
Abstract
Analysis of the genome sequence of Pyrobaculum calidifontis revealed the presence of an open reading frame Pcal_1127 annotated as ribose-5-phosphate pyrophosphokinase. To examine the properties of Pcal_1127 the coding gene was cloned, expressed in Escherichia coli, and the purified gene product was characterized. Pcal_1127 exhibited higher activity when ATP was replaced by dATP as pyrophosphate donor. Phosphate and EDTA activated the enzyme activity and equivalent amount of activity was detected with ATP and dATP in their presence. Recombinant Pcal_1127 could utilize all the four nucleotides as pyrophosphate donors with a marked preference for ATP. Optimum temperature and pH for the enzyme activity were 55 °C and 10.5, respectively. A unique feature of Pcal_1127 was its stability against temperature as well as denaturants. Pcal_1127 exhibited more than 95 % residual activity after heating for 4 h at 90 °C and a half-life of 15 min in the boiling water. The enzyme activity was not affected by the presence of 8 M urea or 4 M guanidinium chloride. Pcal_1127 was a highly efficient enzyme with a catalytic efficiency of 5183 mM−1 s−1. These features make Pcal_1127, a novel and unique ribose-5-phosphate pyrophosphokinase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ribose-5-phosphate pyrophosphokinase (RPPK, EC 2.7.6.1) catalyzes the transfer of pyrophosphoryl group from ATP to C1 hydroxyl group of ribose-5-phosphate resulting in the production of phosphoribosyl pyrophosphate (PRPP) and AMP. PRPP plays a central role in several processes of life, including the synthesis of nucleotides, co-enzyme NAD+, and amino acids histidine and tryptophan (Hove-Jensen 1988). RPPK has been characterized from all the three domains of life, including eukarya, bacteria, and archaea. Among eukaryotes, it has been studied from Saccharomyces cerevisiae (Hove-Jensen 2004), Ashbya gosypii (Jiménez et al. 2008), Arabidopsis thaliana (Krath et al. 1999), spinach (Krath and Hove-Jensen 2001a), mosquito (Hong et al. 2013), rat (Roth et al. 1974, Kita et al. 1989), and human (Nosal et al. 1993). The bacteria from which RPPK has been investigated include Salmonella typhimurium (Switzer 1969), Escherichia coli (Willemoës et al. 2000), Bacillus subtilis (Arnvig et al. 1990), Bacillus amyloliquefaciens (Zakataeva et al. 2012), and Mycobacterium tuberculosis (Lucarelli et al. 2010). Among archaea, the third domain of life, it has been characterized from Thermococcus kodakarensis (Rashid et al. 1997), Methanocaldococcus jannaschii (Kadziola et al. 2005), Thermoplasma volcanium (Cherney et al. 2011), and Sulfolobus solfataricus (Andersen et al. 2015).
Based upon their properties, RPPKs can be divided into three classes. Class I enzymes are limited to ATP and, in some cases, dATP in their specificity for pyrophosphoryl donors. They are allosterically inhibited either by ADP or GDP or both. Furthermore, they are activated by phosphate, which may be regarded as an allosteric activator as phosphate binding competes with ribonucleoside diphosphate binding at the allosteric site (Willemoës et al. 2000). Class II shows broad pyrophosphoryl donor specificity by accepting GTP, CTP, or UTP (in addition to ATP and dATP). Allosteric regulation has not been detected in this class and their activity is independent of phosphate (Krath and Hove-Jensen 2001a, b). Members of class III do not appear to be allosterically regulated by ribonucleoside diphosphate, but activated by phosphate (Kadziola et al. 2005). Thus, the characteristics of this class are a mixture of those of class I and II.
Here, we report cloning and characterization of an efficient RPPK from the hyperthermophilic archaeon Pyrobaculum calidifontis, a facultative aerobe that grows optimally between 90 and 95 °C (Amo et al. 2002). The complete genome of the microorganism has been determined (http://www.ncbi.nlm.nih.gov/nuccore/CP000561.1). Genome search revealed the presence of an open reading frame Pcal_1127 annotated as ribose-5-phosphate pyrophosphokinase. To get functional information, we expressed the gene encoding Pcal_1127 in E. coli and characterized the gene product.
Materials and methods
Chemicals and materials were purchased either from Sigma Aldrich Co. or Thermo-Fisher Scientific Inc. or Fluka Chemical Corp. Cloning vectors, restriction enzymes and DNA purification kits were purchased from Thermo-Fisher Scientific Inc. Gene-specific oligonucleotides were commercially synthesized from Macrogen Inc. Vectors, pTZ57R/T (Thermo-Fisher), and pET-21a (Novagen, Madison, WI, USA) were employed for cloning and expression purposes, respectively. E. coli cells DH5α and BL21-CodonPlus(DE3)-RIL (Stratagene, La Jolla, CA, USA) were used for cloning and expression purposes.
Gene cloning, expression in E. coli, and purification of recombinant Pcal_1127
RPPK gene, Pcal_1127, from P. calidifontis was amplified by polymerase chain reaction (PCR) using sequence-specific forward, Pcal_1127F (5′ CATATGGACAAAATAAATAGCCGTGCTTTGTATACGCC 3′), and reverse, Pcal_1127R (5′ CTATAGCAGTTTCTCCACCTCTC 3′), primers and genomic DNA of P. calidifontis as template. Recognition site for restriction enzyme NdeI (underlined sequence) was introduced in the forward primer. The PCR-amplified DNA fragment was ligated in cloning vector pTZ57R/T using T4 DNA ligase as recommended by the supplier (Thermo-Fisher Scientific Inc). The resulting plasmid was named as pTZ_1127. Pcal_1127 gene was liberated from pTZ_1127 using NdeI (introduced in the forward primer) and Hind III (from multiple cloning sites of pTZ57R/T) restriction enzymes and cloned in pET-21a expression vector utilizing the same sites. The resulting recombinant plasmid was named as pET_1127.
Escherichia coli BL21-CodonPlus(DE3)-RIL were transformed using pET_1127. The transformed cells were cultivated in Luria–Bertani (LB) medium supplemented with 100 μg/mL ampicillin at 37 °C till an optical density of 0.4–0.5 at 660 nm was reached. The expression of the gene was induced by the addition of isopropyl-β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 0.2 mM. After induction, the cells were allowed to grow for 4–6 h at 37 °C and harvested by centrifugation. Cell pellet (3 g wet weight from 1 L culture) was resuspended in 30 mL of 50 mM Tris–Cl buffer of pH 8 containing 0.2 mM phenylmethylsulfonyl fluoride and β-mercaptoethanol. Cells were then lysed by sonication. Soluble and insoluble fractions were separated and supernatant containing recombinant Pcal_1127 was heated at 80 °C for 25 min to denature the heat-labile proteins of E. coli. The denatured proteins were removed by centrifugation at 20,000×g for 20 min. The supernatant containing Pcal_1127 was loaded onto HiTrap Q anion exchange column.
The proteins were eluted using a linear gradient of 0–1 M NaCl. The fractions having the desired protein were pooled, dialyzed, and loaded onto Resource Q anion exchange column. The proteins were eluted in a similar way as described above. Protein concentration was determined spectrophotometrically at every step of purification using Bradford reagent (Bradford 1976).
Molecular mass determination
The molecular mass of recombinant Pcal_1127 was determined by the SDS-PAGE analysis as well by gel filtration chromatography. For gel filtration chromatography, Superdex 200 10/300 GL gel filtration column (GE Healthcare) was equilibrated with 150 mM NaCl in 20 mM Tris–Cl (pH 8). The standard curve was obtained with ferritin (440 kDa), catalase (240 kDa), lactate dehydrogenase (140 kDa), BSA (64.5 kDa), and proteinase K (28.9 kDa). Solutions of the standard and sample proteins were prepared in 20 mM Tris–Cl (pH 8) containing 150 mM NaCl.
Enzyme assay
Enzyme activity of Pcal_1127 was measured by flourometric coupling reaction. Formation of PRPP from ribose 5-phosphate (R-5P) was measured through its utilization by anthranilate phosphoribosyl transferase (TrpD) to form N-5′-phosphoribosyl anthranilate (PRA) as summarized below. The absorption and emission wavelengths for anthranilic acid (AA) are 315 and 390 nm, respectively. Anthranilic acid reacted with PRPP (produced by Pcal_1127) and its utilization resulted in decrease in emission/fluorescence at 390 nm.
Assay mixture contained 1 mM R-5P, 5 mM MgCl2, 2.5 mM ATP, 100 mM Tris–Cl buffer pH 8.5, 25 µM anthranilic acid, 100 µM ZnCl2, 20 µg BSA, 1 mM EDTA, 20 µg of RPPK, and 100 µg of anthranilate phosphoribosyl transferase in a total volume of 2 mL. For estimation of optimal temperature for Pcal_1127, enzyme assays were performed at various temperatures ranging from 40 to 80 °C keeping the pH constant. For estimation of optimal pH, assays were performed at various pH keeping the temperature at 55 °C.
One unit of enzyme activity is defined as the amount of enzyme required to catalyze the synthesis of 1 µmol of PRPP per min which is equivalent to disappearance of 1 µmol of anthranilic acid per min.
For thermostability experiments, Pcal_1127, in 20 mM Tris–HCl, was heated at 90 °C for various intervals of time and the residual activity was examined at 55 °C and pH 10.5 in 50 mM glycine-NaOH buffer.
Circular dichroism analysis
Structural stability of Pcal_1127 was analyzed by circular dichroism (CD) spectroscopy using Chirascan-plus CD Spectrometer (Applied Photophysics, UK). The protein samples were incubated at different temperatures ranging from 50 to 100 °C. The CD spectra of the protein solutions were recorded in 20 mM Tris–Cl pH 8.0 in the far-UV range of 200–260 nm. Solvent spectra were subtracted from those of the protein solutions.
Denaturation studies of Pcal_1127
For denaturation studies, protein samples were prepared in different concentrations of urea (0–8 M final concentration) or guanidinium chloride (0–6 M final concentration) and incubated at room temperature for 30 min. Residual enzyme activity of these samples was examined as described above.
Results
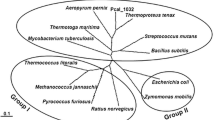
Genome search of P. calidifontis revealed the presence of an open reading frame, Pcal_1127, annotated as ribose-5-phosphate pyrophosphokinase. The gene consisted of 891 nucleotides encoding a polypeptide of 297 amino acids having a theoretical molecular mass of 32,741 Da and an isoelectric point of 6.97. Pcal_1127 displayed highest identity of 76 % with an uncharacterized enzyme from Pyrobaculum neutrophilum. Among the characterized enzymes, Pcal_1127 displayed highest identity of 38 % with RPPK from T. kodakarensis and S. solfataricus. When amino acid sequences of the characterized members of this family were aligned and a phylogenetic tree was constructed, three distinct groups were demarcated. Group I consists of RPPKs from eukarya and bacteria, including S. cerevisiae, human, rat, spinach, A. thaliana, B. subtilis, M. tuberculosis, S. typhimurium, E. coli, and B. amyloliquefaciens. Group II seems to be specific for plants. It comprises two isozymes from spinach and two from A. thaliana. Pcal_1127, along with all the RPPKs characterized from archaea, neither cluster in group I nor in group II. Archaeal RPPKs make a third and relatively dispersed group (Fig. 1). Alignment of amino-acid sequences of all the characterized RPPKs revealed the presence of six highly conserved regions. Regions I to IV contain active-site residues (typed bold in Fig. 2) according to the crystal structures of RPPK from B. subtilis, M. jannaschii, and S. solfataricus. They are involved in substrate binding. In these regions, 41Phe, 43Asp and 45Glu (Region I), 102Arg and 103Gln (Region II), 134Asp and 136His (Region III), and 198Lys and 200Arg (Region IV), according to B. subtilis numbering, are involved in ATP binding. 41Phe, 43Asp, and 45Glu define specificity for adenine base. 198Lys and 200Arg are involved in the binding of triphosphate chain of ATP and stabilization of transition state (Eriksen et al. 2000; Kadziola et al. 2005; Andersen et al. 2015). All these residues are conserved in Pcal_1127. Region V is reported to be involved in substrate (R-5P) binding in the enzymes from above three microorganisms. This region is highly conserved in archaea. In this region, amino-acid residues 224Asp, 225Asp, 231Gly, and 232Thr are conserved in all the characterized RPPKs. Region VI has been reported to play a role in allosteric regulation in RPPKs from B. subtilis and human (Eriksen et al. 2000; Li et al. 2007). This region is found only in class I RPPKs which are allosterically regulated and absent in the characterized members of class II and III (Fig. 2). These results are consistent with the experimental data demonstrating that class II and III enzymes are not allosterically regulated (Krath and Hove-Jensen 2001a; Kadziola et al. 2005).

Phylogenetic tree of Pcal_1127 and all the characterized RPPKs, whose amino-acid sequences are available in the database. Unrooted tree with branch length was constructed using the neighbor-joining method. Bootstrap values and segments corresponding to an evolutionary distance of 0.1 are shown. The tree was constructed using ClustalW provided at http://clustalw.ddbj.nig.ac.jp/. Following are the sequences, with accession numbers, used for the alignment to construct the phylogenetic tree: P. calidifontis (Pcal_1127), ABO08552; S. solfataricus, AAK41307; T. kodakarensis, BAD86424; M. jannaschii, AAB99374; T. volcanium, Q97CA5; B. subtilis, P14193; B. amyloliquefaciens, CBI41177; B. caldolyticus, P42816; S. typhimurium, P0A1V6; E. coli, P0A717; M. tuberculosis, P9WKE3; S. cerevisiae, P38063; Human, P60891; rat, P09330; A. thaliana, Q42581, Q42583, Q93Z66, Q680A5, Q64888; and S. oleracea, Q9XG98, Q9XG99, Q9XGA0, Q9XGA1

Alignment of six conserved regions found in the characterized RPPKs. Regions I to IV contain the amino-acid residues of the active site of M. jannaschii (Kadziola et al. 2005) and B. subtilis (Eriksen et al. 2000). Region V is involved in the ribose 5-phosphate binding. Region VI has been reported to play a role in allosteric regulation which is present only in Class I RPPKs. Names at the left hand side indicate the source organism from which the sequence originated. The numbers show the position of the amino acid in the protein sequence
Production and purification of recombinant Pcal_1127
Analysis of production of recombinant Pcal_1127 in E. coli cells harboring pET-1127, by SDS-PAGE, demonstrated that a high amount of Pcal_1127 was produced in the cells induced with IPTG. Recombinant Pcal_1127 was more than 20 % of the total proteins of the host. When the soluble and insoluble fractions, after lysis, were analyzed, it was found that recombinant Pcal_1127 was produced in the soluble form (Fig. 3). The first step of purification was based on the thermostability of recombinant Pcal_1127. The protein sample was heated at 80 °C which resulted in precipitation and removal of most of the heat-labile proteins of the host. Further purification by ion exchange column chromatography using HiTrap Q and Resource Q columns resulted in a final yield of 17 % with a 4.6-fold-purification (Table 1).

Coomassie brilliant blue stained SDS-PAGE showing the purified recombinant Pcal_1127. Lane 1, soluble fraction of E. coli cells harboring pET-21a; lane 2, soluble fraction of E. coli cells containing pET-1127 plasmid; lane 3, soluble fraction after heat treatment of sample from lane 2; lane 4, Pcal_1127 after HiTrap Q column; lane 5, recombinant Pcal_1127 after Resource Q column; lane M, molecular mass marker
Molecular mass determination
Molecular mass and subunit number of recombinant Pcal_1127 were determined by gel filtration chromatography. Pcal_1127 eluted at a retention volume of 15.8 mL, which corresponded to an approximate molecular mass of 32 kDa on a standard curve obtained from the elution volumes of various standard proteins of known molecular weight (data not shown). This indicated that the recombinant Pcal_1127 existed in a monomeric form similar to RPPK from T. kodakarensis (Rashid et al. 1997). All other RPPKs, characterized either from bacteria, eukarya, or archaea, exist in more than one identical subunits (Roth et al. 1974; Kadziola et al. 2005; Cherney et al. 2011; Andersen et al. 2015; Li et al. 2007; Hove-Jensen and McGuire 2004).
Biochemical characterization of Pcal_1127
The enzyme activity of Pcal_1127 was examined in Britton–Robinson buffer, a universal buffer, at various pH. The optimum pH for Pcal_1127 enzyme activity was 10.5. Activity decreased rapidly at pH above 10.5 (Fig. 4a). Pcal_1127 is the only RPPK that exhibits optimum activity at such a high pH.

Effect of pH and temperature on Pcal_1127 enzyme activity. a Effect of pH. Activity assays were performed in 50 mM Britton–Robinson buffer at 55 °C. b Optimal temperature for enzyme activity. The activity assays were conducted at various temperatures (40–80 °C) in 50 mM Britton–Robinson buffer of pH 10.5. c Thermostability of Pcal_1127. Recombinant Pcal_1127, in 20 mM Tris–HCl, was heated at 90 °C (filled circle), 95 °C (open circle), or in the boiling water (open square) for various intervals of time and the residual activity was examined at 55 °C and pH 10.5 in 50 mM Britton–Robinson buffer. The data are average values of three independent experiments
When we examined the activity of Pcal_1127 at various temperatures, we found that the enzyme exhibited highest activity at 55 °C (Fig. 4b), although the optimal growth temperature of P. calidifontis is between 90 and 95 °C. This characteristic of Pcal_1127 is similar to other RPPKs from hyperthermophilic sources, including T. kodakarensis [50 °C; (Rashid et al. 1997)] and S. solfataricus [60 °C; (Andersen et al. 2015)]. RPPK from M. jannaschii is an exception which displayed its highest activity at 85 °C (Kadziola et al. 2005).
When thermostability of Pcal_1127 was examined by incubating the protein at 90 °C, the optimal growth temperature of P. calidifontis, for various intervals of time and measuring the residual activity, Pcal_1127 was found to be highly thermostable with no significant loss in activity even after an incubation of 240 min. As Pcal_1127 was highly stable at 90 °C; therefore, we heated the enzyme at 95 and 100 °C for various intervals of time and examined the residual activity. Half-life of the enzyme was 50 and 15 min at these temperatures, respectively (Fig. 4c). No reports on thermostability of RPPKs are available in literature except for RPPK from T. kodakarensis which exhibited a half-life of 40 min at 70 °C (Rashid et al. 1997). Based on these facts, we propose that Pcal_1127 is the most thermostable RPPK characterized till now. Structural stability of Pcal_1127 was also confirmed by circular dichroism spectroscopy (Fig. 5).

Circular dichroism studies on Pcal_1127. Far-UV spectrum of Pcal_1127 (200 μg/mL) was analyzed by examining the circular dichroism spectra from 200 to 260 nm at 50 °C (filled circle), 60 °C (open square), 70 °C (filled triangle), 80 °C (open circle), and 90 °C (filled diamond)
Stability of Pcal_1127 was also examined in the presence of denaturants, such as urea and guanidinium chloride. The protein samples, after treatment with these denaturants for 30 min, were examined for enzyme activity. When Pcal_1127 was incubated in the presence of various concentrations of urea, there was no significant difference in the enzyme activity till 8 M, indicating that there was no inactivation of the protein. Similarly, when guanidinium chloride was used, we found that there was no significant difference in the enzyme activity till 4 M. However, the enzyme activity started decreasing above 4 M final concentration of this denaturant and a 50 % residual activity was detected at 6 M guanidinium chloride.
Pcal_1127 displayed enzyme activity without addition of any metal ion. However, a drastic decrease in enzyme activity was observed when 1 mM EDTA was added in the reaction mixture. This result indicated that enzyme activity of Pcal_1127 is metal ion dependent. The activity observed without the addition of any metal ion could be attributed to the metal ions bound to the enzyme during production in E. coli. Addition of various metal ions in the assay mixture at a final concentration of 100 μM resulted in an increase in enzyme activity to a variable amount (Fig. 6a). Although various metal ions could activate Pcal_1127, highest activity was found in the presence of Mn2+ or Mg2+. Therefore, enzyme activity was examined in the presence of various concentrations of Mn2+ and highest activity (a 5-fold increase compared with the activity without the addition of any metal ion) was observed at a concentration of 300 μM. There was no increase in the activity at higher concentrations of Mn2+. Similarly, when enzyme activity was examined in the presence of various concentrations of Mg2+, highest activity (an 8-fold increase compared with the activity without the addition of any metal ion) was found at a final concentration of 1 mM (Fig. 6b). Activation of Pcal_1127 by Mg2+ or Mn2+ is similar to most of the RPPKs from archaeal sources except for the enzyme from T. kodakarensis which shows highest activity in the presence of Co2+ followed by Ni2+ (Rashid et al. 1997).

Effect of various metal ions on the enzyme activity of Pcal_1127. a Enzyme activity in the presence of 100 μM chloride salt of each metal. b A comparison of enzyme activity in the presence of various concentrations of Mg2+ and Mn2+
When pyrophosphate donor specificity of Pcal_1127 was examined, we found that the enzyme could utilize all the four ribonucleoside triphosphates with a marked preference for ATP. In the absence of phosphate ions, highest activity was observed with dATP, a 2-fold higher than that with ATP. However, in the presence of phosphate ions, equivalent amount of activity was observed with dATP and ATP (Fig. 7). Phosphate although activates Pcal_1127 but is not an absolute requirement for the activity of the enzyme. Apart from phosphate ions, the presence of bovine serum albumin, β-mercaptoethanol, and EDTA at lower concentrations (less than 1 mM) enhanced the enzyme activity of Pcal_1127. A combination of these additives resulted in a 4.5-fold increase in enzyme activity. Similar results have been reported for RPPK from rat liver (Roth et al. 1974).

Substrate specificity of Pcal_1127 in the presence (filled bars) and absence (blank bars) of phosphate ions. Each substrate was used at a final concentration of 1 mM
Similar to other RPPKs (Kadziola et al. 2005; Gibson et al. 1982), the enzyme activity of Pcal_1127 was inhibited by ADP. A 40 % decrease in enzyme activity was observed when ATP and ADP were present in the reaction mixture at an equimolar (1 mM) concentration.
Kinetic parameters
Kinetic parameters towards R-5P were measured by varying the concentration of R-5P and keeping ATP constant at 1 mM. Similarly, when these parameters were measured towards ATP, then R-5P concentration was kept constant at 1 mM and ATP concentration was varied. The enzyme followed the Michaelis–Menton equation. Pcal_1127 exhibited a K m value of 60 ± 2 µM towards R-5P and 80 ± 3 µM towards ATP. A V max value of 570 µmol min−1 mg−1 was calculated from Lineweaver‒Burk plot. From the V max (570 µmol min−1 mg−1) and molecular weight (32,741 Da) of Pcal_1127, a k cat value of 311 s−1 was calculated. A comparison of specific activities and K m values of the characterized RPPK is given in Table 2. Pcal_1127 displayed the highest specific activity among all the characterized RPPKs and a very low K m value. These features make Pcal_1127 a highly efficient enzyme. Catalytic efficiency (k cat/K m) of Pcal_1127 was found to be 5183 mM−1 s−1. To the best of our knowledge, this is the highest catalytic efficiency of any RPPK characterized till now.
Discussion
Complete genomes of more than 4200 organisms, including 328 eukaryotes, 3650 bacteria, and 223 archaea, have been sequenced (http://www.genome.jp/kegg/catalog/org_list.html). The sequence information has tremendous contribution in identifying the presence or absence of particular genes or metabolic pathway in a particular organism (Fraser et al. 2000; Nelson et al. 2000; Rashid et al. 2002). When we searched the genome sequence of P. calidifontis for candidate gene encoding RPPK, we found an open reading frame, Pcal_1127, annotated as ribose-5-phosphate pyrophosphokinase. To examine the properties of Pcal_1127, the gene was expressed in E. coli and the gene product was purified. Although Pcal_1127 originates from a hyperthermophile with optimal growth temperature between 90 and 95 °C (Amo et al. 2002), the optimal temperature for the enzyme activity was found 55 °C. This prompted us to examine the thermostability of the enzyme. When we heated the enzyme at 90 °C, the optimal growth temperature of P. calidifontis, for various intervals of time and measured the residual activity, no significant loss of enzyme activity could be observed even after an incubation of 240 min. We, therefore, heated the protein in the boiling water, where it displayed a half-life of 15 min. Low optimal temperature (55 °C) for enzyme activity may be attributed to the instability of the substrate at high temperature. Amino acid composition is considered very relevant in thermostability, as it is related to the hydrophobic interactions (Baldwin 2007; Pace 2009). A comparison of amino-acid composition showed that Pcal_1127 has quite high number of hydrophobic residues L and V which constitute 24.6 % of the protein. Furthermore, there are 34 (11.4 %) alanine residues, the best α-helix former, which may be one of the factors responsible for the thermostability of Pcal_1127. Amino-acid content of thermolabile amino acids, such as C, M, Q (each 1.3 %), and N (2.4 %) was very low. These amino acids tend to be avoided in thermostable enzymes (Hensel 1993; Muir et al. 1995; Russell and Taylor 1995; Russell et al. 1997). High thermostability of Pcal_1127 may be attributed to higher content of α-helix formers along with higher number of hydrophobic residues and lower number of thermolabile amino acids. Recombinant Pcal_1127 exhibited several unique and novel features which include high thermostability, high enzyme activity, and catalytic efficiency. Another unique feature of Pcal_1127 was its highest activity with dATP in the absence of phosphate.
Mostly, the proteins lose their enzyme activities in the presence of high concentrations of denaturants like urea or guanidinium chloride, because these chaotropic agents disturb the native physiological active structure. However, a few proteins from hyperthermophilic archaea are reported to maintain their structures, and hence, the enzyme activities, in the presence of these denaturants (Rasool et al. 2010; Chohan and Rashid 2013; Gharib et al. 2016). We found that there was no inactivation of Pcal_1127 in the presence of even 8 M urea. However, there was a 50 % decrease in activity in the presence of 6 M guanidinium chloride. This can be due to the fact that guanidinium chloride is a salt as well as a denaturant, whereas urea is an uncharged molecule, hence, deficient in ionic strength effects. We could not compare the CD spectra of the protein samples containing urea or guanidium chloride or none due to interference of these chaotropic agents. Structural changes caused by these chaotropic agents are usually studied by recording the fluorescence of the tryptophan residues of the protein samples. Unfortunately, there was no tryptophan residue in the sequence of Pcal_1127; therefore, we could not measure these structural changes, if any.
In conclusion, the results obtained in this study demonstrate that Pcal_1127 exhibits a combination of properties of class I and II RPPKs and is highly stable against temperature and denaturants. Furthermore, the catalytic efficiency reflects that Pcal_1127 is the most efficient RPPK characterized to date.
References
Amo T, Paje ML, Inagaki A, Ezaki S, Atomi H, Imanaka T (2002) Pyrobaculum calidifontis sp. nov., a novel hyperthermophilic archaeon that grows in atmospheric air. Archaea 1:113–121
Andersen RW, Leggio LL, Hove-Jensen B, Kadziola A (2015) Structure of dimeric, recombinant Sulfolobus solfataricus phosphoribosyl diphosphate synthase: a bent dimer defining the adenine specificity of the substrate ATP. Extremophiles 19:407–415
Arnvig K, Hove-Jensen B, Switzer RL (1990) Purification and properties of phosphoribosyl-diphosphate synthetase from Bacillus subtilis. Eur J Biochem 192:195–200
Baldwin RL (2007) Energetics of protein folding. J Mol Biol 371:283–301
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72:248–254
Breda A, Martinelli LKB, Bizarro CV, Rosado LA, Borges CB, Santos DS, Basso LA (2012) Wild-Type phosphoribosylpyrophosphate synthase (PRS) from Mycobacterium tuberculosis: a bacterial class II PRS? PLoS One 7:e39245
Cherney MM, Cherney LT, Garen CR, James MN (2011) The structures of Thermoplasma volcanium phosphoribosyl pyrophosphate synthetase bound to ribose-5-phosphate and ATP analogs. J Mol Biol 413:844–856
Chohan SM, Rashid N (2013) TK1656, a thermostable l-asparaginase from Thermococcus kodakaraensis, exhibiting highest ever reported enzyme activity. J Biosci Bioeng 116:438–443
Eriksen TA, Kadziola A, Bentsen AK, Harlow KW, Larsen S (2000) Structural basis for the function of Bacillus subtilis phosphoribosylpyrophosphate synthetase. Nat Struct Biol 7:303–308
Fraser CM, Eisen JA, Salzberg SL (2000) Microbial genome sequencing. Nature 406:799–803
Gallois R, Prevot JC, Clément A, Jacob JL (1997) Purification and characterization of phosphoribosylpyrophosphate synthetase from rubber tree latex. Plant Physiol 115:847–852
Gharib G, Rashid N, Bashir Q, Gardner QA, Akhtar M, Imanaka T (2016) Pcal_1699, an extremely thermostable malate dehydrogenase from hyperthermophilic archaeon Pyrobaculum calidifontis. Extremophiles 20:57–67
Gibson KJ, Schuber KR, Switzer RL (1982) Binding of the substrates and the allosteric inhibitor adenosine 5′-diphosphate to phosphoribosylpyrophosphate synthetase from Salmonella typhimurium. J Biol Chem 257:2391–2396
Hensel R (1993) Proteins of extreme thermophiles. In: Kates M, Kushner DJ, Matheson AT (eds) The biochemistry of the archaea. Elsevier, Amsterdam, pp 209–221
Hong S, Zhou D, Chen C, Wang W, Lv Y, Ye Y, Zou P, Yv J, Chang X, Yv X, Shi L, Ma Lei (2013) Ribose-phosphate pyrophosphokinase 1 (PRPS1) associated with deltamethrin resistance in Culex pipiens pallens. Parasitol Res 112:847–854
Hove-Jensen B (1988) Mutation in the phosphoribosylpyrophosphate synthetase gene (prs) that results in simultaneous requirements for purine and pyrimidine nucleosides, nicotinamide nucleotide, histidine, and tryptophan in Escherichia coli. J Bacteriol 170:1148–1152
Hove-Jensen B (2004) Heterooligomeric phosphoribosyl diphosphate synthase of Saccharomyces cerevisiae: combinatorial expression of the five PRS genes in Escherichia coli. J Biol Chem 279:40345–40350
Hove-Jensen B, McGuire JN (2004) Surface exposed amino acid differences between mesophilic and thermophilic phosphoribosyl diphosphate synthase. Eur J Biochem 271:4526–4533
Jiménez A, Santos MA, Revuelta JL (2008) Phosphoribosyl pyrophosphate synthetase activity affects growth and riboflavin production in Ashbya gossypii. BMC Biotechnol 8:67
Kadziola A, Jepsen CH, Johansson E, McGuire J, Larsen S, Hove-Jensen B (2005) Novel class III phosphoribosyl diphosphate synthase: structure and properties of the tetrameric, phosphateactivated, non-allosterically inhibited enzyme from Methanocaldococcus jannaschii. J Mol Biol 354:815–828
Kita K, Otsuki T, Ishizuka T, Tatibana M (1989) Rat liver phosphoribosyl pyrophosphate synthetase: existence of the purified enzyme as heterogeneous aggregates and identification of the catalytic subunit. J Biochem 105:736–741
Krath BN, Hove-Jensen B (2001a) Class II recombinant phosphoribosyl diphosphate synthase from spinach. Phosphate independence and diphosphoryl donor specificity. J Biol Chem 276:17851–17856
Krath BN, Hove-Jensen B (2001b) Implications of secondary structure prediction and amino acid sequence comparison of class I and class II phosphoribosyl diphosphate synthases on catalysis, regulation, and quaternary structure. Protein Sci 10:2317–2324
Krath BN, Eriksen TA, Poulsen TS, Hove-Jensen B (1999) Cloning and sequencing of cDNAs specifying a novel class of phosphoribosyl diphosphate synthase in Arabidopsis thaliana. Biochim Biophys Acta 1430:403–408
Li S, Lu T, Peng B, Ding J (2007) Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem J 401:39–47
Lucarelli AP, Buroni S, Pasca MR, Rizzi M, Cavagnino A, Valentini G, Riccardi G, Chiarelli LR (2010) Mycobacterium tuberculosis phosphoribosylpyrophosphate synthetase: biochemical features of a crucial enzyme for mycobacterial cell wall biosynthesis. PLoS One 5:e15494
Muir JM, Russell RJM, Hough DW, Danson MJ (1995) Citrate synthase from the hyperthermophilic archaeon Pyrococcus furiosus. Protein Eng 8:583–592
Nelson KE, Paulsen IT, Heidelberg JF, Fraser CM (2000) Status of genome projects for nonpathogenic bacteria and archaea. Nat Biotechnol 18:1049–1054
Nosal JM, Switzer RL, Becker MA (1993) Overexpression, purification, and characterization of recombinant human 5-phosphoribosyl-1-pyrophosphate synthetase isozymes I and II. J Biol Chem 268:10168–10175
Pace CN (2009) Energetics of protein hydrogen bonds. Nat Struct Mol Biol 16:681–682
Rashid N, Morikawa M, Imanaka T (1997) Gene Cloning and Characterization of recombinant ribose phosphate pyrophosphokinase from a hyperthermophilic archaeon. J Biosci Bioeng 83:412–418
Rashid N, Imanaka H, Kanai T, Fukui T, Atomi H, Imanaka T (2002) A novel candidate for the true fructose-1,6-bisphosphatase in archaea. J Biol Chem 277:30649–30655
Rasool N, Rashid N, Iftikhar S, Akhtar M (2010) N-terminal deletion of Tk1689, a subtilisin-like serine protease from Thermococcus kodakaraensis, copes with its cytotoxicity in Escherichia coli. J Biosci Bioeng 110:381–385
Roth DG, Shelton E, Deuel TF (1974) Purification and properties of phosphoribosyl pyrophosphate synthetase from rat liver. J Biol Chem 249:291–296
Russell RJM, Ferguson JMC, Haugh DW, Danson MJ, Taylor GL (1997) The crystal structure of citrate synthase from the hyperthermophilic archaeon Pyrococcus furiosus at 1.9 Å resolution. Biochemistry 36:9983–9994
Russell RJM, Taylor GL (1995) Engineering thermostability: lessons from thermophilic proteins. Curr Opin Biotechnol 6:370–374
Switzer RL (1969) Regulation and mechanism of phosphoribosylpyrophosphate synthetase I. purification and properties of the enzyme from Salmonella typhimurium. J Biol Chem 244:2854–2863
Switzer RL, Gibson KJ (1978) Phosphoribosylpyrophosphate synthetase (ribose-5-phosphate pyrophosphokinase) from Salmonella typhimurium. Methods Enzymol 51:3–11
Willemoës M, Hove-Jensen B (1997) Binding of divalent magnesium by Escherichia coli phosphoribosyl diphosphate synthetase. Biochemistry 36:5078–5083
Willemoës M, Hove-Jensen B, Larsen S (2000) Steady state kinetic model for the binding of substrates and allosteric effectors to Escherichia coli phosphoribosyl-diphosphate synthase. J Biol Chem 275:35408–35412
Zakataeva NP, Romanenkov DV, Skripnikova VS, Vitushkina MV, Livshits VA, Kivero AD, Novikova AE (2012) Wild-type and feedback-resistant phosphoribosyl pyrophosphate synthetases from Bacillus amyloliquefaciens: purification, characterization, and application to increase purine nucleoside production. Appl Microbiol Biotechnol 93:2023–2033
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Atomi.
Rights and permissions
About this article

Cite this article
Bibi, T., Perveen, S., Aziz, I. et al. Pcal_1127, a highly stable and efficient ribose-5-phosphate pyrophosphokinase from Pyrobaculum calidifontis . Extremophiles 20, 821–830 (2016). https://doi.org/10.1007/s00792-016-0869-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-016-0869-z