Table 4. By analogy with the hydrogen bond distances, the new vibrational modes \( \upsilon_{{\left( {{\rm{Y}} \cdot \cdot \cdot {{\rm{H}}^{\lambda }}} \right)}}^{{Str}} \) and \( \upsilon_{{\left( {{\rm{Y}} \cdot \cdot \cdot {{\rm{H}}^{{\lambda \prime }}}} \right)}}^{{Str}} \)) of the aziridine complexes are generally stronger than those calculated for oxirane and thiirane. Nevertheless, the values of the intermediate stretch frequencies are not similar to those known for the 2HF dimer. In fact, these values increase hugely following complexation of ternary systems, a phenomenon that has also been observed for the hydrogen bond distances. In short, in total agreement with the theory of hydrogen bonding, all of the intermolecular stretch frequency intensities are very weak and accompanied by small absorption intensities [96]. Furthermore, the change in the vibrational infrared spectrum is the most important criterion for characterizing the formation of hydrogen complexes. It is through the shift in stretch frequencies of the proton donors (either downward [97, 98] or upward [99, 100]) and the increase in absorption intensity that the intermolecular complexes are identified, especially those formed by hydrogen bonds, such as the heterorings examined here [46, 74].
Table 4, however, illustrates a fundamental feature. Among all of the heteroring tricomplexes, the strongest redshift effects and most intense absorption intensity ratios were seen for the first hydrogen fluoride Hλ–Fφ, mainly in n and o. The respective \( \Delta \upsilon_{{({H^{\lambda }} - {F^{\varphi }})}}^{{Str}} \) values for these systems are −1713.0 cm−1 and −1893.7 cm−1, and the \( \frac{{I_{{({H^{\lambda }} - {F^{\varphi }}) \cdot C}}^{{Str}}}}{{I_{{({H^{\lambda }} - {F^{\varphi }}) \cdot m}}^{{Str}}}} \) values are 23.7 and 20.3. These spectroscopic results are in excellent agreement with the structural results reported here, which suggests that the clearest evidence of decreased length is found for the Hλ–Fφ bonds. A higher level of charge transfer was computed for Hλ, which indicates that the charge distribution is locally concentrated on Hλ–Fφ, so and their redshift effects are more evident, as is the shortening of the bond lengths, as previously explained. If we consider the new vibrational modes \( \upsilon_{{\left( {Y \cdot \cdot \cdot {H^{\alpha }}} \right)}}^{{Str}} \) and \( \upsilon_{{\left( {F \cdot \cdot \cdot {H^{{\lambda \prime}}}} \right)}}^{{Str}}, \) an increase was found in the latter, due to the high concentration of charge density at the middle of the 2HF dimer. However, we cannot ignore one essential factor: the absence of experimental data relating to the heteroring complexes [101–104]. The literature shows that scaling factors are currently used to correct the vibrational stretch frequencies [105]. However, this is a parameterization procedure, which was not adopted here. Even in this situation, the values of the intermolecular stretch frequencies must be checked theoretically. We believe that these vibrational modes can only be carefully authenticated by measuring the charge concentration.
Electronic density topography: the more strongly bonded complex
Over the last five decades, the desire for an ideal model of atomic behavior has constantly featured in theoretical and experimental debates [106]. In this regard, Bader proposed a way of partitioning the molecular system into atomic fragments on the basis of the electronic density computed at the zero-flux surface. This procedure is supported by quantum mechanical theories and mathematical formulations, and as such, the QTAIM content is formulated by topological properties that originate in the locations of bond critical points (BCP) with coordinates (3, −1). In practice, QTAIM-based analysis of the charge density is considered a routine procedure for studies of intermolecular systems in view of the efficiency of this method for mapping bond paths [107], which can determine whether atoms are bonded or whether they are simply interacting with each other [108]. It is through the computation of the electronic density (ρ) and its Laplacian (\( \nabla_{\rho }^2 \)) that covalent and π bonds or intermolecular contacts such as hydrogen bonds and other types can be identified and classified as shared or closed-shell interactions [109]. Table 5 thus shows all ρ and \( \nabla_{\rho }^2 \) values for the C2H4O···2HF, C2H4S···2HF and C2H5N···2HF tricomplexes. These values form the basis for characterizing the hydrogen bonds, since the Laplacian results are positive but the electron densities are also very small. According to the virial theorem for the charge density [110], a positive Laplacian is a classical QTAIM criterion for characterizing the formation of intermolecular interactions, since kinetic energy is the dominant operator. Charge densities of approximately 0.001 \( e\,/\,\alpha_{\rho }^3 \) suggest that the hydrogen bonds of the tricomplexes are typical closed-shell interactions. Therefore, by establishing the expected bond paths in Fig. 6 (I) and 7 (I), a dual hydrogen bond with the same topological features was observed in g and j, for which the values of ρ and \( \nabla_{\rho }^2 \) are 0.141 \( e\,/\,\alpha_{\rho }^3 \) and 0.027 \( e\,/\,\alpha_{\rho }^3 ,\) as well as 0.147 \( e\,/\,\alpha_{\rho }^5 \) and 0.057 \( e\,/\,\alpha_{\rho }^5 , \) respectively. Comparison with the remaining tricomplexes reveals that aziridine generates the strongest bonds, followed by oxirane and thiirane. This observation has already been reported from a structural viewpoint using hydrogen bond distances, as well as in electronic terms by computing the charge transfer. Furthermore, the topology results for the hydrogen fluoride dimer are also consistent with those obtained from structural and electronic analyses. It should be noted that the electronic densities of the hydrogen bonds \( \left( {{F^{\varphi }}\, \cdot \, \cdot \, \cdot \,{H^{{\lambda \prime}}}} \right) \) are higher than the value of 0.024 \( e\,/\,\alpha_{\rho }^3 \) for the 2HF dimer. Also, once again, the aziridinic tricomplexes are the most strongly bonded, as they are clearly seen to have ρ values of 0.042 \( e\,/\,\alpha_{\rho }^3 \) and 0.046 \( e\,/\,\alpha_{\rho }^3 \) for the hydrogen bond \( \left( {{F^{\varphi }}\, \cdot \, \cdot \, \cdot \,{H^{{\lambda \prime}}}} \right). \) Additionally, all other hydrogen bonds of the aziridinic tricomplexes show higher concentrations of electronic density than those for oxirane and thiirane, for instance.

Bond paths of the three structures of the tricomplex C2H4O···2HF obtained through B3LYP/6-311++G(d,p) calculations

Bond paths of the three structures of the tricomplex C2H4S···2HF obtained through B3LYP/6-311++G(d,p) calculations
Complementary analysis of intermolecular electronic density detected the existence of a bifurcated hydrogen bond in the tricomplexes i and m, which is modeled as \( \left( {{F^{{\varphi \prime}}} \cdot \cdot \cdot {H^{\alpha }}} \right) \) and \( \left( {{F^{{\varphi \prime}}} \cdot \cdot \cdot {H^{\beta }}} \right) \) and depicted by the bond paths illustrated in Figs. 6 (III) and 7 (III). The QTAIM calculations, in fact, determine the electronic densities of these interactions; these values are 0.005 \( e\,/\,\alpha_{\rho }^3 \) and 0.006 \( e\,/\,\alpha_{\rho }^3 \) for i and m, respectively. Throughout this study, many points have been made which demonstrate that i and m are the most stable configurations of the C2H4O···2HF and C2H4S···2HF complexes, respectively. The positive Laplacian values of 0.018 \( e\, / \,\alpha_{\rho }^5 \) and 0.021 \( e\, / \,\alpha_{\rho }^5 \) show quantum-mechanical support for the QTAIM framework which indicates that i and m are cyclic structures that are based on four hydrogen bonds: \( \left( {Y\, \cdot \, \cdot \, \cdot \,{H^{\lambda }}} \right) \), \( \left( {{F^{\varphi }}\, \cdot \, \cdot \, \cdot \,{H^{{\lambda \prime}}}} \right) \), \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\alpha }}} \right) \) and \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\beta }}} \right) \). In view of recent studies in which QTAIM topology revealed the preferential structures of several chemical systems [111], it is prudent to assume that it is essential to identify these hydrogen bonds in order to ensure that, among g, h, i, j, l and m, complexes i and m are in fact the most strongly bonded structures, which corroborates the structural results previously documented here. However, the aziridinic complexes present a novel intermolecular feature. In contrast to the bifurcated hydrogen bonds \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\alpha }}} \right) \) and \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\beta }}} \right) \) in the o complex, only the first hydrogen bond was located using QTAIM calculations, as demonstrated by Fig. 8 (III). Although only the hydrogen bond \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\alpha }}} \right) \) was identified, it can be seen that the electron density of 0.007 \( e\,/\,\alpha_{\rho }^3 \) for o is higher than the values of 0.005 \( e\,/\,\alpha_{\rho }^3 \) for i and 0.006 \( e\,/\,\alpha_{\rho }^3 \) for m, which leads to the conclusion that aziridine exhibits a peculiar feature: an open reaction mechanism [112]. The ring-opening reaction mechanism for aziridine has been extensively explored [113–116]. The results of this study provide some information on the open reaction of aziridine by way of the attack/interaction of two hydrogen fluoride acids. Figure 9 shows that the o-I, o-II and o-III structures constitute the final three steps in the ring-opening reaction. The main difference between o-I and o-II is the lengths of the H–F bonds by which the “novel” acid, Hλ′–Fφ, is finally obtained. Naturally, as the literature reports [1], an excess of acid provokes catalysis of the process, leading to the regeneration of hydrogen fluoride.

Bond paths of the three structures of the tricomplex C2H5N···2HF obtained through B3LYP/6-311++G(d,p) calculations

Illustration of the ring-opening reaction mechanism for aziridine and the insertion of the cyclic tricomplex o-I and the hypothetical intermediates o-II and o-III
However, the most important information contained in the QTAIM results presented in Table 6 is the reduction in the electron density of the H–F bonds of the 2HF dimer upon the formation of tricomplexes n and o, as well as the others. When the strength of the chemical bond and its reduced electronic density is considered, these results indicate a tendency for the hypothetical intermediary o-II to be formed. Thus, on the basis (as always) of intermolecular electronic density, it is clear that the protonation of the nitrogen seems to be preferred, in view of the wider \( \Delta {\rho_{{\left( {{H^{\lambda }} - {F^{\varphi }}} \right)}}} \) variation of −0.106 \( e\,/\,\alpha_{\rho }^3 \). Furthermore, the increase in the Laplacian is also an indication of a tendency for all H–F bonds to form closed-shell interactions, although these are not shared, as is widely believed. In other words, it is suggested here that the first Hλ–Fφ is easily disfigured [117], but this reasoning also applies to the second Hλ′–Fφ′ molecule. Finally, the identification of the intermolecular BCP of the hydrogen bond \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\alpha }}} \right) \) is in agreement with the view of the ring-opening mechanism of the aziridine pictured in Fig. 8, although the bifurcated hydrogen bonds \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\alpha }}} \right) \) and \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\beta }}} \right) \) of the tricomplexes formed by oxirane and thiirane are not. Although the chemical similarity of these heterorings is well known, we are unable to explain this at this point in time. The o-III structure and its regioselectivity was not the focus of this investigation [118], and will not therefore be discussed in this paper.
Electronic topography: validation of structural and spectroscopic parameters
The existence of the novel tertiary hydrogen bonds \( \left( {{F^{{\varphi \prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\alpha }}} \right) \)and \( \left( {{F^{{\varphi \prime\prime}}}\, \cdot \, \cdot \, \cdot \,{H^{\beta }}} \right) \) was demonstrated spectroscopically by means of the stretch frequencies and absorption intensities. However, owing to the lack of experimental values, this analysis is purely theoretical. This is not to dispute the efficiency of B3LYP, since it is well established that this hybrid functional yields excellent results for corroborating and predicting vibrational parameters [119–121]. Without contradicting this, the conclusion here is that an additional analysis should be performed in order to prove and validate the vibrational results reported in this study, such as the stretch frequencies of the whole set of hydrogen bonds characterized using the QTAIM formalism. Figure 10 plots the values of the hydrogen bond distances against the electronic density of the intermolecular BCP. According to the linear regression of Eq. 1, it can be seen that there is a direct relationship between these two parameters, which suggests that QTAIM could be useful for examining the molecular structures of hydrogen complexes, as already documented by Grabowski et al. in many related studies [122–125].

Relationship between the hydrogen bond distances and the intermolecular electronic densities of the whole set of interactions of the tricomplexes
This initial analysis naturally serves as the basis for validating stretch frequencies, but, as the close relationship between structural and vibrational parameters is already widely known [126, 127], the claim here is that topological parameters can also be used for this conjecture. Figure 11 clearly shows that the values of the hydrogen stretch frequencies are closely correlated with the electronic density of the intermolecular BCP; the linear regression equation is shown in Eq. 2:

Relationship between the hydrogen bond stretch frequencies and the intermolecular electronic densities of the whole set of interactions of the tricomplexes
This conclusion, in combination with results we have obtained recently [128], clearly characterizes the hydrogen stretch frequencies, since the use of QTAIM provides a basis for future experimental studies such as the determination of the charge density via X-ray diffraction [129] or the detection of the infrared modes in a variable-temperature environment [130].
Electronic stabilization energy
Hitherto, the lengths of the bifurcated and directional hydrogen bonds, cooperative charge transfers and electronic densities, as well as the vibrational redshifts of the proton donors have been the emblematic criteria that have been used to define the most stable structure among the tricomplexes studied here. There can be no doubt that these molecular parameters are not conclusive, since the interpretation of the electronic energy is absolute. Table 7 lists electronic energies (E) and hydrogen bond energies (ΔE) followed by their respective corrections, ΔZPE and BSSE. According to Parra et al. [131], the cooperative idea is based on the energy distribution being determined by the number of hydrogen bonds. With the exception of the tricomplexes g and j, as the number of hydrogen bond chains increases, the cooperative effect becomes less negative; in other words, the bonding energy per hydrogen bond decreases, reducing bond stability [132]. This is an important proviso, because the reduction in hydrogen bond strength benefits from the equivalent energy distribution. Although the BSSE counterpoises [133] produce smaller energy corrections (ideally due to the large basis set 6-311++G(d,p) [134]), corroborating the structural and vibrational results presented above, the most strongly bonded tricomplexes are those formed by aziridine, which are also the most stable, as can be seen in Table 8. It is natural that the relative energies (ΔE R) of the cyclic systems i and m were caused by the less negative cooperative energies (ΔE C).
Discussion and conclusions
The specialized literature contains many studies that provide theoretical analyses of the reaction mechanism using various approaches. These include geometric characterization of the transition state, computation of atomic point charges, examination of the frontier orbitals (HOMO and LUMO), generally using natural population analysis, although it should be noted that we are not intendingto contribute to the discussion of structural and vibrational parameters here. The great merit of the present study lies in the application of QTAIM calculations to identify and quantify charge density, thus generating important information on chemical bond strength for further discussion. That was the aim of this research, which investigated a single step in the ring-opening reaction mechanisms of the oxirane, thiirane and aziridine heterorings with regard to the hydrogen bonding, and performed characterization using traditional intermolecular concepts. In principle, the main structural parameters were examined, initially considering the shorter hydrogen bond distances of the aziridinic complexes to be the most important evidence for bifurcated hydrogen bonds between the second hydrogen fluoride and the axial hydrogen atoms of the heterorings. In agreement with this, the deviation in linearity was also systematic, although in fact it is the orientations of the n lone pairs of the oxygen and sulfur that govern the formation of multiple hydrogen bonds, such as the bifurcated tertiary. In terms of charge transfer, a cooperative effect was observed for the central hydrogen fluoride molecule, where the highest charge fluxes were measured, although these were more evident in the cyclic tricomplexes formed by the bifurcated hydrogen bonds. Spectroscopically, the redshift effects were characterized, the increase in absorption intensity of the hydrogen fluoride bonds was fully elucidated, and the new vibrational modes—commonly called hydrogen bond stretch frequencies—were identified. Owing to an absence of experimental data, the QTAIM calculations of charge density were used to validate the values of the new vibrational modes, which serve as a basis for predicting the hydrogen bond strength, as many studies have shown. Although the aziridinic tricomplexes—in particular the cyclic one—exhibit a directional rather than a bifurcated hydrogen bond, its strength adequately corroborates the experimental behavior of the ring-opening reaction mechanism. Finally, computation of the electronic energy enabled us to conclude our study by confirming that the cyclic tricomplexes formed via bifurcated tertiary hydrogen bonds are ideally the most stable structures.
References
Acheson RM (1976) An introduction to the chemistry of heterocyclic compounds. Wiley, New York
Katritzky AR, Ramsden CA, Scriven EFV, Taylor RJK (eds)(1995–2007) Comprehensive heterocyclic chemistry III. Elsevier, Amsterdam
Christl M, Leininger H, Brunn E (1982) J Org Chem 47:661–666
Singh MM, Angelici RJ (1984) Inorg Chem 23:2691–2698
Bertani R, Mozzon M, Michelin RA (1988) Inorg Chem 27:2809–2815
Lukevits É (1994) Chem Heterocycl Compd 30:11–12
Salimon J, Salih N, Hussien, Yousif E (2009) Eur J Sci Res 31:256–264
Banks HD, White WE (2001) J Org Chem 66:5981–5986
Araújo OBG, RCMU CAB, Ramos MN (2007) J Theor Comput Chem 6:647–660
Araújo OBG, RCMU CAB, Ramos MN (2009) Struct Chem 20:663–670
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN, Hernandes MZ, Cavalcante KR (2007) J Mol Struct THEOCHEM 802:91–97
Kojić-Prodić B, Molčanov K (2008) Acta Chim Slov 55:692–708
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2009) J Mol Model 15:123–131
Desiraju GR (2010) Angew Chem Int Ed 49:2–10
Custelcean R, Jackson JE (2001) Chem Rev 101:1963–1980
Tubergen MJ, Andrews AM, Kuczkowski RL (1993) J Phys Chem 97:7451–7457
Fowler PW, Legon AC, Thumwood JMA, Waclawik ER (2000) Coord Chem Rev 197:231–247
Antolínez S, Gerbi M, López LC, Alonso JL (2001) Phys Chem Chem Phys 3:796–799
Goswami M, Arunan E (2009) Phys Chem Chem Phys 11:8974–8983
Leung HO, Marshall MD, Drake TL, Pudlik T, Savji N, McCune DW (2009) J Chem Phys 131:204301–204308
Møllendal H, Konovalov A, Guillemin JC (2010) J Phys Chem A 114:5537–5543
Cooke AS, Corlett GK, Legon AC (1998) Chem Phys Lett 291:269–276
Cole CG, Legon AC (2004) Chem Phys Lett 400:419–424
Arunan E, Dev S, Mandal PK (2004) App Spec Rev 39:131–181
Maris A, Ottaviani P, Caminati W (2002) Chem Phys Lett 360:155–160
Oliveira BG, Pereira FS, Araújo RCMU, Ramos MN (2006) Chem Phys Lett 427:181–184
Del Bene JA (1996) Mol Phys 89:47–59
Oliveira BG, Araújo RCMU, Carvalho AB, Lima EF, Silva WLV, Ramos MN, Tavares AM (2006) J Mol Struct THEOCHEM 755:39–45
Gershinowitz H, Eyring H (1935) J Am Chem Soc 57:985–991
Rozenberg BA (1986) Adv Polymer Sci 75:113–165
Jursic BS (1998) J Mol Struct THEOCHEM 434:37–42
Oliveira BG, Araújo RCMU, Chagas FF, Ramos MN (2008) J Mol Model 14:949–955
Legon AC, Thorn JC (1994) Chem Phys Lett 227:472–479
Legon AC (1995) Chem Phys Lett 247:24–31
Oliveira BG, Vasconcellos MLAA (2006) J Mol Struct THEOCHEM 774:83–88
Oliveira BG, Leite LFCC (2009) J Mol Struct THEOCHEM 915:38–42
Legon AC, Wallwork AL, Millen DJ (1991) Chem Phys Lett 178:279–284
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2009) J Mol Model 15:421–432
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793–1873
Bader RFW (1991) Chem Rev 91:893–928
Oliveira BG, Araújo RCMU, Pereira FS, Lima EF, Silva WLV, Carvalho AB, Ramos MN (2008) Quim Nova 31:1673–1679
Rao L, Ke H, Fu G, Xu X, Yan Y (2009) J Theor Comput Chem 5:86–96
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) Chem Phys Lett 433:390–394
Oliveira BG, Santos ECS, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2005) Spectrochim Acta A 60:1883–1887
Bader RFW (1991) Atoms in molecules. A quantum theory. Clarendon, Oxford
Bone RGA, Bader RFW (1996) J Phys Chem 100:10892–10911
Filho EBA, Ventura E, do Monte SA, Oliveira BG, Junior CGL, Rocha GB, Vasconcellos MLAA (2007) Chem Phys Lett 449:336–340
Ren F-D, Cao D-L, Wang W-L, Ren J, Hou S-Q, Chen H-S (2009) J Mol Model 15:515–523
Risikrishna Varadwaj PR (2010) J Mol Model 16:965–974
Oliveira BG, Araújo RCMU, Ramos MN (2008) Struct Chem 19:185–189
Oliveira BG, Vasconcellos MLAA, Olinda RR, Filho EBA (2009) Struct Chem 20:81–90
Oliveira BG, Araújo RCMU, Ramos MN (2008) Struct Chem 20:665–670
Oliveira BG, Vasconcellos MLAA (2009) Inorg Chem Commun 12:1142–1144
Smith DA (1994) ACS Symp Ser 569:1–5
Olovsson I (2006) Z Phys Chem 220:963–978
Ratajczak H, Orville-Thomas WJ, Rao CNR (1976) Chem Phys 17:197–216
Dognon J-P, Durand S, Granucci G, Lévy B, Millié P, Rabbe C (2000) J Mol Struct THEOCHEM 507:17–23
Carbó-Dorca R, Bultinck P (2004) J Math Chem 36:231–239
Breneman CM, Wiberg KB (1990) J Comput Chem 11:361–373
Cioslowski J, Hamilton T, Scuseria G, Hess BA Jr, Hu J, Schaad LJ, Dupuis M (1990) J Am Chem Soc 112:4183–4186
Oliveira BG, Araújo RCMU, Ramos MN (2010) J Mol Struct THEOCHEM 944:168–172
Talaty ER, Simons G (1978) Theor Chim Acta 48:331–335
Grigorenko BL, Nemukhin AV, Apkarian VA (1997) J Chem Phys 108:4413–4425
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery Jr JA, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Rega N, Salvador P, Dannenberg JJ, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA (1998) Gaussian 98W, revision A.1. Gaussian Inc., Pittsburgh
Cioslowski J (1992) Chem Phys Lett 194:73–78
Cioslowski J (1992) Chem Phys Lett 219:151–154
Cioslowski J, Nanayakkara A, Challacombe M (1993) Chem Phys Lett 203:137–142
Biegler-König F (2002) AIM 2000 1.0 program. University of Applied Sciences, Bielefeld
Gilli P, Bertolasi V, Ferretti V, Gilli G (1994) J Am Chem Soc 116:909–915
Grabowski SJ, Sokalski WZ, Leszczynski J (2006) J Phys Chem A 110:4772–4779
Oliveira BG, Santos ECS, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2004) Spectrochim Acta A 60:1883–1887
Oliveira BG, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2005) Spectrochim Acta A 61:491–494
Oliveira BG, Araújo RCMU, Ramos MN, Carvalho AB (2007) J Theor Comput Chem 6:647–660
Grabowski SJ (2009) Croat Chim Acta 82:185–192
Majerz I (2007) Mol Phys 105:2305–2314
Martin TW, Derewenda ZS (1999) Nat Struct Biol 6:403–406
Oliveira BG, Araújo RCMU, Ramos MN, Carvalho AB (2007) Quim Nova 30:1167–1170
Van Meerssche M, Feneau-Dupont J (1976) Introduction à la cristallographie et à la chimie structurale. Oyez é editeur, Leuven
Oliveira BG, Vasconcellos MLAA (2009) Acta Chim Slov 56:340–344
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) Spectrochim Acta A 68:626–631
Pople JA, Frisch MJ, Del Bene JE (1982) Chem Phys Lett 91:185–189
Araújo RCMU, Silva JBP, Ramos MN (1995) Spectrochim Acta A 51:821–830
Araújo RCMU, Ramos MN (1996) J Mol Struct THEOCHEM 366:233–240
Deakyne CA, Cravero JP, Hobson WS (1984) J Phys Chem 88:5975–5981
Parra RD, Bulusu S, Zeng XC (2003) J Chem Phys 118:3499–3509
Karpfen A (1997) Molecular interactions. Wiley, New York
King BF, Weinhold F (1995) J Chem Phys 103:333–348
Suhai S (1994) J Chem Phys 101:9766–9783
Berashevich JA, Chakraborty T (2007) Chem Phys Lett 446:159–164
Oliveira BG, Araújo RCMU (2007) Quim Nova 30:791–796
Ratajczak H (1972) J Phys Chem 76:3000–3004
Ratajczak H, Orville-Thomas WJ (1975) J Mol Struct 26:387–391
Allen AC (1975) Proc Nat Acad Sci USA 72:4701–4705
Nesbitt DJ (1988) Chem Rev 88:843–870
Swanepoel J, Heyns AM (1990) Spectrochim Acta A 46:1629–1638
Araújo RCMU, Ramos MN (1998) J Braz Chem Soc 9:499–505
Oliveira BG, Araújo RCMU, Ramos MN (20097) J Mol Struct THEOCHEM 908:79-83
Hobza P, Havlas Z (2000) Chem Rev 100:4253–4264
Biswal HS, Chakraborty S, Wategaonkar S (2008) J Chem Phys 129:184317–184321
Rozenberg M, Loewenschuss A, Marcus Y (2000) Phys Chem Chem Phys 2:2699–2702
Dinadayalane TC, Leszczynski J (2009) J Chem Phys 130:81101–81105
Cézard C, Rice CA, Suhm MA (2006) J Phys Chem A 110:9839–9848
Borowski P, Pilorz K, Pitucha M (2010) Spectrochim Acta A 75:1470–1475
Freed KF (1971) Ann Rev Phys Chem 22:313–346
Bader RFW (2009) J Phys Chem A 113:10391–10396
Bader RFW (1998) J Phys Chem A 102:7314–7323
Bader RFW (1991) Chem Rev 91:893–928
Bader RFW, Beddall PM, Peslak J Jr (1973) J Chem Phys 58:557–566
Vila A, Mosquera RA (2007) Chem Phys Lett 443:22–28
Gnecco D, Laura Orea F, Galindo A, Enríquez RG, Toscano RA, Reynolds WR (2000) Molecules 5:998–1003
Watson IDG, Yudin AK (2003) J Org Chem 68:5160–5167
Hu XE (2004) Tetrahedron 60:2701–2743
Schneider C (2009) Angew Chem Int Ed 48:2082–2084
Seki K, Yu R, Yamazaki Y, Yamashita Y, Kobayashi S (2009) Chem Commun 5722–5724
Giguere PA, Turrell S (1980) J Am Chem Soc 102:5473–5477
Tamamura H, Yamashita M, Muramatsu H, Ohno H, Ibuka T, Otaka A, Fujii N (1997) Chem Comm 3227–3228
Oliveira BG, Vasconcellos MLAA, Olinda RR, Filho EBA (2009) Struct Chem 20:897–902
Babkov LM, Baran J, Davydova NA, Uspenskiy KE (2006) J Mol Struct 792–793:68–72
Dimitrova Y (2004) Spectrochim Acta A 60:3049–3057
Grabowski SJ (2000) J Mol Struct 553:151–156
Grabowski SJ (2001) J Mol Struct 562:137–143
Wojtulewski S, Grabowski SJ (2002) J Mol Struct 605:235–240
Wojtulewski S, Grabowski SJ (2003) Chem Phys Lett 378:388–394
Savatinova I, Anachkova E (1983) Phys Stat Solidi 120:539–545
Unterderweide K, Engelen B, Boldt K (1994) J Mol Struct 322:233–239
Oliveira BG, Araújo RCMU, Ramos MN (2010) Quim Nova 33:1155–1162
Koritsanszky T (2006) Chapter 12. In: Hydrogen bonds—new insights. Springer, Berlin, pp 441–470
Herrebout WA, Stolov AA, van der Veken BJ (2001) J Mol Struct 563–564:221–226
Parra RD, Furukawa M, Gong B, Zeng XC (2001) J Chem Phys 115:6030–6035
Araújo RCMU, Soares VM, Oliveira BG, Lopes KC, Ventura E, do Monte SA, Santana OL, Carvalho AB, Ramos MN (2006) Int J Quantum Chem 106:2714–2722
Van Duijneveldt FB, van Duijneveldt-van de Rijdt JGCM, van Lenthe JH (1994) Chem Rev 94:1873–1885
Oliveira BG, Araújo RCMU, Leite ES, Ramos MN (2011) Int J Quantum Chem 111:111–116
Acknowledgments
The authors would like to thank the Brazilian funding agencies Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oliveira, B.G., Araújo, R.C.M.U., Carvalho, A.B. et al. Hydrogen bonds determine the structures of the ternary heterocyclic complexes C2H4O···2HF, C2H5N···2HF and C2H4S···2HF: density functional theory and topological calculations. J Mol Model 17, 2847–2862 (2011). https://doi.org/10.1007/s00894-011-0969-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-0969-8