
Abstract
Most of the “repressor, open reading frame, kinase” (ROK) proteins already characterized so far, and exhibiting a kinase activity, take restrictedly d-glucose as substrate. By exploring the sequenced bacterial diversity, 61 ATP-dependent kinases belonging to the ROK family have been identified and experimentally assayed for the phosphorylation of hexoses. These kinases were mainly found to be thermotolerant and highly active toward d-mannose and d-fructose with notable activities toward d-tagatose. Among them, the ATP-dependent kinase from the mesophile Streptococcus mitis (named ScrKmitis) was biochemically characterized and its substrate spectrum further studied. This enzyme possessed impressive catalytic efficiencies toward d-mannose and d-fructose of 1.5 106 s−1 M−1 and 2.7 105 s−1 M−1, respectively, but also significant ones toward d-tagatose (3.5 102 s−1 M−1) and the unnatural monosaccharides d-altrose (1.1 104 s−1 M−1) and d-talose (3.4 102 s−1 M−1). Specific activities measured for all hexoses showed a high stereopreference for d- over l-series. As proof of concept, 8 hexoses were phosphorylated in moderate to good yields, some of them described for the first time like l-sorbose-5-phosphate unusually phosphorylated in position 5. Its thermotolerance, its wide pH tolerance (from 7 to 10), and temperature range (> 85% activity between 40 and 70 °C) open the way to applications in the enzymatic synthesis of monophosphorylated hexoses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phosphorylation of monosaccharides is a highly important stage of carbohydrate metabolism in prokaryotes, mainly for preparing carbohydrates for degradation and transfer reactions but also as a mean of uptake. This reaction is catalyzed by sugar kinases such as glucokinases (EC 2.7.1.2), fructokinases (EC 2.7.1.4), and hexokinases (EC 2.7.1.1). While hexokinases, mostly reported among eukaryotes, exhibit broad substrate range tolerance, other sugar kinases have narrow sugar substrate specificity. Many bacterial sugar kinases are grouped into the ROK family (repressor, open reading frame, kinase), a large family of sugar metabolism-related proteins (Conejo et al. 2010; Larion et al. 2007; Titgemeyer et al. 1994).
Enzymatic syntheses of mono O-phosphorylated sugars have been described in the literature (Wohlgemuth et al. 2017) especially by transketolase- or aldolase-catalyzed C-C bond formation or by selective phosphorylation catalyzed by kinases (Charmantray et al. 2009; Guérard-Hélaine et al. 2014; Hélaine et al. 2015; Sánchez-Moreno et al. 2012; Shaeri et al. 2008; Wohlgemuth et al. 2017). Chiral phosphorylated sugars are of importance as central metabolites in the non-oxidative pentose phosphate pathway, in the Calvin cycle, and of particular interest for the study of various metabolic diseases such as diabetes, cancer, immunodeficiency, atherosclerosis, mucoviscidosis (Desvergnes et al. 2012), as well as unintended virus or microorganism developments (Shaeri et al. 2008, Wohlgemuth 2009, Solovjeva and Kochetov 2008, Schorken 1998). Given the vital roles of such compounds and, more particularly, of phosphorylated hexoses for metabolism studies but also organic chemistry, new kinases allowing their straightforward synthesis are needed. Among established strategies to provide enzymes possessing new or improved properties, exploring protein natural diversity is one of the most efficient (Bastard et al. 2014; Davids et al. 2013; de Berardinis et al. 2017; Guérard-Hélaine et al. 2015; Vergne-Vaxelaire et al. 2013; Zhao et al. 2013). In this study, we explored the sequenced bacterial diversity to find novel ATP-dependent C6 sugar kinases able to phosphorylate unusual various substrates such as d-tagatose, d-psicose and l-sorbose, rare or unnatural hexoses. Among the 61 ATP-dependent kinases found to be active toward various hexoses, a thermotolerant kinase from Streptococcus mitis (ScrKmitis) was selected to be further studied and its biocatalytic capabilities were characterized. Data regarding substrate promiscuity, reaction conditions (pH and temperature), biochemical properties, as well as the structural identification of some phosphorylated compounds, were provided.
Materials and methods
Chemicals and materials
All chemical reagents were purchased from commercial sources and used without additional purification. d-(+)- and l-(−)-allose, d-(+) and l-(−)-altrose, l-(−)-mannose, d-(−)- and l-(+)-gulose, d -(+)- and l-(−)-idose, l-(−)-galactose, d-(+)- and l-(−)-talose, l-psicose, l-(+)-fructose, and l-(+)-tagatose were purchased from Carbosynth (Compton, berkshire, UK). d-(−)-fructose, d-psicose, d-(+)- and l-(−)-sorbose, d-(−)-tagatose, d-(+)- and l-(−)-glucose, d-(+)-mannose, d-(−)- and l-(+)-threose, d-xylulose, d-(−)-lyxose, d-(+)-xylose, d-(−)- and l-(+)-arabinose, d-(−)-ribose, d-(−)- and l-(+)-erythrose, d-(+)-galactose, l-(+)-erythrulose, d-(+)-cellobiose and N-acetyl- d-(+)-glucosamine, adenosine triphosphate (ATP), phosphoenolpyruvic acid monopotassium salt (PEP), β-Nicotinamide adenine dinucleotide reduced (NADH) and β-Nicotinamide adenine dinucleotide (NAD), isopropyl-β- d-thiogalactoside (IPTG), 2-(N-morpholino)ethanesulfonic acid (MES), 2-Amino-2-(hydroxymethyl)-1,3-propanediol, enzymes lactate dehydrogenase (LD), and pyruvate kinase (PK) from rabbit muscle were purchased from Sigma Aldrich (MilliporeSigma, StLouis, USA). Bovine serum albumin was purchased from Bio-Rad. Oligonucleotides were from Sigma-Aldrich (MilliporeSigma, StLouis, USA). E. coli strains BL21-CodonPlus (DE3)-RIPL were from Agilent technologies (Santa Clara, USA). Enzymatic spectrophotometric screenings in microplates were performed on a SpectraMax Plus384 (Molecular Devices, Sunnyvale, USA). Spectrophometric assays of ScrKmitis were recorded on a Safas UVMC2 (Safas, Monaco) thermostated when specified with a refrigerated/heating circulator Corio CD-200F (Jubalo®, Seelbach, Germany) using microcells high-precision cell quartz with 10-mm light path (Hellma Analytics, Müllheim, Germany). Cell crude extracts in 96-microwell plates were heated using a tested thermocycler GeneAmpTM PCR system 9700 (Applied Biosystem, Foster City, USA). NMR spectra were recorded on a Bruker (Bruker, Billerica, USA) 300 or 600 MHz spectrometer (Evry University, France) for 1H and 13C experiments, and on a Bruker 500 MHz (ICSN, France) for 1H-31P experiments. Chemical shifts are expressed in ppm and referenced with residual solvent signals. Coupling constant values (J) are given in hertz. NMR spectra of α form and β form are respectively noted as α and β. UHPLC analyses were performed on a UHPLC U3000 RS 1034 bar (Thermo Fisher Scientific, Waltham, USA) equipped with diode array detector DAD3000 and an evaporative light scattering detector (ELSD) 6100 (Esa Chromachem ®). HPLC purifications were done on a semi-preparative HPLC system prominence LC-20AP (Shimadzu, Kyoto, Japan) equipped with an ELSD-LTII (Shimadzu, Kyoto, Japan) and a fraction collector FRC-10A (Shimadzu, Kyoto, Japan). LC-HRMS analyses were performed on a Thermo Scientific™ Dionex™ UltiMate™ 3000 thermostated column compartment rapid separation (TCC-3000RS) LC system (Thermo Fisher Scientific, Waltham, USA) coupled to ultra-high resolution Orbitrap Elite hybrid mass spectrometer (Thermo Fisher Scientific, Waltham, USA) equipped with electrospray ionization (ESI) source. Ion exchange column purifications were performed on a Dowex® 1X8-200-400 mesh resin purchased from Acros Organics (Thermo Fisher Scientific, Waltham, USA).
Cloning, expression, and purification
Primers were chosen and genes were cloned with a histidine tag in N-terminal part in a pET22b(+) (Novagen) modified for ligation independent cloning as already described (Bastard et al. 2014). All primers and strains of the 90 selected ROK-type kinases are listed in Table S1 and named from KIN1 to KIN90. All the strains along with their identifiers were purchased from DSMZ, CIP, or ATCC collections. When DNA samples corresponding to the gene encoding the selected enzyme was not available, PCR was performed on the DNA of another strain from the same species as noted in Table S1. All sequences were verified. In these cases, when the protein sequence or the strain is not identical to that of the selected enzyme, the sequence was deposited to the European Nucleotide Archive (ENA) with ENA id listed in Table S1. Assigned Uniprot id are A0A2K4X4D6, A0A2K4X4G8, A0A2K4X4D1, A0A2K4X4E5, A0A2K4X4D2, A0A2K4X4E4, A0A2K4X4I2, and A0A2K4X4C6. Each expression plasmid was transformed into Escherichia coli BL21-CodonPlus (DE3)-RIPL. Cell culture, induction of protein production, and cell lysis were conducted as previously published (Bastard et al. 2014). Enzymes were purified in 96 microwells using HisLink™ 96 protein purification system (Promega). The elution buffer was 50 mM glycylglycine (pH 7.5), 50 mM NaCl, 250 mM imidazole, and 10% glycerol. Samples were analyzed by SDS-PAGE using the NuPAGE system (Invitrogen), see Table S2. Large-scale purification of ScrKmitis was conducted from a 400-mL culture as described elsewhere (Kreimeyer et al. 2007). The storage buffer was Tris pH 8, 50 mM, NaCl 50 mM, 1 mM DTT, and glycerol 15%. Purified enzymes were stored at − 80 °C. The sample was analyzed by SDS-PAGE using the NuPAGE system. Protein concentration was determined by the Bradford method, with bovine serum albumin as the standard (Bio-Rad). Size exclusion chromatography was conducted on Superdex using a 200 10/300GL increase column (GE Healthcare Life Sciences).
Enzymatic screening assay
General screenings conducted in 384-microwell plates were done by spectrophotometry analysis (SpectraMax Plus384 (Molecular Devices, Sunnyvale, USA)) at 25 °C in a final volume of 70 μL. ATP-dependent activities for phosphorylation were determined by monitoring the consumption of the NADH spectrophotometrically at A340 using the coupled assay (Jaworek and Welsch 1985) employing pyruvate kinase (PK) and lactate dehydrogenase (LD) according to Beer-Lambert’s law and molar absorptivity of β-NADH (ɛ = 6220 M−1 cm−1). The assay mixture comprised 30 mM substrate, 0.2 mM NADH, 2.5 mM ATP, 5 mM MgCl2, 5 mM PEP, 1 U PK, 2.3 U LD, and 50 mM glycylglycine buffer pH 7.5. The reaction was initiated by addition of 10 μL of purified enzyme (average of 500 μg/mL). The background noise was estimated and removed for each enzyme with a supplemental microplate similarly treated but with a mixture lacking hexose substrate.
ROK signatures analysis
Pairwise sequence alignments were performed using Clustal Omega website (https://www.ebi.ac.uk/Tools/msa/clustalo/). The 61 ROK-type kinases and the ROK-type sugar kinases from the literature (Table S3, S4 and S5) were used for the alignment analysis. ROK signatures were searched according to Conejo’s work (Conejo et al. 2010).
Phylogenetic analyses
A careful inventory of “known” ROK-type sugar kinases was done searching as exhaustive as possible fructo-, glucose-, and hexo-kinases described in the literature and related to experimental data. Using Swissprot database information’s, only proteins sharing the Interpro signature IPR000600 specific of ROK family were selected and listed in Table S4 and S5. These ROK kinases were aligned together with the 61 kinases of this study. Pairwise sequence alignments were calculated with gapped BLASTp and the BLOSUM62 scoring matrix. Phylogenetic analysis was performed using MAFFT (Katoh et al. 2002) for multiple sequence alignment, QUICKTREE (Howe et al. 2002) for tree building, and iTOL (Letunic and Bork 2016) for tree rendering. All the UniprotKB identifiers of the enzyme are reported in the phylogenetic trees. Specific activities (Table S3) of the 61 kinases were quantitatively symbolized and reported in the phylogenetic tree. ROK-type sugar kinases from the literature have been included and the activities described in their corresponding article (Table S4 and Table S5) were qualitatively reported in the phylogenetic tree. In addition, 3 known ROK-type repressors (XylR from Bacillus subtilis (Kreuzer et al. 1989) and Staphylococcus xylosus (Sizemore et al. 1991) and NagC from E. coli (Plumbridge 2001) were added. Respective Uniprot id were P16557, P27159, and P0AF20.
Enzymes thermotolerance
Volumes of 200 μL of cell crude extracts in 96 microwells were incubated at 70 °C for 20 min in thermocycler. The microplates were centrifuged at 4000 rpm for 20 min in a centrifuge 5810R (Eppendorf). Supernatants were transferred in a new microplate, analyzed by SDS-PAGE using the E-PAGE High-Throughput Gel system (Invitrogen), and specific activity measured. Protein concentrations were determined by the Bradford method, with bovine serum albumin as standard (Bio-Rad). ATP-dependent activities for phosphorylation of d-fructose, d-tagatose, and d-glucose were determined by monitoring the consumption of the NADH spectrophotometrically as described for the enzymatic screening assay.
Characterization of ScrKmitis
All specific activities and kinetic parameters were determined from duplicate experiments by monitoring the consumption of the NADH spectrophotometrically as described for the enzymatic screening assay. All reactions (except when notified) were performed on purified ScrKmitis at 25 °C in 100-μL scale using microcuvettes.
-
Protocol A: The assay mixture (100 μL) comprised 50 mM hexose (except for d-fructose 2.5 mM), 0.2 mM NADH, 2.5 mM ATP, 5 mM metal ion, 5 mM phosphoenolpyruvate (PEP), 1 U PK, 2.3 U LD, and 50 mM glycylglycine buffer pH 7.5. The initial slope measured at 340 nm for 10 min after addition of purified ScrKmitis determined the specific activity of the enzyme.
-
Protocol B: Protocol carried out in end-point to ensure the independence of the pyruvate kinase and lactate dehydrogenase assay system previously validated between pH 6.5 and 9 (data not shown). Assay mixture was composed of 2.5 mM d-fructose, 2.5 mM neutralized ATP, 5 mM MgCl2, and 50 mM specified buffer at specified pH. To 100 μL of the assay mixture preincubated 3 min at the specified temperature was added purified ScrKmitis and the reaction was stirred at 400 rpm for 2 min at the specified temperature. After acidic quench to pH = 1 (HCl 4–10 M), neutralization to pH 6.5–9 (KOH 4–10 M), the ADP formed was assayed by pyruvate kinase and lactate dehydrogenase system (0.2 mM NADH, 1 U PK, 5 mM neutralized PEP, 2.3 U LD) by measuring the difference in absorbance at 340 nm after total consummation of ADP.
-
(i)
Specific activities of ScrKmitis toward various substrates. Specific activities of ScrKmitis were determined by using protocol A with MgCl2 as metal ion. Tested substrates were (d- and l-form) allose, altrose, glucose, mannose, gulose, idose, galactose, talose, psicose, fructose, sorbose, tagatose, arabinose, erythrose, and threose. In addition, d-ribose, d-xylose, d-lyxose, d-xylulose, l-erythrulose, d-cellobiose, and N-acetyl-d-glucosamine were also assayed.
-
(ii)
Thermotolerance of ScrKmitis. Thermotolerance of ScrKmitis was confirmed with a cell crude extract obtained as already described (Kreimeyer et al. 2007) from 100 mL of culture. A volume of 8 mL of cell crude extract was incubated 20 min at 70 °C and centrifuged at 8000 rpm 30 min. The clear cell crude extract was analyzed by SDS-PAGE using the Nu-PAGE system (Invitrogen). Protein concentrations were determined by the Bradford method, with bovine serum albumin as standard (Bio-Rad). Specific activity of ScrKmitis was measured with protocol A for d-fructose, d-mannose, and d-glucose.
-
(iii)
Specific activity of ScrKmitis versus pH/buffer solution. The protocol B was used at room temperature with the following buffers: MES buffer (pH 4.5–6.8), TRIS HCl buffer (pH 6.8–9.6), glycylglycine NaOH buffer (pH 7.0–12.4), TRIS NaOH buffer (pH 11.0–12.4).
-
(iv)
Specific activity of ScrKmitis versus temperature. The protocol B was used in glycylglycine buffer pH 7.5 at 15, 20, 30, 40, 45, 50, 55, 60, 65, 70, and 80 °C.
-
(v)
Specific activity of ScrKmitis versus metal ions: the protocol A was used with 2.5 mM of d-fructose. Tested inorganic salt at 5 mM were MgCl2, MnCl2, CoCl2, NiCl2, ZnCl2, CuCl2, or CaCl2.
-
(vi)
Determination of kinetic parameters of ScrKmitis. Kinetic constants were determined on purified ScrKmitis by using the spectrophotometric coupled assay employing pyruvate kinase and lactate dehydrogenase system. Assay mixture was composed of 0.25 mM NADH, 2.5 mM ATP, 5 mM MgCl2, 5 mM PEP, 1 U PK, 2.3 U LD, and 50 mM glycylglycine buffer pH 7.5. The kinetic constants for ATP were determined with 0.25 mM NADH, 10 mM d-fructose, 5 mM MgCl2, 5 mM PEP, 1 U PK, 2.3 U LD, and 50 mM glycylglycine buffer pH 7.5. The following substrate concentrations were used: ATP from 0.005 to 2.5 mM, d-fructose from 0.1 to 10 mM, d-altrose from 1 to 100 mM, d-mannose from 0.01 to 20 mM, d-glucose from 5 to 400 mM, l-sorbose from 5 to 500 mM, d-sorbose from 10 to 1200 mM. 100 μL of the assay mixture was preincubated until the complete consumption of ADP present in commercial ATP, followed by addition of ScrKmitis. Kinetic parameters were determined by fitting data by non-linear analysis of initial rates using SigmaPlot version 9.0 (Systat Software).
-
(vii)
ATPase activity of ScrKmitis. The ATPase activity was determined by using the spectrophotometric coupled assay employing pyruvate kinase and lactate dehydrogenase system. Assay mixture was 0.25 mM NADH, 5 mM MgCl2, 5 mM PEP, 1 U PK, 2.3 U LD and 50 mM glycylglycine buffer pH 7.5, and 2.5 mM ATP. 100 μL of the assay mixture was preincubated until the complete consumption of ADP present in commercial ATP, followed by addition of ScrKmitis. Determination of inorganic phosphate in the enzymatic reaction in presence and absence of D-fructose was followed with the colorimetric assay with molybdate. Assay mixture was composed of 50 mM glycylglycine buffer pH 7.5, 5 mM MgCl2, 12 μg of purified ScrKmitis, and 2.5 mM of ATP in a final volume of 200 μl. In the reaction with hexose, 2.5 mM of D-fructose was added. Control was done without adding ScrKmitis. Aliquot of 50 μl was withdrawn at 0, 15, and 30 min and added to 150 μl of buffer (50% acetone, 25% sulfuric acid 5 M, and 25% ammonium molybdate 10 mM) and 10 μl of 1 M citric acid. The absorbance was immediately measured at 355 nm.
HPLC conditions
HPLC-ELSD analyses were conducted on a Zic-pHilic (Merck, Darmstadt, Germany) column (4.6 × 150 mm; 5 μm) with the following conditions: mixture of NH4HCO2 20 mM pH 3.6/MeCN as eluent with a linear gradient (ratio 20/80 to 60/40 in 15 min, then 60/40 during 5 min); flow 0.5 mL min−1; temperature 30 °C; injection volume 10 μL. ELSD parameters Teva = 70 °C; Tneb = 40 °C; attenuation = 5.
Semi-preparative HPLC purifications were conducted on a Zic-Hilic (Merck, Darmstadt, Germany) column (250 × 10 mm; 5 μm) with the following conditions: mixture of NH4AcO 10 mM pH 7.7/MeCN as eluent with a linear gradient (ratio 20/80 to 60/40 in 15 min, then 60/40 during 5 min); flow 4 mL min−1. ELSD parameters: T = 70 °C; gain = 5: filter = 1 s.
LC-HRMS analyses were performed on a Zic-pHilic (Merck, Darmstadt, Germany) column (4.6 × 150 mm; 5 μm) with the following chromatographic conditions: mixture of (NH4)2CO3 10 mM pH 9.9 (adjusted with NH4OH)/MeCN as eluent with a linear gradient (20/80 for 2 min, 20/80 to 60/40 in 20 min, then 60/40 during 8 min); flow 0.5 mL min−1; temperature 40 °C; injection volume 10 μL. The Orbitrap Elite mass spectrometer was operated in the ESI negative ion mode. For LC-MS and MS/MS (collision induces dissociation, CID) experiments, the resolving power was set at 60000 m/∆m (full width at half maximum, FWHM at m/z = 400). Ion spray (IS) was set at − 4.2 kV and capillary temperature at 275 °C. Sheath gas and auxiliary gas flow rates were set at 60 and 50 arbitrary units (a. u.), respectively. Mass spectra were acquired over an m/z range from m/z 50 up to m/z 1000. Raw data were analyzed using the Qual-browser module of Xcalibur version 2.2 (Thermo Fisher Scientific, Waltham, USA).
Biocatalytic synthesis of phosphorylated hexoses
Conditions A
In a Greiner tube (15 mL) with a screw cap was poured 7.5 mL of a media containing 50 mM of TRIS HCl buffer (375 μL of a 1 M solution at pH 8.1), 20 mM of hexose (150 μmol, 1500 μL of a 100 mM solution), 0.5 mM of ATP (3.75 μmol, 37.5 μL of a 100 mM solution at pH 6), 5 mM of MgCl2 (37.5 μmol, 375 μL of a 100 mM solution), 25 mM of PEP (187.5 μmol, 750 μL of a 250 mM solution at pH 6), 15 U of PK, and the specified quantity of purified ScrKmitis. The reaction mixture was stirred at 400 rpm at 30 °C for the indicated period of time then lyophilized overnight.
Conditions B
In a Greiner tube (50 mL) with a screw cap was poured 15 mL of a media containing 50 mM of TRIS HCl buffer (750 μL of a 1 M solution at pH 8.1), 10 mM of hexose (150 μmol, 1500 μL of a 100 mM solution), 10 mM of ATP (150 μmol, 1500 μL of a 100 mM solution at pH 6), 5 mM of MgCl2 (75 μmol, 75 μL of a 1 M solution), 25 mM of PEP (375 μmol, 1500 μL of a 250 mM solution at pH 6), 150 U of PK, and the specified quantity of purified ScrKmitis. The reaction mixture was stirred at 400 rpm at 30 °C for the indicated period of time then freeze-dried overnight.
Purification by ion exchange chromatography
The freeze-dried product was dissolved in NH4HCO3 buffer 0.012 M and purified by ion exchange chromatography using a Dowex (1X8-200-400 mesh, HCO3−) resin with a non-linear gradient of various NH4HCO3 concentrations: 0.012 M, 0.03 M, 0.06 M, 0.1 M, 0.2 M, 0.3 M, 0.4 M, and 0.6 M (2 vol columns). Fractions containing the compound (HPLC-ELSD analyses 50 μL of eluting fractions +50 μL of HPLC eluent NH4HCO2 20 mM pH 3.6/MeCN 8/2) were combined and freeze-dried to afford the phosphorylated hexose. Three successive freeze-drying (solid recovery, dissolution in water) were necessary to ensure the complete removal of buffer.
Purification by semi-preparative HPLC
The freeze-dried product was dissolved in HPLC eluent NH4AcO 10 mM pH 7.7/MeCN 8/2) and injected on Zic-Hilic semi-preparative column (injection volumes 2–2.5 mL). Positive fractions (ELSD detection) were combined and freeze-dried to afford the phosphorylated hexose. Three successive freeze-drying (solid recovery, dissolution in water) were necessary to ensure the complete removal of buffer.
d-(+)-altrose-6P: The reaction was conducted under conditions A with 0.53 mg of ScrKmitis in 10 h and the product purified by ion-exchange chromatography. Isolated yield: 57% (25 mg of white solid). NMR 13C was identical to the one found in the literature (Franke et al. 1985). HRMS ESI−, m/z calcd. for [C6H12O9P]− = 259.02244; found 259.02214.
d-(+)-glucose-6P: The reaction was conducted under conditions A with 0.38 mg of ScrKmitis in 8 h and the product purified by ion-exchange chromatography. Isolated yield: 56% (22 mg of white solid). Present forms: α-pyranose (35%) and β-pyranose (65%). 1H NMR (300 MHz, D2O) δ 5.12 (d, J = 3.63 Hz, H1α),4.54 (d, J = 7.26 Hz, H1β), 4.00–3.85 (m, H6, H2, H4), 3.85–3.76 (m, H5), 3.62 (m, H3), 3.48 (m, H6), 3.45–3.35 (m, H3, H4), 3.61 (dd, J = 9.21 and 7.99 Hz); 13C NMR (75 MHz, D2O) δ 96.3 (C1β), 92.4 (C1α), 75.6 (C3α), 75.4 (d, J = 7.55 Hz, C5β), 74.4 (C2β), 72.6 (C3α),71.8 (C2α), 71.1 (d, J = 7.71 Hz, C5α), 69.3 (C4), 63.4 (d, J = 4.48 Hz, C6 α and β) (Fig. S5). HRMS ESI−, m/z calcd. for [C6H12O9P]− = 259.02244; found 259.02192.
d-(+)-mannose-6P: The reaction was conducted under conditions A with 0.04 mg of ScrKmitis in 15 h and the product purified by ion-exchange chromatography. For practical reason, the reaction was carried out overnight but the conversion was > 95% after 2 h 30 on 100 μL scale. Isolated yield: 77% (30 mg of white solid). Present forms: α-pyranose (66%) and β-pyranose (34%). 1H NMR (300 MHz, D2O) δ 5.03 (s, H1α), under H2O (H1β), 3.93–3.85 (m, H6), 3.79–3.74 (m, H2), 3.74–3.62 (m, H3α, H4α, H5α), 3.62–3.49 (m, H4β, H3β), 3.28 (m, H5β);13C NMR (75 MHz, D2O) δ 94.4 (C1α), 94.1 (C1β), 75.6 (d, J = 7.69 Hz, C5β), 72.8 (C3β), 72.0 (d, J = 7.36 Hz, C5α), 71.5 (C2β), 70.9 (C2α), 70.1 (C3α), 66.5 (C4α), 66.2 (C4β), 63.1 (d, J = 4.34 Hz, C6αβ) (Fig. S6). HRMS ESI−, m/z calcd. for [C6H12O9P]− = 259.02244; found 259.02194.
d-(+)-talose-6P: The reaction was conducted under conditions A with 0.53 mg of ScrKmitis in 7 h and the product purified by ion-exchange chromatography. Isolated yield: 77% (30 mg of white solid). Present forms: α-pyranose (41%), β-pyranose (36%), α-furanose (13%) and β-furanose (10%). 1H NMR (600 MHz, D2O) δ 5.27 (d, J = 4.41 Hz, H1’β),5.17 (d, J = 1.62 Hz, H1α), 5.14 (m, H1’α), 4.73 (d, J = 1.08 Hz, H1β), 4.36 (m, H3’β), 4.27 (m, H3’α), 4.10 (m, H5’α, H5α), 4.01–4.05 (m, H4’β, H2’β), 3.91 (m, H2’α, H3α), 3.87 (m, H4’α, H6αβ), 3.82–3.84 (m, H4αβ, H6’β), 3.78 (m, H5’β, H2β, H6’α), 3.73 (m, H2α), 3.70 (m, H3β), 3.65 (m, H5β); 13C NMR (150 MHz, D2O) δ 103.0 (C1’α), 96.4 (C1’β), 94.7 (C1β), 94.1 (C1α), 82.6 (C4’β), 82.1 (C4’α), 75.2 (C2’α), 74.3 (d, J = 7.67 Hz, C5β), 71.5 (C2β), 71.0 (d, J = 7.12 Hz, C5’α), 70.9 (C3’β), 70.8 (C2’β), 70.6 (C2α), 70.4 (C3’α), 70.1 (d, J = 7.30 Hz, C5’β), 70.0 (d, J = 7.64 Hz, C5α), 69.0 (C3α), 68.2 (C3α), 68.0 (C4β), 65.4 (d, J = 4.99 Hz, C6’β), 65.3 (d, J = 4.95 Hz, C6’α), 64.9 (C4α), 63.4 (d, J = 4.38 Hz, C6α), 63.1 (d, J = 4.30 Hz, C6β) (Fig. S7). HRMS ESI−, m/z calcd. for [C6H12O9P]− = 259.02244; found 259.02211.
d-psicose-6P: The reaction was conducted under conditions B with 3 mg of ScrKmitis in 4 days and the product purified by ion-exchange chromatography. Isolated yield: 51% (20 mg of white solid). Present forms: α-furanose (35%) and β-furanose (65%). 1H NMR (300 MHz, D2O) δ 4.37–4.29 (m, H4αβ), 4.19–4.04 (m, H3αβ), 4.00–3.66 (m, H5αβ, H6αβ), 3.55–3.45 (m, H1αβ) (Fig. S8); NMR 13C was identical to the one found in the literature (Koerner Jr et al. 1980). HRMS ESI−, m/z calcd. for [C6H12O9P]− = 259.02244; found 259.02206.
D-fructose-6P: The reaction was conducted under conditions A with 0.04 mg of ScrKmitis in 30 min and the product purified by semi-preparative HPLC. Isolated yield: 72% (28 mg of white solid). Present forms: α-furanose (25%) and β-furanose (75%); 13C NMR was identical to those found in the literature (Sánchez-Moreno et al. 2012).
L-sorbose-5P: The reaction was conducted under conditions A with 1.5 mg of ScrKmitis in 24 h and the product purified by semi-preparative HPLC. Isolated yield: 67% (26 mg of white solid). Present form: α-pyranose (> 95%). 1H NMR (500 MHz, D2O) δ 3.82 (m, H5), 3.71 (dd, J = 11.08 and 5.38 Hz, H6a), 3.62 (t, J = 9.15 Hz, H4), 3.55–3.49 (m, H1a, H6b), 3.39 (d, J = 9.50 Hz, H3), 3.36 (d, J = 11.75 Hz, H1b); 13C NMR (125 MHz, D2O) δ 97.5 (C2), 73.5 (d, J = 3.09 Hz,C4), 72.4 (d, J = 4.40 Hz, C5), 70.4 (C3), 63.6 (C1), 60.9 (d, J = 2.93 Hz, C6) (Fig. S9). HRMS ESI-, m/z calcd. for [C6H12O9P]- = 259.02244; found 259.02208.
D-tagatose-6P: The reaction was conducted under conditions A with 0.38 mg of ScrKmitis in 2 h and the product purified by semi-preparative HPLC. Isolated yield: 51% (20 mg of white solid). Present forms: α-furanose (25%) and β-furanose (75%). 1H NMR (300 MHz, D2O) δ 4.37–4.30 (m, H4αβ), 4.20 (m, H3αβ), 4.12–3.82 (m, H5αβ, H6αβ), 3.61 (d, J = 4.29 Hz, H1α), 3.51 (d, J = 3.99 Hz, H1β) (Fig. S10); 13C NMR was identical to the one found in the literature (Koerner Jr et al. 1980). HRMS ESI-, m/z calcd. for [C6H12O9P]- = 259.02244; found 259.02211.
Results
Screening of a kinase collection
A kinase collection was built by a sequence driven approach (Vergne-Vaxelaire et al. 2013) using the fructokinase FrK from Zymomonas mobilis (Weisser et al. 1996; Zembrzuski et al. 1992) as reference. Frk is the unique ROK-type fructokinase (Uniprot ID: Q03417) with enzymatic characterization done on purified enzyme (Table S4) showing moderate activity toward d-mannose in addition to d-fructose. Since little is known about the substrate scope of ROK-type fructokinase enzymes, thus, exploration of their substrate specificity toward sugars is of great interest.
A collection of 90 proteins representative of the diversity among bacteria was selected from the UniProt database. From these candidates, 65 corresponding genes were successfully cloned with a poly-histidine tag in an expression vector (Table S1 and Table S2). After overexpression of recombinant genes in Escherichia coli strain BL21, cells were lyzed and proteins purified by nickel affinity chromatography in 96 microwells (Fig. S1). The 63 successfully purified enzymes were then screened against 4 ketohexoses (d-fructose, d-tagatose, d-sorbose, and d-psicose) and 3 aldohexoses (d-glucose, d-mannose, and d-galactose). The kinase activity of each candidate protein was assessed in 384-microplates by spectrophotometrically monitoring at 340 nm, the consumption of the NADH using the coupled generic assay employing pyruvate kinase, and lactate dehydrogenase (Scheme S1). Sixty-one enzymes very diverse in term of protein sequence (Fig. S11) were found to catalyze the formation of at least one phosphorylated hexose. In our conditions, no activity could be detected toward D-galactose. Two enzymes weakly purified showed no activity against any hexose (no activity could be detected for C5 and C4 monosaccharides too, data not shown). These kinases could be classified in two groups according to their substrate specificity: a small group, gathering proteins active toward d-glucose and a group, bringing together most of the proteins (55 out of 61), exhibiting, at least, activities toward both d-mannose and d-fructose (Fig. 1a). Noteworthy among them, 42 kinases exhibited such a high activity (> 50 U/mg) toward d-mannose and d-fructose that it was not possible to determine their specific activity in the generic screening conditions. Moreover, most of these kinases were also active toward d-tagatose and some of them had low activity toward l-sorbose or d-psicose (Table S3). It may be noted that, in our conditions, most of the fructo- and glucokinases were weakly active toward d-glucose and d-fructose, respectively.

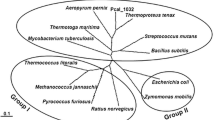
Phylogenetic tree with the 61 ROK-type kinases compared to known ROK-type proteins. Activity of known ROK-type sugar kinases from the inventory (Table S4 and Table S5) are noted with gene name put in parenthesis next to Uniprot identifier. Their activities toward substrates described in the corresponding publications have been symbolized qualitatively with a star and mostly have not been tested in this study. Experimentally data from this study are symbolized by squares whose size is categorized according to specific activities (Table S3 and Table S6). Frk used as reference set for the sequence driven approach is written in blue. The most promiscuous and efficient enzyme in our conditions of screening is written in red. Dotted rectangle gathers known ROK-type repressors whereas green rectangle gathers fructokinases. a Activity toward d-fructose (F), L-sorbose (S), d-tagatose (T), d-psicose (P), d-glucose (G), d-mannose (M). b Activity toward d-cellobiose (C) and N-acetyl-d-glucosamine (N). Bootstrap values superior to 80 have been noted on the branch of the phylogenetic tree
61 ROK-type sugar kinases
Since Frk from Z. mobilis used as reference protein belongs to ROK-type family, we investigated the ROK conserved sequence features (Conejo et al. 2010) among the 61 kinases. From a literature survey, a careful inventory of sugar kinases was done. For each of these kinases, the conserved Interpro domain IPR000600 specific of ROK family was searched allowing the identification of 29 ROK-type sugar kinases related to a publication but not always biochemically characterized. The resulting glucokinases, allokinases, ATP-glucomannokinases, β-glucoside kinases, N-acetyl-d-glucosamine kinases, and fructokinases (listed in Table S4 and Table S5) were added to the multiple sequence alignment of the 61 kinases.
First, the two glycine-rich ROK consensus motifs corresponding to ExGH and a Zn-binding motif (Conejo et al. 2010) were found in all experimentally tested kinases of this study confirming their belonging to ROK family (Fig. S12). Nevertheless, we observed that the ATP/Mg2+ binding site (DXGXT), described as common to ROK-type kinases (Holmes et al. 1993) was shared as expected by glucokinases, allokinases, ATP-glucomannokinases, β-glucoside kinases, and N-acetyl-d-glucosamine kinases but not by fructokinases (Fig. S13). For this sub-family, glutamate is always substituted by aspartate in their ATP/Mg2+ binding site (EXGXT) confirming the predictions based on structural analysis (Conejo et al. 2010). In addition, we also observed that the Thr/Pro/Lys triad was conserved among the fructokinases and absent within other ROK-type sugar kinases (Fig. S14) partitioning clearly these families, corroborating the hypothesis formulated by (Conejo et al. 2010).
To evaluate the distribution of these 61 sugar kinases among the whole ROK family, our experimental data were mapped on a phylogenetic tree (Fig. 1a) built with the 61 characterized kinases of this study supplemented with the known ROK-type kinases (see the inventory in Table S4 and Table S5) and ROK-type repressors (two XylR (Kreuzer et al. 1989; Sizemore et al. 1991) and NagC (Plumbridge 2001)). Glucokinases, including the ones found in this study, are grouped together with the ROK-type repressors, forming a group significantly distinct to the one of the fructokinases. Actually, the 4 known fructokinases (ScrK(Sato et al. 1993), FrK (Scopes et al. 1985) GmuE (Nocek et al. 2011) and Mak (Sproul et al. 2001)) and the experimentally proved fructokinases are not significantly phylogenetically related to other ROK-type sugar proteins. Notably, glucokinases are found clusterized with kinases described to be β-glucoside kinases BglK (Thompson et al. 1992) and N-acetyl-glucosamine kinases NagK (Uehara and Park 2004). In order to test the ability of the sugar kinase collection to phosphorylate glucose derivatives, ATP-dependent activities for phosphorylation of d-cellobiose and N-acetyl-d-glucosamine were assayed (Table S6 and Fig. 1b). The results showed that only two glucokinases were active toward d-cellobiose whereas 5 glucokinases were active toward N-acetyl-d-glucosamine. In our conditions, no fructokinase was found significantly active toward these derivatives.
Guided by potential biotechnological applications of these promiscuous 61 kinases for the synthesis of rare sugars, enzyme thermotolerance was assessed. Enzyme thermotolerance was determined by incubation of cell crude extract in 96-microplates at 70 °C for 20 min. Thirty-six percent of expressed enzymes were significantly visible to the eye on SDS gel, suggesting their thermotolerance (Fig. S2). ATP-dependent activities for phosphorylation of d-fructose, d-tagatose, and d-glucose were determined as previously described (Fig. S4). Twenty-three kinases showed no significant loss of activity (Fig. 1c and Table S7). From these thermotolerant promiscuous kinases, one enzyme from Streptococcus mitis (Uniprot id: E1M3M4) was selected for its ability to phosphorylate most of the tested hexoses.
Purification and biochemical properties of E1M3M4 from Streptococcus mitis NCTC 12261 (DSM 12643)
E1M3M4 shares 72% of identity with the fructokinase ScrK from Streptococcus mutants (ScrKmutants, Uniprot ID: Q07211) involved in the sucrose transport through a phosphoenolpyruvate-dependent phosphotransferase system (PTS) (Sato et al. 1993). An apparent Vmax for d-fructose of 20.0 pmol/min/mg of protein was reported on cell crude extract by (Sato et al. 1993). In addition, the genes encoding for E1M3M4 and ScrKmutants share a similar genomic context (Fig. S15) including a predicted sucrose enzyme II (ScrA) and sucrose-6P hydrolase (ScrB) (Sato et al. 1989) usually involved in disaccharide uptake by phosphoenolpyruvate-dependent phosphotransferase system (PTS system) (Postma et al. 1993). Based on these features, E1M3M4 was named ScrKmitis. Purified ScrKmitis was obtained by nickel affinity chromatography purification (47.5 mg from 100 mL of culture). SDS gel showed a single band corresponding to a calculated molecular mass of 32.8 kDa (Fig. S3). Size exclusion chromatography indicated that ScrKmitis is a homodimer, as previously reported for the fructokinase of Zymomonas mobilis (FrKmobilis) (Scopes et al. 1985). Biochemical properties of this purified enzyme were as follows.
-
(i)
Substrate specificity
To determine the substrate range of ScrKmitis, specific activities were calculated for all hexoketoses and hexoaldoses (Table 1) in the d- and l- series and some C4 and C5 monosaccharides. ScrKmitis showed an unprecedented substrate scope with high activities toward d-fructose (287 U mg−1) and d-mannose (395 U mg−1) (Table 1, entry 10 and 4). Significant specific activities (> 0.1 U mg−1 of protein) were measured for all natural hexoses except d-galactose. ScrKmitis phosphorylated also rare sugars, like for d-psicose, d-allose, and d-gulose, with low specific activities (< 0.1 U mg−1 of protein) (Table 1, entries 9, 1, and 5 respectively). Unnatural monosaccharides were also phosphorylated, d-altrose with high specific activities (105 U mg−1, Table 1 entry 2) and d-talose and d-idose with lower activities (4.7 and < 0.1 U mg−1 respectively, Table 1 entries 8 and 6). Some l-monosaccharides were also phosphorylated such as l-sorbose and l-altrose (Table 1, entries 11 and 2, respectively) even if the activity was generally higher for d-configuration than l-configuration. Moreover, some C4 and C5 monosaccharides revealed to be also substrates, notably d-lyxose (0.16 U mg−1). Traces of activity could be detected for d-erythrose (0.04 U mg−1) and l-erythrulose (0.018 U mg−1). In our reaction conditions d-ribose, d- and l-arabinose, d-xylose, d-xylulose, l-erythrose, d- and l-threose, d-erythrulose were not substrates.
In addition, ScrKmitis showed a low ATPase activity in our conditions (KM = 0.9 mM; kcat/KM = 32 s−1 M−1). Nevertheless, in presence of fructose, no release of Pi was detected using acidified ammonium molybdate assay (Heinonen and Lahti 1981) (Fig. S16). To our knowledge, this feature was only described for sugar kinases from yeast (Rother et al. 2017; Rudolph and Fromm 1971).
-
(ii)
Thermotolerance
To confirm the thermotolerance previously observed in 96-microwells, measurement of enzyme activity after heat treatment was carried out. Heat treatment cell free extract of ScrKmitis at 70 °C for 20 min was then done. Recovered protein mass (42.0 mg from 100 mL of culture) was similar to the one obtained by nickel affinity chromatography purification. SDS gel showed only one band of 32.8 kDa corresponding to the expected mass of ScrKmitis (Fig. S3b) showing that heat treatment can be used as a quick and easy purification method. Specific activities were then measured for d-fructose, d-mannose, and d-glucose, and compared to ones obtained with ScrKmitis purified by nickel affinity chromatography. Loss of activity was estimated between 20 and 30% (Table S8).
-
(iii)
pH and temperature activity profiles
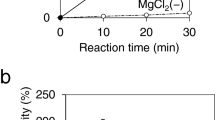
Several parameters influencing the enzymatic activity have been tested. In order to define the optimal pH, specific activities toward d-fructose were determined using various buffer and pH (Fig. 2). Buffers ranging from pH 4.5 to pH 12.4 were used. ScrKmitis displayed a relatively broad pH profile with a maximum activity at basic pH (7 to 10). Activities dropped at pH values below 5.9 and above 11.0.

Effect of pH on the fructokinase activity of ScrKmitis. Error bars represent the standard deviation of three independent experiments
Unlike ROK glucokinases, no ROK fructokinase has been shown to be active at high temperatures. Since ScrKmitis from the mesophilic bacteria Streptococcus mitis was shown to be thermotolerant, we investigated its temperature activity profile (Fig. 3). Assays were carried out toward d-fructose at various temperatures ranging from 15 to 80 °C. Its maximum activity was found at approximately 55 °C, leading to an about two-fold higher activity compared to incubation at room temperature. The activity decreased rapidly at temperature higher than 70 °C but high activity (> 85%) was maintained within a relatively wide temperature range of 40–70 °C.
-
(iv)
Metal ion requirement

Temperature activity profile of ScrKmitis. Error bars represent the standard deviation of three independent experiments
ScrKmitis metal ion activity profile (Fig. 4) conducted with 5 mM of cation and d-fructose as substrate indicated a maximal activity with Mg2+ and Co2+ (relative activity 100 and 98.5%, respectively). The activity remained important with Mn2+ (79.3%) but dropped drastically in the presence of Zn2+, Ca2+, or Ni2+ (24.1, 12.9, and 6.5%, respectively). No activity was detected in presence of 5 mM of Cu2+ or in absence of metal cation.

Effect of the metal ions on the specific activity of ScrKmitis. Error bars represent the standard deviation of three independent experiments
Kinetic constants
In the course of our characterization study, kinetic constants were determined for 8 hexoses for which specific activities were found higher than 0.1 U mg−1 of enzyme (Table 1). Lowest KM were measured for d-mannose and d-fructose with respective values of 150 and 730 μM compared, for example, to 23 mM for d-glucose (Table 2). ScrKmitis was highly effective toward d-mannose and d-fructose (kcat/KM = 1.5 106 and 2.7 105, respectively, Table 2, entries 2 and 3). An about 10-fold lower catalytic efficiency was still recorded for the transformation of the isomer d-altrose (Table 2, entry 4) while it was 1000-fold lower for d-tagatose, d-talose and d-glucose (Table 2, entries 5–7).
Biocatalytic synthesis and characterization of phosphorylated hexoses
To evaluate the ScrKmitis ability for the synthesis of phosphorylated hexoses, biocatalytic transformations of a representative panel of substrates were carried out on a 150-μmol scale. In a first step, the biocatalytic synthesis was developed on d-fructose. Polyphosphate (from 1 to 10 equivalents) was evaluated as low cost phosphate donor but no conversion could be detected. Therefore, ATP was maintained using a recycling system. Regeneration of 0.5 mM of ATP by the PEP/pyruvate kinase system led to a total conversion of 20 mM of substrate in 30 min with only 0.005 mg/mL of enzyme. In the absence of the regeneration system, the conversion reached only 7% in 3 h with one equivalent of ATP or 85% in 5 days with regular addition of ATP (1 eq in total) (data not shown). The corresponding phosphorylated product was purified by semi-preparative HPLC in 72% yield. The same reaction conditions, with the appropriate amount of enzyme ScrKmitis, were applied to the synthesis of the phosphorylated derivatives of aldoses d-altrose, d-glucose, d-mannose, d-talose and ketoses d-psicose, l-sorbose, and d-tagatose. All the phosphorylated products were formed in 30 min to 30 h with at least 90% conversion except for d-psicose for which 4 days with 10 mM of ATP were required to reach 70% conversion (Table 3). The corresponding phosphorylated products were purified by semi-preparative HPLC or ion-exchange chromatography in 51 to 77% yield.
The phosphorylated sugars synthetized were all fully characterized by mass spectrometry and NMR spectroscopy (Fig. 5). The regioselectivity of the hexose phosphorylation catalyzed by ScrKmitis, was determined by NMR. Considering the literature, phosphorylation was expected in position 6 (Conejo et al. 2010). Indeed, 1H and 13C NMR spectra (Fig. S5–S10) confirmed a C6 regioselectivity of the phosphorylation (Table 3, entries 1–2, 4–8) except for L-sorbose (Table 3, entry 3). A position of phosphorylation elsewhere than in position 6 was also reinforced by a fragmentation in FTMS different from those obtained for the other synthesized phosphorylated hexoses (Fig. S17). The observed coupling for C4 and C5 on 13C NMR spectra suggested a phosphorylation at the 5-position which was confirmed by a 2D 1H-31P NMR analysis showing a coupling between 31P and 1H5 (Fig. S18).

Phosphorylated hexoses biocatalytically synthetized in this study
Discussion
The exploration of the protein natural diversity is one of the most efficient strategies to provide enzymes possessing new or improved properties. In this study, we experimentally identified 61 sugar kinases. These enzymes shared the specific signatures of the ROK family originally built from only few members, signatures which were thereby reinforced by the large set of experimentally tested kinases. Notably, we confirmed that the ATP/Mg2+ binding site of fructokinases was EXGXT and not DXGXT (Conejo et al. 2010) as for other ROK-type sugar kinases. However, the biological function of Mak from E. coli (Uniprot ID: P23917) described as fructokinase (Miller and Raines 2004) could be questioned since it is not clusterized with fructokinases in the phylogenetic tree and its ATP/Mg2+ binding site is DXGXT. These features consolidated the signatures predicted to partition fructokinases from other sugar kinases.
The kinases from this study were active toward various sugars and many of them were found thermotolerant. The combined analysis of the phylogeny including a careful inventory of ROK-type kinases, and our experimental data, revealed that the majority of the 61 sugar kinases from this study are mannofructokinases not related to the already known ROK-type sugar kinases, mostly described as glucokinases (21 out of the 27 known ROK-type sugar kinases according to the inventory Table S4 and Table S5). As underlined by Conejo and co-workers, carbohydrate specificity of individual ROK-type kinase is difficult to assign due to the lack of experimental data (Conejo et al. 2010). In this study, we experimentally assigned the substrate specificity of 20 ATP-dependent fructokinases and 5 ATP-dependent glucokinases previously annotated as ROK family proteins. In addition, we experimentally confirmed the predicted annotations for 23 fructokinases and 3 glucokinases. Finally, function could be assigned without ambiguity for 7 “uncharacterized proteins” revealing 6 mannofrucokinases and 1 glucokinase.
Little is known about the substrate scope of ROK-type kinases. It was generally restricted to d-fructose, d-glucose, and d-mannose. Only few were reported for other hexoses, even if some ROK-type glucokinases with broad substrate specificity were described (Table S5). To address this lack of experimental data, we selected one promiscuous kinase (ScrKmitis) and undertook an in-depth kinetic parameters and substrate spectrum study. For this purpose, we determined its specific activities for all hexoses (aldoses and ketoses d- and l- series) and also some C4 and C5 monosaccharides. ScrKmitis was clearly distinguished from those described in the literature by its high catalytic efficiency toward both d-mannose and d-fructose and was found to be the first purified mannofructokinase deeply characterized from the ROK family (Table 4). FrKmobilis, the only one other purified enzyme with characterized manno- and fructokinase activity, shares 38% of sequence identity with ScrKmitis but showed only moderate activity toward d-mannose compared to ScrKmitis. Indeed, even if their Michaelis constants KM for d-fructose and d-mannose were similar, Vmax of ScrKmitis for D-mannose was 500 times higher (Table 4). Furthermore, its catalytic activity toward other hexoses as d-tagatose and unnatural monosaccharides (d-altrose and d-talose) revealed its large substrate scope mostly on hexoses. As far as we know, such substrate promiscuity was never reported for any ROK-type sugar kinase (Table S4 and Table S5).
In addition, as far as we know, ScrKmitis is the only purified fructokinase encoded by an identified gene for which operating conditions have been so extensively studied (Table 4). ScrKmitis kept more than 80% of activity between pH 7 and 10, and still 60% at pH 6–7 and pH 10–11. Such broader pH range seems not shared by the two fructokinases orphan of genes (Table 4). Its metal ion requirement is in accordance with literature reported for ROK-type kinase, exemplified by the clear visualization of the binding mode between the enzyme and divalent cation in crystal structures (Nishimasu et al. 2007; Nocek et al. 2011). Mg2+, Co2+, and Mn2+ were found more effective on fructokinase activity of ScrKmitis than Zn2+, Ca2+, and Ni2+. This tolerance with regard to a wide range of bivalent metal ions was similar to those reported (Table 4). In addition, despite coming from a mesophilic bacterium, it showed a very broad temperature range (> 85% activity between 40 and 70 °C). Maximum activity around 40–50 °C was reported for kinases from mesophilic bacteria, like hexokinase from the Arthrobacter sp. (Mukai et al. 2003) (45 °C) and glucokinase from Streptomyces peucetius var. caesius (Ramos et al. 2004) (42 °C) but without data above 50 °C reported (Table S5). ScrKmitis seemed to distinguish from these reported enzymes by its conserved activities at 60–70 °C and from kinases from thermophilic organisms, which generally exhibit a restricted maximum activity temperature range (70 °C to 100 °C) (Dorr et al. 2003; Hansen et al. 2002; Qian et al. 2013). Interestingly, ScrKmitis thermotolerance allowed its purification by a simple cell crude extract heat treatment with however 20 to 30% loss of activity. By comparison, ScrK from Lactobacillus lactis is completely inactivated by heating treatment within 3 min at 65 °C (Thompson et al. 1991). It is noteworthy that from an industrial point of view, enzymatic processes at elevated temperature offer many advantages, such as higher reaction rate, improved solubility of organic substrates, and higher tolerance toward nonconventional media (Choi et al. 2015; Elleuche et al. 2014). In addition, thermostable enzymes offer better resistance against protein destabilizing factors during protein engineering processes (Littlechild 2015).
We demonstrated the biocatalytic applicability of this sugar kinase from Streptococcus mitis by synthesizing 8 phosphorylated hexoses through simple ATP-dependent phosphorylation and in situ regeneration of ATP by PEP/PK system. All the substrates were regioselectively phosphorylated in position 6 except l-sorbose which gave the 5-derivative. One hypothesis could be that the reacting conformational form in the active site is the pyranose one which eliminates any possibility of phosphorylation in position 6, supported by structures of gluco- and fructokinases showing closed conformational forms of the substrates in the active sites (Nishimasu et al. 2007; Nocek et al. 2011). In addition, Balieu and collaborators have observed the preferred pyranose form in water for L-sorbose (Zebiri et al. 2011). We observed a trend that the enzyme better converts aldoses with S-configuration at position 2 (α- or β –pyranose configuration) with preferentially R configuration at positions 4 (specific activity(D-altrose and D-mannose) > specific activity(D-talose)), as well as ketoses with S configuration at position 3, with preferentially R configuration at position 4 (specific activity(D-fructose) > specific activity(D-tagatose)).
All these results revealed ScrKmitis as an “easy enzyme” (Bornscheuer 2016), efficient and versatile biocatalyst for the synthesis of phosphorylated sugars, compounds of particular interest in the study of various metabolic diseases (Wohlgemuth et al. 2017).
References
Bastard K, Smith AA, Vergne-Vaxelaire C, Perret A, Zaparucha A, De Melo-Minardi R, Mariage A, Boutard M, Debard A, Lechaplais C, Pelle C, Pellouin V, Perchat N, Petit JL, Kreimeyer A, Medigue C, Weissenbach J, Artiguenave F, De Berardinis V, Vallenet D, Salanoubat M (2014) Revealing the hidden functional diversity of an enzyme family. Nat Chem Biol 10(1):42–49. https://doi.org/10.1038/nchembio.1387
Bornscheuer UT (2016) Biocatalysis: successfully crossing boundaries. Angew Chem Int Ed Eng 55(14):4372–4373. https://doi.org/10.1002/anie.201510042
Charmantray F, Hélaine V, Legeret B, Hecquet L (2009) Preparative scale enzymatic synthesis of d-sedoheptulose-7-phosphate from β-hydroxypyruvate and d-ribose-5-phosphate. J Mol Catal B Enzym 57(1–4):6–9. https://doi.org/10.1016/j.molcatb.2008.06.005
Choi JM, Han SS, Kim HS (2015) Industrial applications of enzyme biocatalysis: current status and future aspects. Biotechnol Adv 33:1443–1454. https://doi.org/10.1016/j.biotechadv.2015.02.014
Conejo MS, Thompson SM, Miller BG (2010) Evolutionary bases of carbohydrate recognition and substrate discrimination in the ROK protein family. J Mol Evol 70(6):545–556. https://doi.org/10.1007/s00239-010-9351-1
Davids T, Schmidt M, Böttcher D, Bornscheuer UT (2013) Strategies for the discovery and engineering of enzymes for biocatalysis. Curr Opin Chem Biol 17(2):215–220. https://doi.org/10.1016/j.cbpa.2013.02.022
de Berardinis V, Guérard-Hélaine C, Darii E, Bastard K, Hélaine V, Mariage A, Petit JL, Poupard N, Sánchez-Moreno I, Stam M, Gefflaut T, Salanoubat S, Lemaire M (2017) Expanding the reaction space of aldolases using hydroxypyruvate as a nucleophilic substrate. Green Chem 19:519–526. https://doi.org/10.1039/C6GC02652D
Desvergnes S, Courtiol-Legourd S, Daher R, Dabrowski M, Salmon L, Therisod M (2012) Synthesis and evaluation of malonate-based inhibitors of phosphosugar-metabolizing enzymes: class II fructose-1,6-bis-phosphate aldolases, type I phosphomannose isomerase, and phosphoglucose isomerase. Bioorg Med Chem 20(4):1511–1520. https://doi.org/10.1016/j.bmc.2011.12.050
Dorr C, Zaparty M, Tjaden B, Brinkmann H, Siebers B (2003) The hexokinase of the hyperthermophile Thermoproteus tenax. ATP-dependent hexokinases and ADP-dependent glucokinases, two alternatives for glucose phosphorylation in Archaea. J Biol Chem 278(21):18744–18753. https://doi.org/10.1074/jbc.M301914200
Elleuche S, Schröder C, Sahm K, Antranikian G (2014) Extremozymes—biocatalysts with unique properties from extremophilic microorganisms. Curr Opin Biotechnol 29:116–123. https://doi.org/10.1016/j.copbio.2014.04.003
Franke FP, Kapuscinski M, Lyndon P (1985) Synthesis and n.m.r. characterization of intermediates in the l-type pentose phosphate cycle. Carbohydr Res 143:69–76. https://doi.org/10.1016/S0008-6215(00)90696-7
Guérard-Hélaine C, Debacker M, Clapés P, Szekrenyi A, Hélaine V, Lemaire M (2014) Efficient biocatalytic processes for highly valuable terminally phosphorylated C5 to C9 d-ketoses. Green Chem 16(3):1109–1113. https://doi.org/10.1039/c3gc42140f
Guérard-Hélaine C, de Berardinis V, Besnard-Gonnet M, Darii E, Debacker M, Debard A, Fernandes C, Hélaine V, Mariage A, Pellouin V, Perret A, Petit J-L, Sancelme M, Lemaire M, Salanoubat M (2015) Genome mining for innovative biocatalysts: new dihydroxyacetone aldolases for the chemist’s toolbox. ChemCatChem 7(12):1871–1879. https://doi.org/10.1002/cctc.201500014
Hansen T, Reichstein B, Schmid R, Schönheit P (2002) The first Archaeal ATP-dependent glucokinase, from the hyperthermophilic crenarchaeon Aeropyrum pernix, represents a monomeric, extremely thermophilic ROK glucokinase with broad hexose specificity. J Bacteriol 184(21):5955–5965. https://doi.org/10.1128/jb.184.21.5955-5965.2002
Heinonen JK, Lahti RJ (1981) A new and convenient colorimetric determination of inorganic orthophosphate and its application to the assay of inorganic pyrophosphatase. Anal Biochem 113(2):313–317. https://doi.org/10.1016/0003-2697(81)90082-8
Hélaine V, Mahdi R, Sudhir Babu GV, de Berardinis V, Wohlgemuth R, Lemaire M, Guérard-Hélaine C (2015) Straightforward synthesis of terminally phosphorylated L-sugars via multienzymatic cascade reactions. Adv Synth Catal 357(8):1703–1708. https://doi.org/10.1002/adsc.201500190
Holmes KC, Sander C, Valencia A (1993) A new ATP-binding fold in actin, hexokinase and Hsc70. Trends Cell Biol 3(2):53–59. https://doi.org/10.1016/0962-8924(93)90161-S
Howe K, Bateman A, Durbin R (2002) QuickTree: building huge neighbour-joining trees of protein sequences. Bioinformatics 18(11):1546–1547. https://doi.org/10.1093/bioinformatics/18.11.1546
Jaworek D, Welsch J (1985) Adenosine 5-diphosphate and adenosine 5-monophosphate. In: Bergmeyer HU, Bergmeyer J, Graßl M (eds) Methods Enz Anal, vol 7, pp 365–370
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066
Koerner Jr TA, Voll RJ, Cary LW, Younathan ES (1980) Carbon-13 nuclear magnetic resonance studies and anomeric composition of ketohexose phosphates in solution. Biochemistry 19(12):2795–2801. https://doi.org/10.1021/bi00553a040
Kreimeyer A, Perret A, Lechaplais C, Vallenet D, Medigue C, Salanoubat M, Weissenbach J (2007) Identification of the last unknown genes in the fermentation pathway of lysine. J Biol Chem 282(10):7191–7197. https://doi.org/10.1074/jbc.M609829200
Kreuzer P, Gartner D, Allmansberger R, Hillen W (1989) Identification and sequence analysis of the Bacillus subtilis W23 xylR gene and xyl operator. J Bacteriol 7(171):3840–3845. https://doi.org/10.1021/bi00553a040
Larion M, Moore LB, Thompson SM, Miller BG (2007) Divergent evolution of function in the ROK sugar kinase superfamily: role of enzyme loops in substrate specificity. Biochemistry 46(47):13564–13572. https://doi.org/10.1021/bi700924d
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44(W1):W242–W245. https://doi.org/10.1093/nar/gkw290
Littlechild JA (2015) Enzymes from extreme environments and their industrial applications. Front Bioeng Biotechnol 3:161. https://doi.org/10.3389/fbioe.2015.00161
Miller BG, Raines RT (2004) Identifying latent enzyme activities: substrate ambiguity within modern bacterial sugar kinases. Biochemistry 43(21):6387–6392. https://doi.org/10.1021/bi049424m
Mukai T, Kawai S, Matsukawa H, Matuo Y, Murata K (2003) Characterization and molecular cloning of a novel enzyme, inorganic polyphosphate/ATP-glucomannokinase, of Arthrobacter sp. strain KM. Appl Environ Microbiol 69(7):3849–3857. https://doi.org/10.1128/aem.69.7.3849-3857.2003
Nishimasu H, Fushinobu S, Shoun H, Wakagi T (2007) Crystal structures of an ATP-dependent hexokinase with broad substrate specificity from the hyperthermophilic archaeon Sulfolobus tokodaii. J Biol Chem 282(13):9923–9931. https://doi.org/10.1074/jbc.M610678200
Nocek B, Stein AJ, Jedrzejczak R, Cuff ME, Li H, Volkart L, Joachimiak A (2011) Structural studies of ROK fructokinase YdhR from Bacillus subtilis: insights into substrate binding and fructose specificity. J Mol Biol 406(2):325–342. https://doi.org/10.1016/j.jmb.2010.12.021
Plumbridge J (2001) DNA binding sites for the Mlc and NagC proteins: regulation of nagE, encoding the N-acetylglucosamine-specific transporter in Escherichia coli. Nucleic Acids Res 29(2):506–514. https://doi.org/10.1093/nar/29.2.506
Postma PW, Lengeler JW, Jacobson GR (1993) Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 3(57):543–594
Qian Z, Zhao J, Bai X, Tong W, Chen Z, Wei H, Wang Q, Liu S (2013) Thermal stability of glucokinases in Thermoanaerobacter tengcongensis. Biomed Res Int 2013:646539. https://doi.org/10.1155/2013/646539
Ramos I, Guzman S, Escalante L, Imriskova I, Rodriguez-Sanoja R, Sanchez S, Langley E (2004) Glucose kinase alone cannot be responsible for carbon source regulation in Streptomyces peucetius var. caesius. Res Microbiol 155(4):267–274. https://doi.org/10.1016/j.resmic.2004.01.004
Rother C, Gutmann A, Gudiminchi R, Weber H, Lepak A, Nidetzky B (2017) Biochemical characterization and mechanistic analysis of the levoglucosan kinase from Lipomyces starkeyi. Chembiochem 19:596–603. https://doi.org/10.1002/cbic.201700587
Rudolph FB, Fromm HJ (1971) A study on the kinetics and mechanism of D-lyxose and D-xylose activation of the adenosine triphosphatase activity associated with yeast hexokinase. J Biol Chem 7(246):2104–2110
Sánchez-Moreno I, Hélaine V, Poupard N, Charmantray F, Légeret B, Hecquet L, García-Junceda E, Wohlgemuth R, Guérard-Hélaine C, Lemaire M (2012) One-pot cascade reactions using fructose-6-phosphate aldolase: efficient synthesis of D-arabinose 5-phosphate, D-fructose 6-phosphate and analogues. Adv Synth Catal 354(9):1725–1730. https://doi.org/10.1002/adsc.201200150
Sato Y, Poy F, Jacobson GR, Kuramitsu HK (1989) Characterization and sequence analysis of the scrA gene encoding enzyme IIScr of the Streptococcus mutans phosphoenolpyruvate-dependent sucrose phosphotransferase system. J Bacteriol 1(171):263–271. https://doi.org/10.1128/JB.171.1.263-271
Sato Y, Yamamoto Y, Kizaki H, Kuramitsu HK (1993) Isolation, characterization and sequence analysis of the scrK gene encoding fructokinase of Streptococcus mutans. J Gen Microbiol 139(5):921–927. https://doi.org/10.1099/00221287-139-5-921
Schorken U (1998) Thiamin-dependent enzymes as catalysts in chemoenzymatic syntheses. Biochim Biophys Acta 1385(E 2):229–243
Scopes RK, Testolin V, Stoter A, Griffiths-Smith K, Algar EM (1985) Simultaneous purification and characterization of glucokinase, fructokinase and glucose-6-phosphate dehydrogenase from Zymomonas mobilis. Biochem J 228(3):627–634
Shaeri J, Wright I, Rathbone EB, Wohlgemuth R, Woodley JM (2008) Characterization of enzymatic D-xylulose 5-phosphate synthesis. Biotechnol Bioeng 101(4):761–767. https://doi.org/10.1002/bit.21949
Sizemore C, Buchner E, Rygus T, Witke C, Gotz F, Hillen W (1991) Organization, promoter analysis and transcriptional regulation of the Staphylococcus xylosus xylose utilization operon. Mol Gen Genet 227(3):377–384. https://doi.org/10.1007/BF00273926
Solovjeva ON, Kochetov GA (2008) Enzymatic synthesis of d-xylulose 5-phosphate from hydroxypyruvate and d-glyceraldehyde-3-phosphate. J Mol Catal B Enzym 54(3–4):90–92. https://doi.org/10.1016/j.molcatb.2007.12.016
Sproul AA, Lambourne LT, Jean-Jacques DJ, Kornberg HL (2001) Genetic control of manno(fructo)kinase activity in Escherichia coli. PNAS 98(26):15257–15259. https://doi.org/10.1073/pnas.211569798
Thompson J, Sackett DL, Donkersloot JA (1991) Purification and properties of fructokinase I from Lactococcus lactis. Localization of scrK on the sucrose-nisin transposon Tn5306. J Biol Chem 33(266):22626–22633
Thompson J, Nguyen NY, Robrish SA (1992) Sucrose fermentation by Fusobacterium mortiferum ATCC 25557: transport, catabolism, and products. J Bacteriol 10(174):3227–3235
Titgemeyer F, Reizer J, Reizer A, Saier MH Jr (1994) Evolutionary relationships between sugar kinases and transcriptional repressors in bacteria. Microbiology 140(Pt 9):2349–2354. https://doi.org/10.1099/13500872-140-9-2349
Uehara T, Park JT (2004) The N-acetyl-D-glucosamine kinase of Escherichia coli and its role in murein recycling. J Bacteriol 186(21):7273–7279. https://doi.org/10.1128/JB.186.21.7273-7279.2004
Vergne-Vaxelaire C, Bordier F, Fossey A, Besnard-Gonnet M, Debard A, Mariage A, Pellouin V, Perret A, Petit J-L, Stam M, Salanoubat M, Weissenbach J, De Berardinis V, Zaparucha A (2013) Nitrilase activity screening on structurally diverse substrates: providing biocatalytic tools for organic synthesis. Adv Synth Catal 355(9):1763–1779. https://doi.org/10.1002/adsc.201201098
Weisser P, Kramer R, Sprenger GA (1996) Expression of the Escherichia coli pmi gene, encoding phosphomannose-isomerase in Zymomonas mobilis, leads to utilization of mannose as a novel growth substrate, which can be used as a selective marker. Appl Environ Microbiol 11(62):4155–4161
Wohlgemuth R (2009) C2-Ketol elongation by transketolase-catalyzed asymmetric synthesis. J Mol Catal B Enzym 61(1–2):23–29. https://doi.org/10.1016/j.molcatb.2009.04.001
Wohlgemuth R, Liese A, Streit W (2017) Biocatalytic phosphorylations of metabolites: past, present, and future. Trends Biotechnol 35(5):452–465. https://doi.org/10.1016/j.tibtech.2017.01.005
Zebiri I, Balieu S, Guilleret A, Reynaud R, Haudrechy A (2011) The chemistry of l-Sorbose. Eur J Org Chem 2011(16):2905–2910. https://doi.org/10.1002/ejoc.201001578
Zembrzuski B, Chilco P, Liu XL, Liu J, Conway T, Scopes R (1992) Cloning, sequencing, and expression of the Zymomonas mobilis fructokinase gene and structural comparison of the enzyme with other hexose kinases. J Bacteriol 11(174):3455–3460
Zhao S, Kumar R, Sakai A, Vetting MW, Wood BM, Brown S, Bonanno JB, Hillerich BS, Seidel RD, Babbitt PC, Almo SC, Sweedler JV, Gerlt JA, Cronan JE, Jacobson MP (2013) Discovery of new enzymes and metabolic pathways using structure and genome context. Nature 502(7473):698–702. https://doi.org/10.1038/nature12576
Acknowledgments
The authors would like to thank Jean-François Gallard (ICSN, France) for 1H-31P NMR experiments, Olek Maciejak and Marie-Jeanne Clément (SABNP, INSERM U 1204—Université d’Evry Val-d’Essonne, Université Paris-Saclay, France) for assistance in 1H and 13C NMR experiments, the Region Ile de France for financial support of the 600 MHz NMR spectrometer, Alain Perret for fruitful discussions and proofreading of the manuscript, and Christine Pelle and Peggy Sirvain for nickel affinity chromatography purification of ScrKmitis. We thank C. Rivoire (the Swiss-Prot Group at the SIB Swiss Institute of Bioinformatics) for earlier Uniprot identifier assignments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 2580 kb)
Rights and permissions
About this article

Cite this article
Vergne-Vaxelaire, C., Mariage, A., Petit, JL. et al. Characterization of a thermotolerant ROK-type mannofructokinase from Streptococcus mitis: application to the synthesis of phosphorylated sugars. Appl Microbiol Biotechnol 102, 5569–5583 (2018). https://doi.org/10.1007/s00253-018-9018-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9018-1