
Abstract
This theoretical study presents a comparative analysis of the molecular properties of heterocyclic (C2H4O⋯HF and C2H5N⋯HF) and homocyclic (C3H6⋯HF) hydrogen-bonded complexes. Initially, the equilibrium geometries of these complexes were analyzed in detail at the B3LYP/6–311++G(d,p) level of theory. Subsequently, the interaction energies and polarizabilities were also evaluated, as well as the infrared stretch frequencies and absorption intensities. In addition, by combining intermolecular criteria and charge density concepts, calculations of Bader’s theory of atoms in molecules were used to determine the maxima and minima for electron density in order to measure the strength of the n⋯H and pπ⋯H hydrogen bonds. Finally, the possibility of an F⋯Hα secondary interaction between the fluoride (F) of hydrogen fluoride and the axial hydrogen atoms (Hα) of the C2H4O and C2H5N heterocyclic rings was explored.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It has long been known that hydrogen bonds belongs to a non-covalent interaction category often investigated by experimentalists and theoreticians [1]. Nowadays, the hydrogen bond plays a fundamental role in important scientific research, e.g., the study of solvation in the biological medium [2], characterization of the building blocks of protein chains, amino acids and enzymes [3], as well as in the development of new pharmacological drugs [4]. A historical survey reveals a large number of reports of new insights into the molecular parameters of hydrogen-bonded complexes [5–6].
A commonly used experimental technique is Fourier Transform Microwave Spectroscopy (FTMS) [7], which estimates the equilibrium geometry of intermolecular systems by analyzing their inertial moments and rotational constants [8–10]. On the other hand, the study of hydrogen-bonded complexes has also been carried out using theoretical calculations [11–14], mainly in special cases where some of the classic parameters of weakly bound systems, such as attraction energies, infrared stretch frequencies, absorption intensities, and obviously the charge transfer [15], are difficult to explore experimentally. For this reason, Rozas et al. [16] and Tang et al. [17] have shown that the C3H6⋯HF homocyclic hydrogen-bonded complex is formed and stabilized by means of a pseudo (p) π⋯H hydrogen bond, which is manifested by an interaction of proton donors aligned exactly at the middle of the C–C bond of the cyclopropane (C3H6) moiety. In our recent studies [18–20], however, the chemical nature of C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes was discussed in terms of their n⋯H hydrogen bonds, which originate from the interaction of hydrogen fluoride (HF) with the n-lone pairs of oxygen and nitrogen of the three-membered rings (C2H4O and C2H5N) [21–22]. In fact, intermolecular cyclic systems (heterocyclic and homocyclic) have been widely studied by many research groups all over the world [23–24], although it has to be said that some questions about these systems, such as those of an electronic or vibrational nature [25], still require further investigation.
Nevertheless, an interesting conclusion of our studies on hydrogen-bonded complexes is that the charge transfer mechanism may be useful in the interpretation of observable parameters, e.g., polarizability and infrared stretching modes [15]. Not surprisingly, however, it has been widely reported that charge transfer also plays an essential role in explaining some effects inherent to molecular energy [26–31], although it should be noted that the charge transfer phenomenon is essentially non-observable because the quantification of atomic charges is not based on physical arguments [32]. In other words, atomic charges are not computed directly from ab initio wave functions, which usually leads to some divergence in interpretation if atomic charge partitions derived from distinct approaches are used [15]. To avoid this, it is recommended to use a method that has been implemented by means of purely physical parameters, for example electron density. Such an objective can thus be effectively associated with the quantum theory of atoms in molecules (QTAIM) [33–35], whose efficiency is well-known in the scientific community, particularly in studies that characterize intermolecular interactions by measuring electron density, as well as in the interpretation of surface operators [26–41].
In this work, we have performed a theoretical study of the molecular properties of the C3H6⋯HF, C2H4O⋯HF and C2H5N⋯HF hydrogen-bonded complexes using density functional theory (DFT) [42–43], as well as by analysis of molecular descriptors embodied in QTAIM topological integrations. Besides structural, electron and vibrational parameters, by taking into account QTAIM descriptors such as electronic density (ρ), Laplacian (∇2 ρ) and charge transfer (δQ(Ω)), our main goal is that of examining charge density maxima and minima, thereby predicting the strength of pπ⋯H and n⋯H hydrogen bonds. We have also investigated the existence of a secondary F⋯Hα interaction between the fluoride (F) of the hydrogen fluoride and the axial hydrogen atoms (Hα) of the C2H4O and C2H5N heterocyclic rings. Thereby, we expect to demonstrate the reactivity of the C2H4O and C2H5N heterocyclic compounds in comparison with the C3H6 homocyclic system.
Computational methods
The optimized geometries of the C2H4O⋯HF, C2H5N⋯HF and C3H6⋯HF hydrogen-bonded complexes were obtained at the B3LYP/6–311++G(d,p) level of theory [44–46] with calculations performed using the GAUSSIAN 98W program [47]. To obtain the QTAIM data, all topological calculations were executed using GAUSSIAN software [48–50], although some integrations were also processed using the program AIM 2000 1.0 [51]. The values of interaction energies (ΔE) of the hydrogen-bonded complexes (A⋯B) were determined as follows [52]:
According to Eq. 2, the ΔE results were corrected by zero point vibrational energy (ZPE) [53] and basis sets superposition error (BSSE) [54].
Results and discussion
Geometry
Hydrogen bond distance
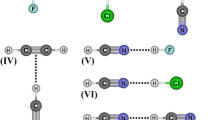
Taking the B3LYP/6–311++G(d,p) calculations as a basis, the optimized geometries of the C2H4O⋯HF, C2H5N⋯HF and C3H6⋯HF hydrogen-bonded complexes are illustrated in Fig. 1. Some time ago, Legon et al. [55] reported FTMS data on the experimental structure of the C2H4O⋯HF complex, and determined a value for the R(n⋅⋅⋅H) hydrogen bond distance of 1.700 Å. In view of the data collected in Table 1, it can be seen that our R(n⋅⋅⋅H) theoretical result of 1.662 Å is in satisfactory agreement with the experimental value mentioned above [55]. In the C3H6⋯HF homocyclic complex, the R(pπ⋯H) hydrogen bond distance presents a result of 2.080 Å [8], which is longer than the values of 1.662 Å and 1.623 Å for the corresponding C2H4O⋯HF and C2H5N⋯HF systems. One explanation for this is that the low charge density on the C–C bond of the cyclopropane results in a low quantity of intermolecular charge being transferred to the HF molecule. Thereby, C3H6⋯HF is thus considered to be a weakly bound complex. So, the large R(pπ⋯H) hydrogen bond distance provides some structural justification of the lower reactivity of the homocyclic system in comparison with the heterocyclic complexes.

Optimized geometries of C3H6⋯HF, C2H4O⋯HF and C2H5N⋯HF hydrogen-bonded complexes using the B3LYP/6–311++G(d,p) calculations
Non-linearity in the hydrogen bonds
The electronic structure of C2H4O and C2H5N is composed of oxygen and nitrogen atoms that have n-lone pairs of electrons orthogonally aligned to the trigonal surface of these heterorings. Besides the formation of n⋯H hydrogen bonds, another interesting structural aspect can be seen regarding the F⋯Hα secondary interaction between the fluoride and axial hydrogen atoms (Hα) of the three-membered rings [56]. It can be hypothesized that the F⋯Hα secondary interaction shows some structural distortions in the C2H4O⋯HF and C2H5N⋯HF hydrogen-bonded complexes, such as the linearity deviation (θ) on the n⋯H hydrogen bonds. From the B3LYP/6–311++G(d,p) results listed in Table 1, the respective θ values of 9.2° and 4.0° for C2H4O⋯HF and C2H5N⋯HF suggest the existence of linearity deviation in their hydrogen bonds. However, the R(F⋅⋅⋅Hα) theoretical distances of 3.273 Å and 3.394 Å for the C2H4O⋯HF and C2H5N⋯HF hydrogen complexes are longer than the van der Waals radii for hydrogen (H = 1.2 Å) and fluoride (F = 1.35 Å) atoms, which add up to a combined length of 2.55 Å [57]. Thus, although FTMS [55] analysis suggests the existence of F⋯Hα, the R(F⋅⋅⋅Hα) results indicate that this secondary interaction is unlikely to form.
Important structural changes
By further comparing the cyclic structures, an elongation of the rHF bond length can be detected (see Table 1), whose δrHF variations are 0.025 Å, 0.045 Å and 0.010 Å for the C2H4O⋯HF, C2H5N⋯HF and C3H6⋯HF complexes, respectively. It should be noted that δrHF cannot be considered the only evidence of cyclic complexation, because significant variations are observed on the rCY “heterobond” (notation for CO and CN bonds of C2H4O and C2H5N, respectively), whose values for the C2H4O⋯HF and C2H5N⋯HF hydrogen-bonded complexes are 0.012 Å and 0.032 Å, respectively. These “heterobond” enhancements are an indication of the relaxation of the C2H4O and C2H5N strained molecules. On the other hand, it should be noted that δrCC does not deform significantly in view of the results of −0.001 Å and −0.002 Å.
Infrared harmonic spectrum
Vibrational modes of three-member rings
In the context of the structural results presented in this study, we have already discussed the fact that variations of rCY “heterobonds” constitute important evidence regarding the formation of heterocyclic hydrogen-bonded complexes. Hence, for the sake of accuracy, it is very important to identify the υCY-i stretch modes of the CO and CN bonds of the C2H4O and C2H5N monomers prior to the formation of the heterocyclic hydrogen-bonded complexes. Thus, according to the B3LYP/6–311++G(d,p) results listed in Table 2, the values of 1,298 cm−1 and 1,266 cm−1 correspond to the υCO-i and υCN-i stretch frequencies. Moreover, these values are in satisfactory agreement with the available experimental data of 1,250 cm−1 and 1,200 cm−1 for the C2H4O [58] and C2H5N [59] monomers, respectively. In fact, a knowledge of the variations in the stretch modes for CO and CN bonds is essential to explain the strain relaxation phenomenon on the C2H4O and C2H5N rings [60]. This trend is confirmed by the ΔυCY frequency shifts, whose values are −3.7 cm−1 and −20.6 cm−1 (Table 3). Of course, the infrared variations on the CO and CN bonds can also be described by an increase in their absorption intensities, whose (ACY,c/ACY-i) ratios are 1.4 and 2.4 for the C2H4O⋯HF and C2H4N⋯HF hydrogen-bonded complexes, respectively.
Shift modes on the HF acid
Of the many changes observed in the infrared spectrum of hydrogen-bonded complexes, the most pronounced effect is related to the stretch frequencies of the proton donors, which are shifted downwards, while their absorption intensities are increased drastically. According to the B3LYP/6–311++G(d,p) results presented in Table 3, the ΔυHF shifts of the C2H4O⋯HF, C2H5N⋯HF and C3H6⋯HF complexes are −538.2 cm−1, −960.2 cm−1 and −222 cm−1, and the (AHF,c/AHF-I) absorption intensity ratios are 9.7, 8.3 and 5.6, respectively. In short, it can be seen that the strongest n⋯H hydrogen bonds cause large shifts in the stretch modes of HF. However, the formation of hydrogen bond complexes is one phenomenon responsible for manifestation of the hydrogen bond stretch frequencies, whose modes are characterized at low values of spectrum with weak absorption intensities. Table 2 therefore also lists the υ(n⋯H) hydrogen bond stretch frequencies of the C2H4O⋯HF and C2H5N⋯HF heterocyclic complexes, whose values are 254.3 cm−1 and 281.1 cm−1, respectively. In terms of absorption intensities, the A(n⋅⋅⋅H) values of 23.3 km mol−1 and 15.0 km mol−1 are lower than those observed for hydrogen fluoride, although by analyzing the new stretch mode υ(pπ⋯H) of 127.2 cm−1 and its absorption intensity A(pπ⋯H) of 1.62 km mol−1, it can be seen that these modest values are in fact indications of how weakly bound the C3H6⋯HF homocyclic complex actually is.
Electronic structure
Charge transfer and polarizability
Electrostatic interaction and charge transfer [1] are the principal factors used to determine molecular energy and hence predict intermolecular interactions, in particular hydrogen bonds. The values of the charge transfers (δQHF (Ω)) and variations in dipole moments (δμ) for the C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes are listed in Table 4. First, it can be seen that C2H5N⋯HF is the strongest hydrogen-bound system because of the higher calculated charge transfer δQHF (Ω) of −0.087 e.u.. By way of comparison, the charge transfer value of −0.030 is one more indication that C3H6⋯HF is a weakly bound complex. For notation purposes only, the results of atomic charges (Q) and consequently charge transfers (δQ(Ω)) were obtained through the QTAIM calculations as follows:
Note that all δQHF (Ω) values are negative, which indicates a dominant charge donation from the highest occupied molecular orbital (HOMO) represented by n-lone electron pairs of oxygen and nitrogen to the hydrogen σ* lowest unoccupied molecular orbital (LUMO) of the HF molecule. This leads to an electrical rearrangement of the molecular structure, which can be interpreted as an increase in the dipole moment, δμ. The positive values for δμ indicate that the polarizability of the heterocyclic hydrogen-bonded complexes is larger than the vector sum of the respective isolated molecules, especially in the case of the C2H5N⋯HF complex, whose value of 1.43 D lead us to affirm that C2H5N is more reactive with HF than C2H4O or C3H6.
Interaction energy
Table 4 also presents results for corrected interaction energies (ΔEC), uncorrected interaction energies (ΔE), BSSE quantities and ZPE thermodynamic corrections. Initially, however, it is worth stressing that the values of 31.20 kJ mol−1 and 51.90 kJ mol−1 of the C2H4O⋯HF and C2H5N⋯HF complexes are higher than those of van der Waals contacts (∼8 kJ mol−1) [63] or biological interactions (∼35 kJ mol−1) [64]. The C3H6⋯HF complex presents the lowest interaction energy, whose value of 9.55 kJ mol−1 shows that cycloprane yields a less stable intermolecular system. In determining ΔEC values, ZPE contributed the most and BSSE the least. Note that BSSE values range from 0.46 kJ mol−1 to 1.90 kJ mol−1 for C3H6⋯HF homocyclic and C2H4O⋯HF heterocyclic complexes, respectively. By taking into account the conclusions reported by Rozas et al. [16], the B3LYP/6–311++G(d,p) level of theory is in fact sufficient to determine minima for BSSE contributions.
QTAIM molecular topology
Characterization of n⋯H and pπ⋯H hydrogen bonds
It is evident that specific features of hydrogen bonding cannot be interpreted only in terms of intermolecular energy, but may also be adequately defined quantum mechanically and computed by using QTAIM theory. The QTAIM methodology performs a numerical integration of electron density, whereby a number of topological parameters, such as electron density (ρ) and the Laplacian (∇2 ρ) descriptor are calculated. Tables 5 and 6 present the values of electron density and Laplacian descriptors for the isolated molecules, as well as for the C2H4O⋯HF, C2H5N⋯HF, and C3H6⋯HF hydrogen-bonded complexes.
According to the scheme elaborated by Koch and Popelier [65], the characterization of intra- and/or inter-molecular interactions is based on bond critical points (BCP) [33] located between neighboring atoms, according to which two essential criteria must be obeyed: (1) besides the proton donor, the acceptor center for positive charge must contain a minimum level of electron density, and (2) typical values for the Laplacian descriptor of charge density must be carefully interpreted. The Laplacian descriptors computed lead us to consider that ∇2 ρ < 0 and ∇2 ρ > 0 when covalent bonds (shared) or intermolecular/intramolecular (closed-shell) contacts are examined [66–70], respectively. Undoubtedly, these conditions are satisfied for the C2H4O⋯HF and C2H5N⋯HF heterocyclic complexes, where their BCP (see Fig. 2) describes the proton donor as being HF, whereas the proton acceptors are the oxygen and nitrogen atoms of the C2H4O and C2H5N rings, respectively.

Bond paths and bond critical points (BCP) of the C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes obtained through quantum theory of atoms in molecules (QTAIM) integrations. BCP are represented by the smallest spheres
For n⋯H hydrogen bonds, the electron density values are 0.046 e/a0 3 and 0.062 e/a0 3 for the C2H4O⋯HF and C2H5N⋯HF complexes (ρ = 10–3 e/a0 3 is typical for van der Waals complexes) [16], respectively. Moreover, the ∇2 ρ (n⋅⋅⋅H) positive values of 0.149 e/a0 5 and 0.116 e/a0 5 indicate that the n⋯H hydrogen bonds possess the typical characteristic of closed-shell interactions. In other words, the charge density is concentrated on separated nuclei, i.e., in the O⋯H and N⋯H hydrogen bonds. In fact, the n⋯H hydrogen bond of the C2H5N⋯HF complex shows a higher concentration of electron density, which demonstrates the greater susceptibility of the C2H5N heterocyclic to bind with HF. For the C3H6⋯HF homocyclic complex, the hydrogen bond presents the lowest values of ρ (pπ⋯H) and ∇2 ρ (pπ⋯H), of 0.017 e/a0 3 and 0.058 e/a0 5, respectively.
F⋯Hα secondary interaction versus QTAIM parameters
One of the most important objectives of this study was the identification of the F⋯Hα secondary interaction between fluoride (F) of the HF acid and the axial hydrogen atoms (Hα) of the C2H4O and C2H5N rings. In light of the QTAIM results, the secondary interaction was not characterized because neither BCP was found between the F and Hα atoms [71–74]. There is a debate in the field of structural analysis where it is admitted that the R(F⋅⋅⋅Hα) distance must be either longer than or equal to the van der Waals radii. Our QTAIM results suggest that formation of this interaction is unlikely as there are no other interactions in the C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes besides the n⋯H hydrogen bonds.
Concentration of charge density on heterocyclic hydrogen-bonded complexes
Table 4 also shows the topological parameters of HF. As can be clearly seen in the relief maps (3-D) and contour line plots (2-D) graphically illustrated in Fig. 3, the electron density in HF is higher that of the n⋯H hydrogen bonds. Due the accumulated concentration of electron density, the HF covalent bond was characterized by AIM postulates as a shared interaction by means of the Laplacian values of −2.432 e/a0 5 and −2.110 e/a0 5 on the C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes, respectively. As observed in the analysis of structural parameters and vibrational modes, the formation of the C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes causes a significant deformation of the molecular properties of the isolated molecules, principally the CY “heterobonds” of the three-membered rings. Indeed, analysis of the AIM results presented in Tables 5 and 6 shows that the electron density on the CY bond is diminished, which confirms the tendency towards strain relaxation on the C2H4O and C2H5N rings upon the formation of the heterocyclic hydrogen-bonded complexes. Moreover, an increase in the Laplacian values for the CO and CN bonds can be observed, which is similar to the electron density results discussed above. For instance, the ∇2 ρ CY values of −0.358 e/a0 5 and −0.467 e/a0 5 for isolated three-membered rings (C2H4O and (C2H5N) are more negative compared with −0.294 e/a0 5 and −0.456 e/a0 5 when the respective C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes are formed.

Representation of relief maps (3-D) and contour line plots (2-D) of the electron density of the C2H4O⋯HF and C2H5N⋯HF heterocyclic hydrogen-bonded complexes using the QTAIM calculations. The charge concentration indicated by ∇2 ρ < 0 identifies the fluorine atoms of the HF acid. In contrast, ∇2 ρ > 0 indicates a depletion of electron density, which is represented by n⋯H hydrogen bonds
Conclusions
Based on B3LYP/6–311++G(d,p) calculations, it has been demonstrated that C2H4O⋯HF and C2H5N⋯HF heterocyclic complexes are more strongly bound than the C3H6⋯HF homocyclic complex. The chemistry of the C2H4O and C2H5N monomers was discussed by focusing on the relaxation of these strained rings, which was described using rupture trends on the CY “heterobonds”. Furthermore, a detailed investigation on HF was carried out, from which some interesting results were obtained, such as the increase in the length of the rHF bond, variation in ρ HF electron density quantified using QTAIM calculations, and vibrational red-shift effects. In addition, an exploratory investigation based on QTAIM topology was performed for the purpose of identifying the maximum and minimum electron density and Laplacian shapes. It was demonstrated that heterocyclic complexes have a greater concentration of intermolecular charge density, which is in accord with the higher values of interaction energies. Although formation of an F⋯Hα secondary interaction between the fluoride of HF acid and the Hα atoms of the C2H4O and C2H5N rings was previously indicated by FTMS analysis, our results did not confirm the existence of F⋯Hα because no connection between the F and Hα atoms was identified using QTAIM calculations. To corroborate this result, it is very important to point out that this secondary interaction was not possible because the R(F⋯Hα) distances are longer than the van der Waals radii. Finally, the main conclusion of this study is that C2H4O⋯HF and C2H5N⋯HF heterocyclic complexes are stabilized by only one hydrogen bond, n⋯H, which is formed by the interaction between the n-lone pairs of oxygen and nitrogen atoms and the HF molecule.
References
Larsen RW, Suhm MA (2006) J Chem Phys 125:154314–154319 doi:10.1063/1.2358349
Smallwood CJ, McAllister MA (1997) J Am Chem Soc 119:11277–11281 doi:10.1021/ja972517p
Grabowski SJ (2001) J Mol Struct 562:137–143 doi:10.1016/S0022-2860(00)00863-2
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN, Hernandes MZ, Cavalcante KR (2007) J Mol Struct THEOCHEM 802:91–97 doi:10.1016/j.theochem.2006.09.002
Hobza P, Sponer J (1999) Chem Rev 99:3247–3276 doi:10.1021/cr9800255
Delanoye SN, Herrebout WA, van der Veken B (2002) J Am Chem Soc 124:11854–11855 doi:10.1021/ja027610e
Suenram RD, Grabow JU, Zuban A, Leonov I (1999) Rev Sci Instrum 70:2127–2135 doi:10.1063/1.1149725
Legon AC, Aldrich PD, Flygare WH (1981) J Chem Phys 75:625–630 doi:10.1063/1.442079
Legon AC, Aldrich PD, Flygare WH (1982) J Am Chem Soc 104:1486–1490 doi:10.1021/ja00370a007
Cole GC, Legon AC (2004) Chem Phys Lett 400:419–424 doi:10.1016/j.cplett.2004.10.138
Araújo RCMU, da Silva JBP, Ramos MN (1995) Spectrochim Acta [A] 51:821–830 doi:10.1016/0584-8539(94)00194-G
Araújo RCMU, Ramos MN (1996) J Mol Struct THEOCHEM 366:233–240 doi:10.1016/0166-1280(96)04536-8
Araújo RCMU, Ramos MN (1998) J Braz Chem Soc 9:499–505
Silva JBP, Silva MR Jr, Ramos MN (2005) J Braz Chem Soc 16:844–850
Oliveira BG, Araújo RCMU (2007) Quim Nova 30:791–796
Rozas I, Alkorta I, Elguero J (1997) J Phys Chem A 101:9457–9463 doi:10.1021/jp971893t
Zhang Y-H, Hao J-K, Wang X, Zhou W, Tang T-H (1998) J Mol Struct THEOCHEM 455:85–99 doi:10.1016/S0166-1280(98)00247-4
Oliveira BG, Santos ECS, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2004) Spectrochim Acta [A] 60:1883–1887 doi:10.1016/j.saa.2003.10.006
Oliveira BG, Duarte EM, Araújo RCMU, Ramos MN, Carvalho AB (2005) Spectrochim Acta [A] 61:491–494 doi:10.1016/j.saa.2004.04.023
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) Spectrochim Acta [A] 68:626–631 doi:10.1016/j.saa.2006.12.038
Cremer D, Kraka E (1985) J Am Chem Soc 107:3800–3810 doi:10.1021/ja00299a009
Greenberg A, Liebman JF (1978) Strained organic compounds. Academic, New York
Castaneda JP, Denisov GS, Kucherov SY, Schreiber VM, Shurukhina AV (2003) J Mol Struct 660:25–40 doi:10.1016/j.molstruc.2003.07.010
Chandra AK, Minh TN, Uchimaru T, Zeegers-Huyskens T (2000) J Mol Struct 555:61–66 doi:10.1016/S0022-2860(00)00587-1
Dimitrova Y (1999) Rec Res Dev Phys Chem 3:133–148
Tretiak S, Mukamel S (2002) Chem Rev 102:3171–3212 doi:10.1021/cr0101252
Bredas JL, Beljonne D, Coropceanu V, Cornil J (2004) Chem Rev 104:4971–5004 doi:10.1021/cr040084k
Rubtsov IV, Redmore NP, Hochstrasser RM, Therien MJ (2004) J Am Chem Soc 126:2684–2685 doi:10.1021/ja0305499
Bredenbeck J, Helbing J, Hamm P (2004) J Am Chem Soc 126:990–991 doi:10.1021/ja0380190
Sun MT, Chen YH, Song P, Ma FC (2005) Chem Phys Lett 413:110–117 doi:10.1016/j.cplett.2005.07.070
Sun D, Fang J, Yu G, Ma F (2007) J Mol Struct THEOCHEM 806:105–112 doi:10.1016/j.theochem.2006.11.015
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926 doi:10.1021/cr00088a005
Bader RFW (1991) Chem Rev 91:893–928 doi:10.1021/cr00005a013
Bader RFW (1990) Atoms in molecules. A quantum theory. Clarendon, Oxford, UK
Popelier PLA (2000) Coord Chem Rev 197:169–189 doi:10.1016/S0010-8545(99)00189-7
Oliveira BG, Pereira FS, Araújo RCMU, Ramos MN (2006) Chem Phys Lett 427:181–184 doi:10.1016/j.cplett.2006.06.019
Oliveira BG, Vasconcellos MLAA (2006) J Mol Struct THEOCHEM 774:83–88 doi:10.1016/j.theochem.2006.06.018
Oliveira BG, Araújo RCMU, Carvalho AB, Lima EF, Silva WLV, Ramos MN, Tavares AM (2006) J Mol Struct THEOCHEM 775:39–45 doi:10.1016/j.theochem.2006.06.028
Bader RFW, MacDougall PJ, Lau CD (1984) J Am Chem Soc 106:1594–1605 doi:10.1021/ja00318a009
Bader RFW (1980) J Chem Phys 73:2871–2883 doi:10.1063/1.440457
Bone RGA, Bader RFW (1996) J Phys Chem 100:10892–10911 doi:10.1021/jp953512m
Hohenberg P, Kohn W (1964) Phys Rev B 136:864–871 doi:10.1103/PhysRev.136.B864
Kohn W, Sham LJ (1965) Phys Rev A 140:1133–1138 doi:10.1103/PhysRev.140.A1133
Becke AD (1997) J Chem Phys 107:8554–8560 doi:10.1063/1.475007
Becke AD (1993) J Chem Phys 98:5648–5652 doi:10.1063/1.464913
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789 doi:10.1103/PhysRevB.37.785
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Rega N, Salvador P, Dannenberg JJ, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA (1998) GAUSSIAN 98W (Revision A.1). Gaussian, Pittsburgh PA
Cioslowski J (1992) Chem Phys Lett 194:73–78 doi:10.1016/0009-2614(92)85745-V
Cioslowski J (1992) Chem Phys Lett 219:151–154 doi:10.1016/S0009-2614(94)87001-2
Cioslowski J, Nanayakkara A, Challacombe M (1993) Chem Phys Lett 203:137–142 doi:10.1016/0009-2614(93)85377-Z
AIM (2000) 1.0 program designed by Biegler-König F, University of Applied Sciences. Bielefeld, Germany
van Duijneveldt FB, Murrell JN (1967) J Chem Phys 46:1759–1767 doi:10.1063/1.1840932
McQuarrie DA (1973) Statistical thermodynamics. Harper and Row, New York
Boys SB, Bernardi F (1970) Mol Phys 19:553–566 doi:10.1080/00268977000101561
Legon AC, Kisiel Z, Georgiou AS, Millen DJ (1989) Chem Phys Lett 155:447–454 doi:10.1016/0009-2614(89)87184-2
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) J Theor Comp Chem 6:647–660
Pauling L (1960) The nature of the chemical bond, 3rd edn. Cornell University, New York
Lord RC, Nolin B (1956) J Chem Phys 24:656–658 doi:10.1063/1.1742592
Shagidullin RR, Grechkin NP (1969) Chem Heter Comp 3:232–235 doi:10.1007/BF01172558
Dudev T, Lim C (1998) J Am Chem Soc 120:4450–4458 doi:10.1021/ja973895x
Prichard DG, Nandi RN, Muenter JS (1988) J Chem Phys 89:115–123 doi:10.1063/1.455513
Bishop DM, Cheung LM (1982) J Phys Chem Ref Data 11:119–133
Carbó R, Klobukowski M (1990) Self consistent field: theory and applications. Elsevier, Amsterdam
Stryer L (1995) Biochemistry. Freeman, New York
Koch U, Popelier PLA (1995) J Phys Chem 99:9747–9754 doi:10.1021/j100024a016
Grabowski SJ (2004) J Phys Chem A 108:1806–1812 doi:10.1021/jp036770p
Grabowski SJ, Sokalski WA, Leszczynski J (2006) Chem Phys Lett 432:33–39 doi:10.1016/j.cplett.2006.10.069
Grabowski SJ (2007) Chem Phys Lett 436:63–67 doi:10.1016/j.cplett.2007.01.041
Grabowski SJ (2007) J Phys Chem A 111:13537–13543 doi:10.1021/jp076990t
Grabowski SJ (2007) J Phys Chem A 111:3387–3393 doi:10.1021/jp070530i
Oliveira BG, Araújo RCMU, Chagas FC, Carvalho AB, Ramos MN (2008) J Mol Model 14:949–955 doi:10.1007/s00894-008-0337-5
Oliveira BG (2007) J Arg Chem Soc 95:59–69
Oliveira BG, Araújo RCMU, Pereira FS, Lima EF, Silva WLV, Carvalho AB, Ramos MN (2008) Quim Nova (in press)
Oliveira BG, Araújo RCMU, Carvalho AB, Ramos MN (2007) Quim Nova 30:1167–1170
Acknowledgments
Thanks to Brazilian funding agencies CAPES and CNPq.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Optimized geometries of the C2H4O⋯HF, C2H5N⋯HF, and C3H6⋯HF hydrogen-bonded complexes using B3LYP/6–311++G(d,p) calculations (DOC 29.5 KB)
Rights and permissions
About this article
Cite this article
Oliveira, B.G., Araújo, R.C.M.U., Carvalho, A.B. et al. The molecular properties of heterocyclic and homocyclic hydrogen-bonded complexes evaluated by DFT calculations and AIM densities. J Mol Model 15, 123–131 (2009). https://doi.org/10.1007/s00894-008-0380-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-008-0380-2