
Abstract
The genome of the hyperthermophilic archaeon Pyrobaculum calidifontis contains an open reading frame, Pcal_1032, annotated as glucokinase. Amino acid sequence analysis showed that Pcal_1032 belonged to ROK (repressor, open reading frame, and kinase) family of sugar kinases. To examine the properties of Pcal_1032, the coding gene was cloned and expressed in Escherichia coli. However, expression of the gene was low resulting in a poor yield of the recombinant protein. A single site directed mutation in Pcal_1032 gene, without altering the amino acid sequence, resulted in approximately tenfold higher expression. Purified recombinant Pcal_1032 efficiently phosphorylated various hexoses with a marked preference for glucose. ATP was the most preferred phosphoryl group donor. Optimum temperature and pH for the glucokinase activity of Pcal_1032 were 95 °C and 8.5, respectively. Catalytic efficiency (k cat/K m) towards glucose was 437 mM−1 s−1. The recombinant enzyme was highly stable against temperature with a half-life of 25 min at 100 °C. In addition, Pcal_1032 was highly stable in the presence of denaturants. There was no significant change in the CD spectra and enzyme activity of Pcal_1032 even after overnight incubation in the presence of 8 M urea. To the best of our knowledge, Pcal_1032 is the most active and highly stable glucokinase characterized to date from archaea, and this is the first description of the characterization of a glucokinase from genus Pyrobaculum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucokinase (EC 2.7.1.2) is the enzyme which catalyzes the conversion of glucose into glucose 6-phosphate utilizing ATP, ADP, or PPi as phosphoryl group donor, the first irreversible phosphorylation reaction of glycolysis.
Based on amino acid sequence, microbial glucokinases are classified into three groups (Lunin et al. 2004). The group I consists of ATP- and ADP-dependent glucokinases from eukaryotes (Ronimus and Morgan 2004) and archaeal phylum euryarchaeota (Sakuraba et al. 2004). Group II glucokinases are found in Gram-negative bacteria, cyanobacteria, and amitochondriate protists (Wu et al. 2001). Group III glucokinases are present in archaeal phylum crenarchaeota (Hansen et al. 2002) and bacteria (Hansen and Schonheit 2003). This group possesses two conserved sequence motifs which include ROK (repressor, open reading frame, and kinase) motif (Titgemeyer et al. 1994) and a cysteine-rich motif (CXCGXXGCXE). Mutational study of Bacillus subtilis glucokinase revealed the functional importance of the conserved cysteine residues in the cysteine-rich motif. Moreover, these residues are involved in glucose binding (Mesak et al. 2004).
We are interested in sugar kinases from hyperthermophiles and we have previously studied novel ribose-5-phosphate pyrophosphokinases from Thermococcus kodakarensis (Rashid et al. 1997), an anaerobic hyperthermophilic archaeon (Morikawa et al. 1994; Atomi et al. 2004) and P. calidifontis (Bibi et al. 2016), a facultative aerobic hyperthermophilic archaeon (Amo et al. 2000). Complete genomes of both the microorganisms have been determined (Fukui et al. 2005; http://www.ncbi.nlm.nih.gov/nuccore/CP000561.1). The genome sequence of P. calidifontis contained an open reading frame, Pcal_1032, annotated as glucokinase. To understand the functional properties, the gene encoding Pcal_1032 was cloned and expressed in Escherichia coli. Here, we report enhanced gene expression and functional characterization of the enzyme.
Materials and methods
Chemicals and materials were purchased either from Sigma-Aldrich or Thermo Fisher Scientific or Fluka Chemical Corp. Cloning vectors, restriction enzymes, and DNA purification kits were purchased from Thermo Fisher Scientific. Gene-specific oligonucleotides were commercially synthesized from Macrogen. Plasmid vectors, pTZ57R/T (Thermo Fisher) and pET-28a(+) (Novagen), were employed for cloning and expression purposes, respectively. E. coli cells DH5α and BL21-CodonPlus (DE3)-RIL (Stratagene) were used for cloning and expression purposes.
Gene cloning and expression in E. coli
Glucokinase gene, Pcal_1032, from P. calidifontis was amplified by polymerase chain reaction (PCR) using sequence specific forward, Pcal_1032F (5ʹ-CCATGGCGAAGTACTTGGGGATAG-3ʹ) and reverse, Pcal_1032R (5ʹ-CTATCGGGGATACCCAAACTTCTTC-3ʹ), primers and genomic DNA of P. calidifontis as template. Recognition site for restriction enzyme NcoI (underlined sequence) was introduced in the forward primer, Pcal_1032F. The PCR amplified DNA fragment was ligated in cloning vector pTZ57R/T using T4 DNA ligase as recommended by the supplier (Thermo Fisher Scientific). The resulting plasmid was named pTZ_1032. Pcal_1032 gene was liberated from pTZ_1032 using NcoI (introduced in the forward primer) and HindIII (from multiple cloning site of pTZ57R/T) restriction enzymes and cloned in pET-28a(+) expression vector utilizing the same sites. The resulting recombinant plasmid was named pET_1032.
Escherichia coli BL21-CodonPlus (DE3)-RIL cells were transformed using pET_1032. The transformed cells were cultivated in Luria–Bertani (LB) medium supplemented with 50 μg/mL kanamycin at 37 °C until an optical density of 0.4–0.5 at 660 nm was reached. Expression of the gene was induced by the addition of isopropyl-β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 0.2 mM. After induction, the cells were allowed to grow for 4–6 h at 37 °C and harvested by centrifugation at 6000×g.
Codon modification
For codon modification, a new forward primer (Pcal_1032FM 5ʹ-CCATGGCGAAATACTTGGGGATAG-3ʹ) was designed and used to amplify the Pcal_1032 gene. Nucleotide ‘G’ at position 11 was replaced by ‘A’ (shown in bold) in Pcal_1032FM primer. The gene with the modified codon was named Pcal_1032M. PCR amplified gene was cloned in pET-28a expression vector using the same strategy as described above.
Production in E. coli and purification of recombinant Pcal_1032
Cell pellet (4 g wet weight from 1 L culture) was suspended in 40 mL of 50 mM Tris–HCl buffer of pH 8.0 containing 0.2 mM each of phenylmethylsulfonyl fluoride and β-mercaptoethanol. Cells were then lysed by sonication. Soluble and insoluble fractions were separated by centrifugation at 12,000×g and 4 °C for 10 min. Supernatant containing recombinant Pcal_1032 was heated at 80 °C for 25 min to denature the heat-labile proteins of E. coli. The denatured proteins were removed by centrifugation at 20,000×g for 20 min. The supernatant containing Pcal_1032 was loaded onto HiTrap QFF (GE Healthcare) anion-exchange column. The proteins were eluted using a linear gradient of 0–1 M NaCl. Fractions showing high activity were pooled, dialyzed, and applied to Resource Q column (GE Healthcare). The procedure described for HiTrap QFF was exactly followed for Resource Q column. Again, the fractions showing high activity were pooled, dialyzed, equilibrated with 1.2 M ammonium sulphate, and loaded onto hydrophobic column Butyl FF (GE Healthcare) column. The proteins were eluted by gradually lowering the ammonium sulphate concentration from 1.2 to 0 M. Fractions at each step were examined by SDS-PAGE as well as activity analysis. Protein concentration was determined spectrophotometrically at every step of purification using Bradford reagent.
Molecular mass and subunit determination
The molecular mass of recombinant Pcal_1032 was determined by SDS-PAGE analysis and subunits were determined by gel filtration chromatography. For gel filtration, chromatography Superdex 200 10/300 GL gel filtration column (GE Healthcare) was equilibrated with 150 mM NaCl in 20 mM Tris–HCl (pH 8.0). The standard curve was obtained with ferritin (440 kDa), catalase (240 kDa), lactate dehydrogenase (140 kDa), BSA (64.5 kDa), and proteinase K (28.9 kDa). Solutions of the standard and sample proteins were prepared in 20 mM Tris–HCl (pH 8.0) containing 150 mM NaCl.
Enzyme assays
Glucokinase activity of Pcal_1032 was measured in a coupled assay by monitoring the reduction of NADP+ into NADPH at 340 nm by glucose 6-phosphate dehydrogenase (Sigma-Aldrich). Activity was measured either by continuous assay at or below 50 °C or by discontinuous assay at higher temperatures. The coupling reaction is shown below:
The assay mixture (1 mL) for the continuous assay contained 2 mM glucose, 2 mM MgCl2, 2 mM ATP, 100 mM Tris–HCl buffer pH 8.5, 1 mM NADP+, purified Pcal_1032, and 1 U glucose-6-phosphate dehydrogenase. The reaction scheme for discontinuous assay was the same as described above. However, the first step of conversion of glucose into glucose 6-phosphate was performed at higher temperature (60–90 °C) for a specific time interval, and then, the reaction mixture was quenched in ice water. The second step of conversion of glucose 6-phosphate to 6-phospho-d-gluconate was performed at 30 °C.
One unit of glucokinase was defined as the amount of enzyme required to convert 1 µmol of glucose to glucose-6-phosphate per min.
Activity with different substrates was measured following the pyruvate kinase/lactate dehydrogenase (PK/LDH) assay. ADP generated from ATP by the activity of kinase was measured by the oxidation of NADH into NAD+ in a UV spectrophotometer at 340 nm. The PK/LDH coupling reaction is shown below:
The assay mixture (1 mL) contained 100 mM Tris–HCl, pH 8.5, 2 mM of substrate, 2 mM MgCl2, 0.3 mM NADH, 2 mM ATP, 50 mM KCl, 1 mM phosphoenolpyruvate, 1.4 U pyruvate kinase, 2.8 U lactate dehydrogenase, and purified Pcal_1032. The assay was performed at 50 °C. Protein concentration was estimated through Bradford reagent and used in calculation of specific activity of the protein.
Circular dichroism analysis
Structural stability of Pcal_1032 was analyzed by circular dichroism (CD) spectroscopy using Chirascan-plus CD Spectrometer (Applied Photophysics). The protein samples were incubated at different temperatures ranging from 50 to 90 °C. The CD spectra of the protein solutions were recorded in 20 mM Tris–HCl pH 8.0 in the far-UV range of 190–260 nm. Solvent spectra were subtracted from those of the protein solutions.
Denaturation studies
For denaturation studies, Pcal_1032 protein samples were prepared in different concentrations of urea (0–8 M final concentration) or guanidinium chloride (0–6 M final concentration) and incubated at room temperature for various interval of time. Residual glucokinase activity of these samples was examined as described above.
Results
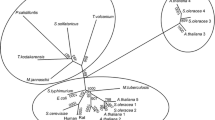
Genome search of P. calidifontis revealed the presence of an open reading frame, Pcal_1032, annotated as glucokinase. The gene consisted of 891 nucleotides encoding a polypeptide of 296 amino acids having a theoretical molecular mass of 31,573 Da and an isoelectric point of 6.46. Pcal_1032 displayed the highest homology of 74% with uncharacterized glucokinases from Pyrobaculum islandicum and Pyrobaculum aerophilum. Among the characterized enzymes, it exhibited 62% identity to its counterpart from Thermoproteus tenax (Dörr et al. 2003) followed by 39 and 34% with glucokinases from Aeropyrum pernix (Hansen et al. 2002) and Thermotoga maritima (Hansen and Schonheit 2003), respectively. When amino acid sequences of the characterized members of this family were aligned and a phylogenetic tree was constructed, three distinct groups were demarcated. Group I consisted of ADP-dependent glucokinases from eukaryotes and euryarchaeota including Rattus norvegicus, Methanocaldococcus jannaschii, Thermococcus litoralis, and Pyrococcus furiosus (Fig. 1). Group II included ATP-dependent glucokinases from E. coli and Zymomonas mobilis. In group I and II enzymes, ROK motif is absent. Pcal_1032 along with all the characterized glucokinases containing ROK motif from archaea and bacteria neither cluster in group I nor in group II. They made a third group. Alignment of amino acid sequences of the characterized members of group III demonstrated three highly conserved regions. Region I is reported to contain ATP-binding site, region II represents ROK motif, and region III is cysteine-rich conserved motif involved in glucose binding (Mesak et al. 2004). When we performed amino acid sequence alignment of cysteine-rich motif found in ATP-dependent ROK glucokinases from archaea and bacteria, we found that the third cysteine in this region was replaced by histidine in all archaeal sequences (Fig. 2). This region is reported to be involved in glucose binding; therefore, it is interesting to study the substrate specificity and compare it with the enzymes from bacterial sources.

Phylogenetic tree of Pcal_1032 and characterized glucokinases, whose amino acid sequences are available in the database. Unrooted tree was constructed using the neighbor-joining method. Segments corresponding to an evolutionary distance of 0.1 are shown. The tree was constructed using ClustalW provided at http://clustalw.ddbj.nig.ac.jp/. Following are the sequences, with accession numbers, used for the alignment to construct the phylogenetic tree: P. calidifontis (Pcal_1032), ABO08457; A. pernix, BAA81102; T. tenax, CCC80737; T. maritima, NP_229269; S. mutans, NP_720979; B. subtilis, P54495; E. coli, NP_416889; Z. mobilis, AAV88993; T. litoralis, EHR79075; P. furiosus, AAL80436; M. jannaschii, Q58999; R. norvegicus, NP_001094193; and Mycobacterium tuberculosis, NP_217218

Alignment of cysteine-rich motif found in ATP-dependent ROK glucokinases. Three conserved cysteines are typed bold. Following are the sequences, with accession numbers, used for alignment: Archaeoglobus fulgidus, AIG98959; Halogeometricum pallidum, ELZ30084; Haloprofundus marisrubri, KTG10816; Haloarcula vallismortis, AAV45565; Halorhabdus tiamatea, CCQ33691; Staphylothermus hellenicus, WP_013143234; Staphylothermus marinus, ABN70603; Thermogladius cellulolyticus, AFK51279; Ignisphaera aggregans, ADM27308; Thermosphaera aggregans, ADG90761; Desulfurococcus amylolyticus, AFL66479; Pyrobaculum islandicum, ABL88155; P. calidifontis, ABO08457; P. aerophylum, AAL64914; Pyrobaculum ferrireducens, AET32063; Pyrobaculum arsenticum, ABP51423; Pyrobaculum oguniense, AFA38285; Pyrobaculum neutrophilum, ACB40229; Thermoproteus uzoniensis, AEA13339; T. tenax, CCC80737; Vulcanisaeta thermophila, WP_069807370; Vulcanisaeta distributa, WP_013337289; Caldivirga maquilingensis, WP_012185382; A. pernix, BAA81102; Aeropyrum camini, BAN90779; Caldisphaera lagunensis, WP_015232398; Thermofilum pendens, ABL78302; Thermotoga neapolitana, ACM23200; Thermotoga naphthophila, ADA67425; Fervidobacterium pennivorans, ACM2320; Fervidobacterium islandicum, AMW32186; Fervidobacterium gondwanense, SHN56637; Fervidobacterium changbaicum, SDH58233; Fervidobacterium thailandensis, ODN31033; Thermosipho melanesiensis, ABR30996; Thermosipho affectus, ONN26936; Thermosipho africanus, ACJ75889; Thermosipho atlanticus, SHH58051; Thermotoga calidifontis, WP_041076719; Kosmotoga pacifica, AKI97915; Defluviitoga tunisiensis, CEP79132; Streptococcus mutans, AFM81004; Streptococcus troglodytae, BAQ24837; Streptococcus devriesei, WP_027975760; Streptococcus orisasini, WP_057490687; Streptococcus macacae, WP_003079242; Streptococcus uberis, CAR42827; Streptococcus phocae, KPJ22469; Streptococcus equi, CAW92910; Streptococcus iniae, AGM98339; Streptococcus porci, WP_027974469; Streptococcus caballi, WP_018365135; Streptococcus pneumonia, ABJ55324; B. subtilis, NP_390365; Bacillus tequilensis, KOS70873; Bacillus mojavensis, WP_024122056; Bacillus axarquiensis, KUP41914; Bacillus vallismortis, KYD02120; Bacillus licheniformis, AAU24173; Brevibacterium halotolerans, OEC78071; Petrotoga mobilis, ABX31202; Geotoga petraea SDC47876; and Pseudothermotoga thermarum, AEH50428
Production of Pcal_1032 in E. coli
When the gene encoding Pcal_1032 was expressed in E. coli by inducing the cells carrying pET_1032 plasmid with 0.2 mM IPTG, the expression level was very low. Higher or lower concentrations of IPTG did not affect the expression level. We then analyzed the folding and secondary structures of mRNA predicted by mfold web server (http://unafold.rna.albany.edu/?q=mfold) using the ribosomal binding site sequence in the vector and a few nucleotides at the start of Pcal_1032 gene. A hair-pin loop formation was predicted, as shown in Fig. 3a, with a ∆G value of − 2.8. This loop was formed due to the hydrogen binding between cytosine of the second amino acid alanine (GCG) and guanine of the third amino acid lysine (AAG). Furthermore, E. coli has biased in codon usage and AAG is not a preferred codon. To avoid hair-pin loop formation, we designed Pcal_1032FM primer for PCR amplification of the gene replacing AAG codon by AAA, a preferred codon in E. coli. This resulted in a dual advantage. The hair-pin loop formation was avoided (Fig. 3b) and a rare codon was replaced by a preferred codon. Nucleotide change was confirmed by DNA sequencing after cloning.

Prediction of secondary structure of mRNA of Pcal_1032 using mfold software. a Native sequence and b codon-modified sequence. The modification is shown by a circle
Analysis of production of recombinant Pcal_1032 in E. coli cells demonstrated that tenfold higher Pcal_1032 was produced in cells harboring pET_1032M plasmid compared to the cells containing pET_1032 plasmid under similar conditions (Fig. 4). Recombinant Pcal_1032 was approximately 30% of the total proteins of the host. When the soluble and insoluble fractions, after lysis of the cells, were analyzed, it was found that recombinant Pcal_1032 was produced in the soluble form. The first step of purification was based on the thermostability of recombinant Pcal_1032. The protein sample was heated at 80 °C for 30 min which resulted in precipitation and removal of most of the heat-labile proteins of the host. Further purification by ion exchange and hydrophobic column chromatographies resulted in purified recombinant Pcal_1032 to apparent homogeneity on SDS-PAGE (Fig. 4).

Coomassie brilliant blue stained 14% SDS-PAGE showing expression of Pcal_1032 and Pcal_1032 M genes. Lane M, protein ladder; lane 1, uninduced cells carrying pET_1032; lane 2, induced cells carrying pET_1032; lane 3, induced cells carrying pET_1032 M; and lane 4, purified Pcal_1032
Molecular mass determination
Molecular mass and subunit number of recombinant Pcal_1032 were determined by gel filtration chromatography. Pcal_1032 eluted at a retention volume of 15.05 mL, which corresponded to an approximate molecular mass of 31 kDa on a standard curve obtained from the elution volumes of various standard proteins of known molecular weight (data not shown). This indicated that the recombinant Pcal_1032 existed in a monomeric form.
Biochemical characterization
The thermostability of Pcal_1032 was examined by incubating the protein at 80 °C, for various intervals of time and measuring the residual activity, and Pcal_1032 was found highly thermostable with no significant loss in activity even after an incubation of 240 min. As Pcal_1032 was highly stable at 80 °C, therefore, we heated the enzyme at 90 and 100 °C for various intervals of time in screw-capped tubes and examined the residual activity. Half-life of the enzyme was 90 and 25 min at 90 and 100 °C, respectively (Fig. 5a).

Thermostability of Pcal_1032 and effect of temperature and pH on glucokinase activity. All the readings are average of three independent experiments. a Thermostability of Pcal_1032. The protein was incubated at 80 (closed circles), 90 (closed squares), and 100 °C (closed diamonds) for various intervals of time and residual activity was examined in 50 mM Tris–HCl buffer (pH 8.5) at 50 °C. b Effect of temperature. Glucokinase activity of Pcal_1032 was examined at various temperatures keeping the pH constant at 8.5. pH of the buffers was adjusted at room temperature. c Effect of pH. Glucokinase activity of Pcal_1032 was examined at various pH keeping the temperature constant. Following buffers were used: sodium phosphate buffer pH 6.5–7.5 (filled squares), Tris–HCl buffer pH 7.5–9.0 (open squares), and glycine–NaOH buffer pH 9.0–10.5 (closed circles)
When we examined the activity of Pcal_1032 at various temperatures keeping the pH constant at 8.5, we found that the glucokinase activity of Pcal_1032 increased with the increase in temperature and the highest activity was found at 95 °C (Fig. 5b) which is in accordance with the optimal growth temperature of P. calidifontis (90–95 °C).
To examine the effect of pH, the glucokinase activity of Pcal_1032 was examined in different buffers of various pH range. The optimum pH for Pcal_1032 glucokinase activity was 8.0‒8.5. Activity decreased rapidly below pH 7.5 and above 9.5 (Fig. 5c).
Pcal_1032 displayed significant glucokinase activity without the addition of any metal ion. However, a drastic decrease in enzyme activity was observed when 2 mM EDTA was added in the reaction mixture. This result indicated that enzyme activity of Pcal_1032 is metal ion dependent. The activity observed without the addition of any metal ion could be attributed to the metal ions bound to the enzyme during production in E. coli. Prior to examine the effect of metal ions, purified recombinant Pcal_1032 was incubated with 2 mM EDTA for 1 h and then extensively dialyzed. The addition of various metal ions in the assay mixture at a final concentration of 100 μM resulted in an increase in enzyme activity to a variable amount. Although various metal ions could activate Pcal_1032, the highest activity (a tenfold increase) was found in the presence of 100 μM of Mg2+. We, therefore, measured the glucokinase activity in the presence of various concentrations of Mg2+. Glucokinase activity of Pcal_1032 increased with the increase in Mg2+ and the highest activity was found in the presence of 1 mM (Fig. 6). Higher than 1 mM Mg2+ did not affect the enzyme activity.

Effect of various concentrations of Mg2+ in the assay mixture. Glucokinase activity was examined at 50 °C in 50 mM Tris–HCl buffer (pH 8.5)
When phosphoryl donor specificity of Pcal_1032 was examined using glucose as substrate, we found that the enzyme could utilize ATP, CTP, GTP, and UTP with a tenfold higher activity with ATP. No activity could be detected when ADP or AMP was used as phosphoryl group donor indicating that Pcal_1032 is nucleoside triphosphate dependent.
Determination of substrate specificity of Pcal_1032 indicated that it can phosphorylate all the hexoses tested. No pento kinase, phosphopento kinase, or phosphohexo kinase activity could be detected. The highest kinase activity was found with glucose (100%) followed by fructose (70%), mannose (30%), galactose (11%), and sorbitol (10%).
Structural stability
Recombinant Pcal_1032 was found highly thermostable; therefore, secondary structure of the protein was analyzed by CD spectroscopy. The results showed that there was no change in the CD spectra up to 90 °C, indicating that the enzyme maintains its secondary structure even at 90 °C (Fig. 7).

Circular dichroism analysis of Pcal_1032. Far-UV spectrum of Pcal_1032 (500 μg/mL) was analyzed by examining the circular dichroism spectra from 190 to 260 nm at 50 °C (closed circles), 60 °C (open squares), 70 °C (closed triangles), 80 °C (open circles), and 90 °C (closed squares)
Proteins usually lose their enzyme activities in the presence of high concentrations of denaturants such as urea and guanidinium chloride. However, Pcal_1032 was found highly stable in the presence of these denaturants. When residual activity was examined after overnight incubation of Pcal_1032 in the presence of 8 M urea or 2 M guanidinium chloride, the protein was found fully functional indicating its structural stability against these denaturants (Fig. 8). However, at higher concentrations of guanidinium chloride, the enzyme activity was significantly lost.

Fluorescent spectra of Pcal_1032 after overnight incubation with various concentrations of a urea and b guanidinium chloride
Kinetic parameters
Kinetic parameters towards glucose were measured by varying the concentration of glucose and keeping ATP constant at 2 mM. Similarly, when these parameters were measured towards ATP, then, glucose concentration was kept constant at 2 mM and ATP concentration was varied. Pcal_1032 exhibited a K m value of 660 ± 7 μM towards glucose and 900 ± 10 μM towards ATP. A V max value of 550 ± 5 μmol min−1 mg−1 was calculated from Lineweaver‒Burk plot (Fig. 9). From the V max (550 μmol min−1 mg−1) and molecular weight (31,573 Da) of Pcal_1032, a k cat value of 289 s−1 was calculated. Catalytic efficiency (k cat/K m) of Pcal_1032 was found to be 437 mM−1 s−1. Comparison of characteristics of the different ROK glucokinases demonstrated that Pcal_1032 is highly active enzyme of this group (Table 1).

Lineweaver–Burk plots of initial reaction rates for glucokinase activity of Pcal_1032 against glucose (a) and ATP (b)
Discussion
The genome sequence of P. calidifontis revealed the presence of an open reading frame, Pcal_1032, annotated as glucokinase. Phylogenetic analysis placed Pcal_1032 in the ROK glucokinase group. All characteristically conserved sequence motifs of this group were present in Pcal_1032. Cysteine-rich motif (CXCGXXGCXE) was found with a slight modification. The third Cys in this region was replaced by His. An alignment of cysteine-rich motifs of ROK glucokinase of archaeal and bacterial origins demonstrated that the third Cys was replaced by His in all the archaeal sequences. Two of the archaeal ROK glucokinases from A. pernix (Hansen et al. 2002) and T. tenax (Dörr et al. 2003) have been characterized and found to utilize multiple substrates as phosphoryl group acceptor similar to Pcal_1032. This region is reported to be involved in substrate (glucose) binding (Mesak et al. 2004). We can speculate that this amino acid change may be responsible for broad substrate specificity of the archaeal enzymes. A substitution of this His to Cys will be interesting and may confirm this speculation.
Escherichia coli expression system is the most popular expression system and high levels of recombinant proteins are produced. However, occasionally, no expression or very low expression is reported due to codon bias or hair-pin loop formation in mRNA secondary structure which restricts translation. Similar was the case with Pcal_1032 gene. When the gene was expressed in E. coli, a very low level of expression was observed. When the secondary structure of mRNA was analyzed using mfold, there was a prediction of formation of a hair-pin loop with a ∆G value of − 2.8. To remove this predicted loop, AAG codon for lysine was replaced by AAA. This silent mutation resulted in removal of hair-pin loop when analyzed by mfold and ultimately resulted in high expression of the gene.
Pcal_1032 was found to be highly thermostable. The amino acid composition of a protein seems quite relevant to support thermostability as it is related to the hydrophobic interactions (Baldwin 2007; Pace 2009). A comparison of amino acid composition showed that Pcal_1032 contains quite high number of hydrophobic residues Leu and Val which constitute 20.9% of the protein similar to 18% in its counterpart from hyperthermophilic bacterium T. maritima (NP_229269), and in contrast to 13.7% in the mesophilic counterpart from B. subtilis (accession # NP_390365). Furthermore, there are 34 (11.5%) alanine residues, the best α-helix former, which may be one of the factors responsible for the thermostability of Pcal_1032. Thermolabile amino acids tend to be avoided in thermostable enzymes (Hensel 1993; Muir et al. 1995; Russell and Taylor 1995; Russell et al. 1997). Thermolabile amino acids such as Cys, Met, Gln, and Asn were very low (5.7%) in Pcal_1032 similar to 5.3% in its counterpart from hyperthermophilic bacterium T. maritima, and in contrast to 10% in the mesophilic counterpart from B. subtilis. High thermostability of Pcal_1032 may be attributed to higher content of α-helix formers along with high number of hydrophobic residues and low number of thermolabile amino acids.
Proteins usually lose their enzyme activities in the presence of high concentrations of denaturants such as urea and guanidinium chloride, because these chaotropic agents disturb the native physiological active structure. However, there are some reports demonstrating stability of some proteins from hyperthermophilic archaea against these denaturants (Rasool et al. 2010; Chohan and Rashid 2013; Gharib et al. 2016). Pcal_1032 was also found highly stable in the presence of these denaturants. When the residual glucokinase activity of Pcal_1032 was measured after overnight incubation in the presence of 8 M urea or 2 M guanidinium chloride, no significant loss in enzyme activity was observed. However, the glucokinase activity of Pcal_1032 was decreased at higher concentrations of guanidinium chloride. A 55% loss in activity was observed after 2 h of incubation in the presence of 4 M guanidinium chloride. This may be due to the fact that guanidinium chloride is a salt as well as denaturant, whereas urea is an uncharged molecule, hence deficient in ionic strength effects.
In conclusion, our results demonstrate that P. calidifontis possesses a hexokinase, for phosphorylation of sugars at elevated temperatures, which belongs to ROK family and exhibits a broad substrate specificity. The enzyme is highly resistant to temperature and denaturants. Pcal_1032 is capable of phosphorylation of various hexoses with a marked preference for glucose. Furthermore, a single-nucleotide substitution resulted in high expression of Pcal_1032 gene in E. coli. Similar substitutions can be extended to other cloned genes which have low levels of expression in E. coli.
References
Amo T, Paje ML, Inagaki A, Ezaki S, Atomi H, Imanaka T (2000) Pyrobaculum calidifontis sp. nov., a novel hyperthermophilic archaeon that grows in atmospheric air. Archaea 1:113–121
Atomi H, Fukui T, Kanai T, Morikawa M, Imanaka T (2004) Description of Thermococcus kodakaraensis sp. nov., a well studied hyperthermophilic archaeon previously reported as Pyrococcus sp. KOD1. Archaea 1:263–267
Baldwin RL (2007) Energetics of protein folding. J Mol Biol 371:283–301
Bibi T, Perveen S, Aziz I, Bashir Q, Rashid N, Imanaka T, Akhtar M (2016) Pcal_1127, a highly stable and efficient ribose-5-phosphate pyrophosphokinase from Pyrobaculum calidifontis. Extremophiles 20:821–830
Chohan SM, Rashid N (2013) TK1656, a thermostable l-asparaginase from Thermococcus kodakaraensis, exhibiting highest ever reported enzyme activity. J Biosci Bioeng 116:438–443
Dörr C, Zaparty M, Tjaden B, Brinkmann H, Siebers B (2003) The hexokinase of the hyperthermophile Thermoproteus tenax ATP-dependent hexokinases and ADP-dependent glucokinases, two alternatives for glucose phosphorylation in archaea. J Biol Chem 278:18744–18753
Fukui T, Atomi H, Kanai T, Matsumi R, Fujiwara S, Imanaka T (2005) Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res 15:352–363
Gharib G, Rashid N, Bashir Q, Gardner QT, Akhtar M, Imanaka T (2016) Pcal_1699, an extremely thermostable malate dehydrogenase from hyperthermophilic archaeon Pyrobaculum calidifontis. Extremophiles 20:57–67
Goward CR, Hartwell R, Atkinson T, Scawen MD (1986) The purification and characterization of glucokinase from the thermophile Bacillus stearothermophilus. Biochem J 237:415–420
Han B, Liu H, Hu X, Cai Y, Zheng D, Yuan Z (2007) Molecular characterization of a glucokinase with broad hexose specificity from Bacillus sphaericus strain C3-41. Appl Environ Microbiol 73:3581–3586
Hansen T, Schonheit P (2003) ATP-dependent glucokinase from the hyperthermophilic bacterium Thermotoga maritima represents an extremely thermophilic ROK glucokinase with high substrate specificity. FEMS Microbiol Lett 226:405–411
Hansen T, Reichstein B, Schmid R, Schonheit P (2002) The first archaeal ATP-dependent glucokinase from the hyperthermophilic crenarchaeon Aeropyrum pernix, represents a monomeric, extremely thermophilic ROK glucokinase with broad hexose specificity. J Bacteriol 184:5955–5965
Hensel R (1993) Proteins of extreme thermophiles. New Comp Biochem 26:209–221
Hsieh PC, Shenoy BC, Samols D, Phillips NFB (1996) Cloning, expression and characterization of polyphosphate glucokinase from Mycobacterium tuberculosis. J Biol Chem 271:4909–4915
Imriskova I, Arreguin-Espinosa R, Guzman S, Rodriguez-Sanoja R, Langley E, Sanchez S (2005) Biochemical characterization of the glucose kinase from Streptomyces coelicolor compared to Streptomyces peucetius var. caesius. Res Microbiol 156:361–366
Kengen SW, Tuininga JE, de Bok FA, Stams AJ, de Vos WM (1995) Purification and characterization of a novel ADP-dependent glucokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem 270:30453–30457
Koga S, Yoshioka I, Sakuraba H, Takahashi M, Sakasegawa S, Shimizu S, Ohshima T (2000) Biochemical characterization, cloning, and sequencing of ADP-dependent (AMP-forming) glucokinase from two hyperthermophilic archaea, Pyrococcus furiosus and Thermococcus litoralis. J Biochem 128:1079–1085
Labes A, Schönheit P (2003) ADP-dependent glucokinase from the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus strain 7324. Arch Microbiol 180:69–75
Lunin VV, Li Y, Schrag JD, Iannuzzi P, Cygler M, Matte A (2004) Crystal Structures of Escherichia coli ATP-dependent glucokinase and its complex with glucose. J Bacteriol 186:6915–6927
Mesak LR, Mesak FM, Dahl MK (2004) Bacillus subtilis GlcK activity requires cysteines within a motif that discriminates microbial glucokinases into two lineages. BMC Microbiol 4:1–10
Morikawa M, Izawa Y, Rashid N, Hoaki T, Imanaka T (1994) Purification and characterization of a thermostable thiol protease from a newly isolated hyperthermophilic Pyrococcus sp. Appl Environ Microbiol 60:4559–4566
Muir JM, Russell RJ, Hough DW, Danson MJ (1995) Citrate synthase from the hyperthermophilic archaeon, Pyrococcus furiosus. Protein Eng 8:583–592
Pace CN (2009) Energetics of protein hydrogen bonds. Nat Struct Mol Biol 16:681–682
Porter EV, Chassy BM, Holmlund CE (1982) Purification and kinetic characterization of a specific glucokinase from Streptococcus mutans OMZ70 cells. Biochim Biophys Acta 709:178–186
Rashid N, Morikawa M, Imanaka T (1997) Gene cloning and characterization of recombinant ribose phosphate pyrophosphokinase from a hyperthermophilic archaeon. J Biosci Bioeng 83:412–418
Rasool N, Rashid N, Iftikhar S, Akhtar M (2010) N-terminal deletion of Tk1689, a subtilisin-like serine protease from Thermococcus kodakaraensis, copes with its cytotoxicity in Escherichia coli. J Biosci Bioeng 110:381–385
Ronimus RS, Morgan HW (2004) Cloning and biochemical characterization of a novel mouse ADP-dependent glucokinase. Biochem Biophys Res Commun 315:652–658
Russell RJ, Taylor GL (1995) Engineering thermostability: lessons from thermophilic proteins. Curr Opin Biotechnol 6:370–374
Russell RJ, Ferguson JMC, Haugh DW, Danson MJ, Taylor GL (1997) The crystal structure of citrate synthase from the hyperthermophilic archaeon Pyrococcus furiosus at 1.9 Å resolution. Biochemistry 36:9983–9994
Sakuraba H, Yoshioka I, Koga S, Takahashi M, Kitahama Y, Satomura T, Kawakami R, Ohshima T (2002) ADP-dependent glucokinase/phosphofructokinase, a novel bifunctional enzyme from the hyperthermophilic archaeon Methanococcus jannaschii. J Biol Chem 277:12495–12498
Sakuraba H, Mitani Y, Goda S, Kawarabayasi Y, Ohshima T (2003) Cloning, expression, and characterization of the first archaeal ATP-dependent glucokinase from aerobic hyperthermophilic archaeon Aeropyrum pernix. J Biochem 133:219–224
Sakuraba H, Goda S, Ohshima T (2004) Unique sugar metabolism and novel enzymes of hyperthermophilic archaea. Chem Rec 3:281–287
Titgemeyer F, Reizer J, Reizer A, Saier MH Jr (1994) Evolutionary relationships between sugar kinases and transcriptional repressors in bacteria. Microbiology 140:2349–2354
Tu J, Tuch BE (1996) Glucose regulates the maximal velocities of glucokinase and glucose utilization in the immature fetal rat pancreatic islet. Diabetes 45:1068–1075
Wu G, Henze K, Müller M (2001) Evolutionary relationships of the glucokinase from the amitochondriate protist, Trichomonas vaginalis. Gene 264:265–271
Acknowledgements
This work was partly supported by an NRPU Grant No. 20-2024 to NR from Higher Education Commission of Pakistan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Rights and permissions
About this article

Cite this article
Bibi, T., Ali, M., Rashid, N. et al. Enhancement of gene expression in Escherichia coli and characterization of highly stable ATP-dependent glucokinase from Pyrobaculum calidifontis . Extremophiles 22, 247–257 (2018). https://doi.org/10.1007/s00792-017-0993-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-017-0993-4