
Abstract
B3LYP/6-311++G(d,p) calculations were employed in order to examine the molecular parameters of the C2H3XS···NH3 heterocyclic hydrogen-bonded complexes with X = H, F and CH3. Intermolecular criteria were taken into account when studying the formation of these hydrogen-bonded complexes, such as geometry analysis, charge density quantification and interpretation of the harmonic vibrational spectrum, in which case the appearance of red-shift and blue-shift effects was discussed. It was assumed from the outset that many hydrogen bond types may exist in these systems, and these were investigated using the results of topological integrations from the quantum theory of atoms in molecules (QTAIM) and intermolecular charge transfer calculations using the ChelpG scheme. The proton donor/acceptor behavior of C2H3XS was interpreted in terms of hydrogen bond energies, whose values were corrected using the basis sets superposition error (BSSE) and zero point energy (ZPE).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In organic chemistry, it is known that there are an enormous variety of nucleophilic mechanisms [1], and that, in these, the heterocyclic compounds are important intermediaries owing to their high reaction capacity [2], which is essentially a function of the strained ring energy [3]. Thiirane (C2H4S), in particular, is a three-member heteroring possessing properties internally related to nucleophilic reaction mechanisms [4]. In order to understand the functionality of C2H4S in nucleophilic reactions, one of the most widely used courses of investigation is the characterization of transition state based on interaction between C2H4S and a number of well-known nucleophiles [5], such as ammonia (NH3). According to Banks [6], elucidation of the reactivity of thiirane with ammonia has been reported, according to which structures of the transition state have been examined through the application of sophisticated computational methods. It is therefore argued that NH3 interacts with thiirane specifically on carbons of its CH2 methyl groups undergoing a SN2 generalized reaction mechanism. Nevertheless, it is also known that small compounds such as C2H4S and similar heterorings have been widely studied with regard to hydrogen-bonded complexes [7, 8]. In these cases, it has been assumed that, instead of a SN2 nucleophilic mechanism, it is possible for there to be an electrophilic interaction of haloacids with n lone pairs of the sulphur atom. This kind of examination has been validated using theoretical results and insights based on available experimental data [9].
However, except in cases reported by Banks and White [10], it should be stressed that in terms of intermolecular parameters, such as those routinely used for studying hydrogen-bonded complexes, no study of the interaction of nucleophiles such as NH3 and C2H4S has been reported until now. It is naturally assumed that hydrogen bonding criteria can be used to investigate nucleophilic interactions of NH3 with C2H4S. In other words, it is conceivable that the C2H4S···NH3 hydrogen-bonded complex is formed. Taking into account the importance of heterocyclic compounds and with reference to oxirane and aziridine [11], for instance, our proposal in this study is to understand the interactions between nucleophilic species and small heterorings [12]. Furthermore, we believe that it may be of great interest for theoreticians and experimenters who specialize in SN2 mechanisms, not only for thiirane but also for investigation of the reaction capacity or interaction strength of thiirane derivatives, such as fluorine-thiirane (C2H3FS) and methyl-thiirane (C2H3CH3S). To achieve this objective, efficient theoretical methodologies are employed, even though a computational strategy needs to be carefully designed. First, we suggest that density functional theory [13] calculations may be a useful computational method, where an efficient functional must be chosen. Using this procedure, we expect that geometrical and electronic parameters, as well as the interpretation of the vibrational harmonic spectrum, will be satisfactorily described and that the intermolecular properties of hydrogen-bonded complexes will be able to be extracted in its essence, i.e., the characterization of vibrational red-shift effects on proton donor bonds [14].
Nevertheless, there can be no doubt that if we wish to characterize hydrogen bonds or any other kind of intermolecular interaction in the C2H3XS···NH3 heterocyclic hydrogen-bonded complexes (with X = H, F and CH3), this objective can in fact be achieved through the application of the quantum theory of atoms in molecules (QTAIM) [15]. In overview, the utilization of the QTAIM theory and its topological parameters serves to locate bond critical points (BCP) [16] either on each chemical bond or on intermolecular contact, demonstrating whether there is a possibility of covalency [17] or interaction between separated nuclei [18]. In addition, employing an atomic charge algorithm may also yield interesting results, especially if the interaction between the hydrogen of the N–H bond of NH3 and the n lone pair of the sulphur in C2H4S can be identified, in addition, if possible, to other intermolecular interactions, such as the secondary ones [19–22]. We thus accept that ChelpG [23] charge partition may be able to describe the electrostatic potential and electron population on the C2H3XS···NH3 set of hydrogen-bonded complexes studied here and thereby quantify the charge transfer on the formation of the hydrogen bonds in these systems.
Another central concern of this study regards the function of C2H4S, which may act as a proton donor or acceptor when it interacts with NH3. Based on valence-shell electron pair repulsion (VSEPR) [24], it is well known that the electronic structure of C2H4S is formed by two n lone electron pairs orthogonally adjusted to the heteroring plane (see Scheme 1). By taking this into account, we are admitting the strong possibility of the formation of multiple interactions within the C2H3XS···NH3 complexes, since it is perfectly acceptable that the hydrogen atoms of NH3 interact with the n lone electron pairs of sulphur atoms, besides the preferential hydrogen bonds formed by the interaction between the nitrogen of NH3 and hydrogen atoms of the CH2 groups of the thiirane ring. Given this, the present study will assess whether the interaction in which thiirane and its derivatives act as proton donors or acceptor donors is the stronger. Finally, we hope this study will be able to serve as a guide for theoreticians and, above all, experimenters who are interested in new insights regarding nucleophilic interactions involving C2H4S and its derivatives, C2H3FS and C2H3CH3S.

Illustration of the n-lone electrons pairs of the thiirane
Computational details
The optimized geometries of the C2H3XS···NH3 heterocyclic hydrogen-bonded complexes with X = H, F and CH3 were obtained using the GAUSSIAN 98W program [25], all calculations being carried out on a single personal computer. Due to the efficiency of B3LYP for describing intermolecular interactions [26], this hybrid functional was chosen, in conjunction with 6-311++G(d,p) basis sets. On the basis of the optimized geometries, the QTAIM calculations were carried out using the AIM 2000 1.0 program [27], although some determinations were also obtained by means of the GAUSSIAN 98W by activating the “AIM = ALL” keyword. The values of the ChelpG atomic charges were also computed using the GAUSSIAN 98W program. For each complex, the interaction energy ΔE at the B3LYP/6-311++G(d,p) level of theory was determined using the supermolecule approach [28] as governed by Eq. 1.
Corrections were thus developed for these interaction energies based on basis sets of superposition error (BSSE) [29] (see Eq. 2), whereby the contribution of the energies of the monomers (m) in comparison with their energies within the (c) complex is quantified.
Finally, the values of the ΔE C corrected interaction energies were determined as follows:
Results and discussion
Optimized geometries
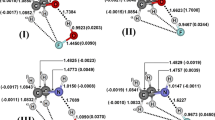
The optimized geometries of the C2H4S···NH3 (a), C2H4FS···NH3 (b) and C2H3CH3S···NH3 (d) hydrogen-bonded complexes are illustrated in Fig. 1. Analysis of these structures reveals a new aspect: systems (c) and (e). The generation of (c) and (e) was anticipated, since it is possible to form more stable complexes when the nucleophilic attack of NH3 occurs on the same side where the F and CH3 substituent groups are located. The structural results for the NH3, C2H4S, C2H4FS, and C2H3CH3S monomers obtained from B3LYP/6-311++G(d,p) calculations are listed in Table 1; and those for the (a), (b), (c), (d) and (e) complexes, the distance values for their main bonds and increments are listed in Table 2. As it is through the intermolecular interactions that the (a), (b), (c), (d) and (e) complexes are formed, the results of their hydrogen bond distances will be discussed here separately in more detail.

Optimized geometries of the C2H4S···NH3 (a), C2H3FS···NH3 (b, c) and C2H3CH3S···NH3 (d, e) hydrogen-bonded complexes using B3LYP/6-311++G(d,p) calculations
Complex (a)
The attack of NH3 on C2H4S is widely regarded by organic chemists as a regioselective process [5]. According to this view, the interaction between NH3 and C2H4S occurs in such a way as to form two hydrogen-bond types: (N···H1) and (H···S). Considering the results of 2.9214 Å and 2.7575 Å for these two hydrogen bonds, it is clear that the shorter distance for (H···S) suggests that NH3 is acting as a proton donor and C2H4S as an acceptor. As for the main deformation caused by NH3 on C2H4S monomers subsequent to the formation of (a), the (N···H1) hydrogen bond does not lead to alterations in C2H4S, i.e., the (C–H1) and (C–H2) bond lengths of 1.0834 Å in (a) are equal to those computed for the monomer. On the other hand, the (N–H) bond is slightly elongated, their distance values being 1.0146 Å and 1.0180 Å before and after formation of the (a) complex respectively. Structurally, these alterations to the proton donor bonds following complexation constitute one of the most important effects verified upon the formation of hydrogen-bonded complexes [30, 31].
Complexes (b)–(c)
At the first sight, the hydrogen bonds on (b) and (c) complexes seem to be directly related to the fluoride substituent, although there may be some doubt about this. In the (b) complex only one hydrogen bond is formed, (N···H2), whose length calculated at the B3LYP/6-311++G(d,p) level of theory is 2.3361 Å. In (c), however, two hydrogen bonds are formed, (N···H1) and (H···F1), whose values are 2.3788 Å and 2.5276 Å, respectively. Note that, although two hydrogen bonds are formed in (c), it is important to take into account the fact that (N···H1) and (H···F1) present longer distance values, which leads us to affirm that (b) is the preferential configuration, because its intermolecular strength is shorter than that of (c). Therefore, in this case, NH3 is a proton acceptor, since C2H4S is a donor. Once again, on analysis of the modifications on bonds after formation of both (b) and (c) complexes, it can be found that (F1C–H2) yields a significant increase in length, varying from 1.0836 to 1.0867 Å when (b) is formed from C2H3FS. Furthermore, a variation was observed in the (N–H) bond, of 1.0146 and 1.0162 Å for the C2H3FS monomer and the (c) complex, respectively. Nevertheless, (C–H1) also presents slight modifications with values of 1.0840 and 1.0870 Å after formation of (c).
Complexes (d)–(e)
The (d) and (e) complexes present multiple interactions, which need to be carefully examined. Initially, the hydrogen bonds (H···S) present values of 2.7990 and 2.8034 Å for the (d)–(e) complexes, respectively. However, the distance values of 2.6667 and 2.8235 Å for the (N···H2) and (N···H1) hydrogen bonds indicate that (d) is more strongly bonded than (e). In fact, even though a tertiary interaction recognized as (N···Hb) has been identified in (e), the value of 2.9046 Å is the longest interaction distance. As complement to this, it is expected that the traditional relationship between the interaction strength and the alterations on proton donor bonds can be obtained [32–34]. Given this, the distance values of 1.0180 and 1.0179 Å of the (N–H) bonds are slightly augmented in comparison with 1.0146 Å for the NH3 monomer. There is one further interesting feature related to (C–H1) and (C–H2): the first bond presents a negligible variation in its length, whereas the second undergoes a reduction, of 1.0843 and 1.0841 for the C2H3CH3S monomer and the (e) complex, respectively. The systematic tendency computed for the remaining bonds was similar: a reduction in bond lengths for the (C1–Hb), (C–C1) and (C1C–H2) of the (d) and (e) complexes. Clearly these changes are not generally so evident, but it should be borne in mind that some consequences may be important: for instance, the alterations in stretching frequencies and absorption intensities of the infrared spectrum [35].
From a structural point of view, evidence emerged of the formation of hydrogen bonds on the (a), (b), (c), (d) and (e) complexes, although, at this level of analysis, only empirical criteria, such as the tabulated values of the van der Waals’s radii [36] are taken into account. All of the hydrogen bonds examined in this study have been considered for potential interaction, because a single criterion was decisive: the distance of the intermolecular contact must be shorter or at least coequal to van der Waals’s radii [37]. It is widely known that the van der Waals’s radii constitute an important chemical parameter and this has often been used to study the formation of intermolecular systems. This study aims to achieve this by analysis of interaction energy or hydrogen bond strength and, principally, topological analysis based on the QTAIM approach, the results of which will be discussed below.
QTAIM parameters
The QTAIM theory can serve as a guide to identification of which kind of intermolecular interactions are formed in the (a), (b), (c), (d) and (e) complexes. It is through analysis of topological parameters such as electron density (ρ) and Laplacian (∇2ρ) that it is possible to determine whether closed-shell interactions may be considered as hydrogen bonds [38–40], in addition to whether they can facilitate the task of determining which of the complexes covered by this study is the more stable. Table 3 duly reports the values of ρ and ∇2ρ computed using the QTAIM approach for all intermolecular interactions which may be hydrogen bonds. It is worth, first, pointing out that the (N···H1) hydrogen bonds were characterized in the (a), (c) and (e) complexes, whose ρ and ∇2ρ value lie in the range of 0.0050–0.0133 e.u. and 0.0163–0.0383 e.u., respectively. Furthermore, the (N···H2) hydrogen bonds of the (b) and (d) complexes were characterized using ρ values of 0.0147 e.u. and 0.0078 e.u., which are higher than those calculated for (N···H1). At first sight, it would appear to be unjustified to affirm that the (b) and (d) complexes are more stable solely on account of the intermolecular electron density; the contrary would appear to be the case, since other intermolecular contacts beyond (N···H1) and (N···H2) can be identified. For example, in (d) a (H···S) hydrogen bond with a higher intermolecular electronic density of 0.0090 e.u. was identified. On the other hand, the (H···S) hydrogen bond is also observed in (a) and (e), which suggests that C2H4S and C2H3CH3S may be either proton donors or acceptors. Moreover, the (e) complex has in its structure the formation of three hydrogen bonds: (N···H1), (H···S) and (N···Hb). By way of illustration the bond critical points (BCP) are presented in Fig. 2. For these hydrogen bonds cited above, the lone electron pair of nitrogen forms two hydrogen bonds (N···H1) and (N···Hb), or more correctly speaking, what is essentially a bifurcated hydrogen bond. Even though the value of 0.0046 e.u. is the lowest electron density, the Laplacian of 0.0133 e.u. is proof of the existence of (N···Hb). The QTAIM analysis presented here was then extremely important in understanding and, above all, visualizing all hydrogen bonds on the (a), (b), (c), (d) and (e) complexes. However, the computation of the electronic density does not provide sufficient evidence to affirm whether the C2H4S, C2H3FS, and C2H3CH3S rings are proton donors or acceptors. Nevertheless, it is well known that hydrogen strength can be predicted using the electronic density calculated at the intermolecular BCP, but quantification of the hydrogen bond energies and charge transfer can provide more accurate results regarding the proton donor–acceptor behavior for thiirane and its derivatives.

Illustration of the electronic density pathways and BCP of the C2H4S···NH3 (a), C2H3FS···NH3 (b, c) and C2H3CH3S···NH3 (d, e) hydrogen-bonded complexes using QTAIM calculations
Hydrogen bond energies and charge transfer
Table 4 presents the uncorrected values for hydrogen bond energies (ΔE), corrected hydrogen bond energies (ΔE C), BSSE and ZPE corrections, and the intermolecular ChelpG charge transfers (ΔQ ChelpG) of the (a), (b), (c), (d) and (e) complexes, all these data being gathered from the results of the B3LYP/6-311++G(d,p) calculations. Although it is well known that large and complete basis sets yield small BSSE errors, it can, in fact, be seen that the ZPE has contributed most to correction of the hydrogen bond energies ΔE. Thus, by taking into account the corrected values ΔE C, the hydrogen bond energies reveal that (b) is the more stable complex, followed by (c). In comparison to other systems, (c) is more stable at least at 1.0 kJ mol−1. However, it should be stressed that this can be observed, not only through the application of BSSE and ZPE, but also from the fact that the uncorrected hydrogen bond energies likewise indicate that (b) and (c) are the most stable complexes. In terms of charge transfer, the positive ΔQ ChelpG results of 0.055 e.u. and 0.012 e.u. suggest that NH3 is partially losing its electronic density. It is also worth noting, in passing, that the ΔQ ChelpG charge transfer was computed only in terms of NH3; in other words, it confirms only the loss or gain in electronic charge on NH3 due to the formation of the (a), (b), (c), (d) and (e) complexes. If there is a loss of charge transfer on NH3, this shows that C2H3FS is acting as a proton donor because the H2 hydrogen in (b), in addition to the H1 hydrogen in (c) receives the charge from NH3. However, in the case of (c), it is not so simple a matter to affirm that that C2H3FS is a proton donor, owing to the existence of the (H···F1) hydrogen bond, which was identified by the QTAIM analysis. On the other hand, the (a), (d) and (e) complexes present negative ΔQ ChelpG charge transfer results, of −0.032 e.u., −0.027 e.u., and −0.027 e.u., respectively. In these situations, C2H4S and C2H3CH3S are typically proton acceptors because NH3 is the charge transfer acceptor.
Analysis of the infrared spectrum
The main stretching frequencies and absorption intensities, based on analysis of the infrared spectrum at the B3LYP/6-311++G(d,p) level of theory, are listed in Table 5. Throughout this study, the main purpose has been to identify hydrogen bonds and the extent to which these interactions may be useful for understanding the proton/acceptor behavior of the C2H4S, C2H3FS, and C2H3CH3S rings on formation of the (a), (b), (c), (d) and (e) complexes. In corroboration of the QTAIM results, analysis of the infrared spectrum also confirmed the existence of stretch frequencies for the (N···H1) hydrogen bonds in (a), (b) and (c) complexes, whose υ(N···H1) values are 78.4, 130.3 and 26.9 cm−1, respectively. Furthermore, the υ(N···H2) frequencies of the (b) and (c) complexes have already been well characterized, at values of 109 and 61.1 cm−1, respectively. Moreover, the values of 100.5, 97.4 and 90.8 cm−1 for the υ(H···S) stretch frequency of the (a), (d) and (e) complexes are the strongest vibrational modes, although the absorption intensities are not the highest, their values being in the region of 0.5–3.7 km mol−1.
Nevertheless, it is well established that the main vibrational mode that occurs as a result of the formation of hydrogen-bonded complexes is not determined by their intermolecular stretching modes, but by the changes on the bonds involving proton donors/acceptors observed after complexation. Table 6 presents one of these changes typically related to hydrogen complexes, which is the displacement of stretching frequencies in proton donors. First, the changes in (N–H) bonds in (a), (c), (d) and (e) complexes are red-shifts because the value of 3605 cm−1 is set at lower values: −154, −133, −33 and −153 cm−1, respectively. As such, the absorption intensity ratios also indicate the existence of red-shifts [41], mainly in (d), whose I(N–H),c/I(N–H), m value is 779. However, another type of vibrational displacement was identified: the so-called blue-shift effect [42]. In converse fashion to red-shift, the blue-shift phenomenon is characterized by changes of proton donor frequencies to higher values, which leads to a strengthening of the bond. In the light of this, the values of +105, +84 and +7 cm−1 indicate that blue-shifts due to the formation of the (N···H1) hydrogen bond were identified on the (C–H1) bonds of the (a), (c) and (e) complexes. With regard to the Δυ(C1–Hb) and Δυ(C–H2) shifts on the (d) and (e) complexes, only slight vibrational deformations were calculated, whose values of −1 and −2 cm−1 are practically invariable wave numbers. In fact, it is through QTAIM analysis that (N···H2) and (N···Hb) can be identified as the weakest hydrogen bonds, although it was not expected that major alterations in (C1–Hb) and (C–H2) bonds would be observed. Finally, the Δυ(C–H2) value of +10 cm−1 in (b) confirms that this complex is formed by a blue-shifting hydrogen bond. In other words, spectroscopically, the proton donor/acceptor behavior of C2H3FS can be understood in terms of changes in (C–H2) bonds, which function as proton donor centers.
Conclusions
In this study, we have put forward a theoretical proposal for identifying the proton donor/acceptor of thiirane derivatives and their ability to form hydrogen-bonded complexes with ammonia. Using B3LYP/6-311++G(d,p) calculations, analysis of ChelpG charge transfer, as well as topographic description based on the QTAIM approach, it has been argued that there are many hydrogen bonds on C2H3XS···NH3 heterocyclic hydrogen-bonded complexes with X = H, F and CH3. In these systems, a large number of hydrogen-bond types have been confirmed, including those characterized by red- and blue-shift effects. In terms of hydrogen-bond energies and charge transfer, fluorine-thiirane (C2H3FS) has been shown to tend to be a proton donor, since the (N···H1), (N···H2) and (N···F1) interactions led to the donation of charge transfer to the ammonia molecule as a whole. However, the infrared spectrum revealed the existence of both red- and blue-shift effects on C2H3XS···NH3, and especially on C2H3FS, where the blue-shift on the (C–H2) bond is an indication that fluorine-thiirane functions as a proton donor center.
References
Burnett JF, Zahler RE (1951) Chem Rev 49:273–412. doi:10.1021/cr60153a002
Bobylev VA, Koldobskii SG, Tereshchenko GF, Gidaspov BV (1988) Chem Heter Comp 24:947–959. doi:10.1007/BF00474036
Skancke A, Van Vechten D, Liebman JF, Skancke PN (1996) J Mol Struct 376:461–468. doi:10.1016/0022-2860(95)09062-2
Fokin AV, Kolomiets AF, Rudnitskaya LS, Shevchenko VI (1975) Russ Chem Bull 24:582–584. doi:10.1007/BF00927483
Siri D, Gaudel-Siria A, Pons J-M, Liotard D, Rajzmann M (2002) J Mol Struct (THEOCHEM) 588:71–78. doi:10.1016/S0166-1280(02)00133-1
Banks HD (2003) J Org Chem 68:2639–2644. doi:10.1021/jo0268411
Oliveira BG, Santos ECS, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2004) Spectrochim Acta [A] 60:1883–1887. doi:10.1016/j.saa.2003.10.006
Oliveira BG, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2005) Spectrochim Acta [A] 61:491–494. doi:10.1016/j.saa.2004.04.023
Cosléou J, Lister DG, Legon AC (1994) Chem Phys Lett 231:151–158. doi:10.1016/0009-2614(95)90571-5
Banks HD, White WE (2001) J Org Chem 66:5981–5986. doi:10.1021/jo001719s
Parker RE, Isaacs NS (1959) Chem Rev 59:737–799. doi:10.1021/cr50028a006
Holubka JW, Bach RD, Andrés JL (1992) Macromolecules 25:1189–1192. doi:10.1021/ma00029a028
Hohenberg P, Kohn W (1964) Phys Rev B 136:864–871. doi:10.1103/PhysRev.136.B864
Oliveira BG, Araújo RCMU, Ramos MN (2008) J Mol Model 14:949–955. doi:10.1007/s00894-008-0337-5
Bader FRW (1990) Atoms in molecules. A quantum theory. Oxford University Press, Oxford
Hati S, Datta D (1992) J Comput Chem 13:912–918. doi:10.1002/jcc.540130716
Bone RGA, Bader RFW (1996) J Phys Chem 100:10892–10911. doi:10.1021/jp953512m
Alkorta I, Rozas I, Elguero J (1998) Struct Chem 9:243–247. doi:10.1023/A:1022424228462
Legon AC (1995) Chem Phys Lett 247:24–31. doi:10.1016/0009-2614(95)01172-9
Oliveira BG, Araújo RCMU, Carvalho AB, Lima EF, Silva WLV, Ramos MN, Tavares AM (2006) J Mol Struct (THEOCHEM) 775:39–45. doi:10.1016/j.theochem.2006.06.028
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) J Theor Comput Chem 6:647–660. doi:10.1142/S0219633607003362
Oliveira BG, Vasconcellos MLAA (2007) J Mol Struct (THEOCHEM) 774:83–88. doi:10.1016/j.theochem.2006.06.018
Wiberg KB, Breneman CM (1990) J Am Chem Soc 112:8765–8775. doi:10.1021/ja00180a019
Gillespie RJ (1978) Molecular geometry. Van Nostrand-Reinhold, London
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Rega N, Salvador P, Dannenberg JJ, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA, Gaussian 98 W (Revision A.11.2), Gaussian, Inc., Pittsburgh PA, 2001
Kolboe S, Svelle S (2008) J Phys Chem A 112:6399–6400. doi:10.1021/jp8027879
AIM 2000 1.0 designed by Biegler-König F, University of Applied Sciences, Bielefeld, Germany
van Duijneveldt FB, Murrell JN (1967) J Chem Phys 46:1759–1767. doi:10.1063/1.1840932
Boys SB, Bernardi F (1970) Mol Phys 19:553–566. doi:10.1080/00268977000101561
Oliveira BG, Araújo RCMU, Ramos MN (2008) Struct Chem 19:185–189. doi:10.1007/s11224-007-9269-4
Oliveira BG, Araújo RCMU, Ramos MN (2008) Struct Chem 19:665–670. doi:10.1007/s11224-008-9344-5
Grabowski SJ (2007) J Phys Chem A 111:3387–3393. doi:10.1021/jp070530i
Grabowski SJ, Sokalski WA, Leszczynski J (2006) Chem Phys Lett 432:33–39. doi:10.1016/j.cplett.2006.10.069
Oliveira BG, Pereira FS, Araújo RCMU, Ramos MN (2006) Chem Phys Lett 427:181–184. doi:10.1016/j.cplett.2006.06.019
Araújo RCMU, Ramos MN (1996) J Mol Struct (THEOCHEM) 366:233–240. doi:10.1016/0166-1280(96)04536-8
Pauling L (1960) The nature of the chemical bond, 3 edn. Cornell University, USA
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) J Theor Comput Chem 6:647–660
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2009) J Mol Model 15:123–131. doi:10.1007/s00894-008-0380-2
Oliveira BG, Vasconcellos MLAA, Olinda RR, Filho EBA (2009) Struct Chem 20:81–90. doi:10.1007/s11224-008-9401-0
Grabowski SJ, Leszczynski J (2009) Chem Phys 355:169–176. doi:10.1016/j.chemphys.2008.12.011
Araújo RCMU, Ramos MN (1998) J Braz Chem Soc 9:499–505
Hobza P, Havlas Z (2000) Chem Rev 100:4253–4264. doi:10.1021/cr990050q
Acknowledgments
The authors gratefully acknowledge partial financial support to CAPES and CNPq Brazilian Funding agencies.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Oliveira, B.G., Araújo, R.C.M.U., Carvalho, A.B. et al. Small heterocyclics as hydrogen bond acceptors and donors: the case of the C2H3XS···NH3 complexes (X = H, F and CH3). Struct Chem 20, 663–670 (2009). https://doi.org/10.1007/s11224-009-9458-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-009-9458-4