
Abstract
Two hydrothermal springs (AI: 51 °C, pH 3; AIV: 92 °C, pH 8) were analysed to determine prokaryotic community composition. Using pyrosequencing, 93,576 partial 16S rRNA gene sequences amplified with V2/V3-specific primers for Bacteria and Archaea were investigated and compared to 16S rRNA gene sequences from direct metagenome sequencing without prior amplification. The results were evaluated by fluorescence in situ hybridization (FISH). While in site AIV Bacteria and Archaea were detected in similar relative abundances (Bacteria 40 %, Archaea 35 %), the acidic spring AI was dominated by Bacteria (68 %). In spring AIV the combination of 16S rRNA gene sequence analysis and FISH revealed high abundance (>50 %) of heterotrophic bacterial genera like Caldicellulosiruptor, Dictyoglomus, and Fervidobacterium. In addition, chemolithoautotrophic Aquificales were detected in the bacterial community with Sulfurihydrogenibium being the dominant genus. Regarding Archaea, only Crenarchaeota, were detected, dominated by the family Desulfurococcaceae (>50 %). In addition, Thermoproteaceae made up almost 25 %. In the acidic spring (AI) prokaryotic diversity was lower than in the hot, slightly alkaline spring AIV. The bacterial community of site AI was dominated by organisms related to the chemolithoautotrophic genus Acidithiobacillus (43 %), to the heterotrophic Acidicaldus (38 %) and to Anoxybacillus (7.8 %). This study reveals differences in the relative abundance of heterotrophic versus autotrophic microorganisms as compared to other hydrothermal habitats. Furthermore, it shows how different methods to analyse prokaryotic communities in complex ecosystems can complement each other to obtain an in-depth picture of the taxonomic composition and diversity within these hydrothermal springs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
With regard to their physicochemical conditions, terrestrial hydrothermal springs are highly diverse habitats, offering a wide range of ecological niches. These niches exhibit extreme conditions, such as high temperature, low or high pH, and the presence of toxic ions. Their extreme features are expected to lead to limited biodiversity, making hydrothermal habitats ideal model systems to study principles of community structure and function. Prokaryotic diversity in hydrothermal ecosystems has been extensively studied, in particular at Yellowstone National Park repeatedly revealing characteristic taxonomic groups like Aquificales and specific groups of Crenarchaeota, in particular (among others Hamamura et al. 2012; Meyer-Dombard et al. 2005; Spear et al. 2005).
The abundant, often predominant occurence of Aquificales has led to the hypothesis that H2-based chemolithoautotrophic processes play a dominant role in hot-temperature ecosystems, in which photosynthesis is no longer possible (Spear et al. 2005; Inskeep et al. 2010; Hugenholtz et al. 1998). Aquificales often form the basis for chemolithotrophic biofilm or mat structures, e.g. the pink filaments of Thermocrinis ruber and the white sausage-shaped filaments found in sulphur-turf mats of Japanese hot springs (Blank et al. 2002; Yamamoto et al. 1998).
Estimates on community composition, however, have been performed mainly on the basis of clone libraries with the known bias of cloning-based technologies and relatively low sample size taken into account (among others Blank et al. 2002; Hugenholtz et al. 1998; Reysenbach et al. 1994; Swingley et al. 2012). With next generation sequencing methods the analysis of larger 16S rRNA gene-data sets has become feasible, making it possible to increase the extent of sequence-based biodiversity studies, leading to a higher degree of reliability. Analysis of large sets of short rRNA gene fragments comprising variable regions has been applied to infer community composition and biodiversity estimates in terrestrial and marine habitats (Huse et al. 2008; Miller et al. 2009; Nacke et al. 2011; Roesch et al. 2007; Will et al. 2010; Youssef et al. 2009). However, the analysis is usually based on PCR-amplified DNA fragments, which still suffers from potential primer bias. Direct sequencing of metagenomic DNA can circumvent this bias, reducing, however, the amount of data available for analysis since non-16S rRNA gene sequences are also generated (Inskeep et al. 2010). To obtain an in-depth picture of prokaryotic hydrothermal spring communities, we combined both sequencing of short variable rRNA gene fragments and phylogenetic analysis of the metagenome, evaluating it with fluorescence in situ hybridization (FISH), which allows direct quantification of specific microbial lineages in the sample, directly. Our study was conducted with samples from the Furnas Valley, situated on São Miguel, Azores.
The Azores are a group of geographically isolated islands in the Atlantic Ocean. They are almost exclusively of volcanic origin, with the main hydrothermal area being the Furnas Valley on the Island of Sao Miguel. However, compared to other hydrothermal zones like Yellowstone National Park and Iceland the overall amount of data available is still limited. Brock and Brock (1967) gave a detailed description of the hot springs of Furnas Valley pointing out the thermal and chemical complexity of the Furnas springs and a major difference with regard to other terrestrial hydrothermal areas: in Furnas “… the largest spring is at the highest elevation and is alkaline, whereas some of the lower springs are smaller and more acidic. This is completely opposite to the situation found in Yellowstone National Park and Iceland… (Allen and Day 1935; Barth 1950)”. Here the higher springs are small and acidic and the lower ones large and alkaline. The hot springs of Furnas Valley have been a valuable source for thermophilic microorganisms and thermostable enzymes with useful features for biotechnological applications (among others Albuquerque et al. 2008, 2010, 2012; Franca et al. 2006; Riessen and Antranikian 2001; Friedrich and Antranikian 1996). However, only little data are available on overall diversity (Hamamura et al. 2012).
Applying different 16S rRNA gene-based and metagenomic approaches, we provide an in-depth study of bacterial and archaeal diversity as a quantitative basis for understanding ecological interactions within the prokaryotic community. The study reveals the wealth of biocatalytic potential for enzymes from heterotrophic organisms still waiting to be recovered from these challenging extreme habitats.
Materials and methods
Study site and sampling
Sediment, biofilm, and water samples were collected from various hydrothermal springs at Furnas Valley, São Miguel, Azores, in September 2010. All necessary permits were obtained for the described field studies from the regional government of the Azores (LICENCA No 107/2010/DRA). A description of the different sampling sites is provided in Table 1. Several samples were collected at a solfataric field (sampling site A) (Fig. 1). Additional samples were collected from the hydrothermal vents Caldeirão (sampling site B) and Caldeira do Asmodeu (sampling site C). With the exception of the solfatara sample AIV (water sample) mainly sediments, muds or biofilms were collected and supplemented with water from the respective hydrothermal spring. Sediments and mud were characterized by brownish, ochre, grey, black or green appearance. The biofilms were black, white or light-grey, and the microbial mats encountered along the effluent stream of one hydrothermal vent exhibited green, orange, yellowish or brownish colours (Fig. 1).

Solfataric field (study site A) with sulphur deposits and various hydrothermal springs of temperatures up to 97 °C and pH values varying between 2.5 and 8. The rising gases (mainly water vapour and hydrogen sulphide) originate from a solfatara, Caldeira do Esguicho (red arrow, sample AIV). Several sediment and water samples were collected at this site. The green arrow depicts site AI
Each sample was transferred to a sterile serum bottle, preventing exposure to air as far as possible. After sampling, the bottles were closed immediately with a butyl rubber stopper and a screw cap and 0.01 % w/v (final concentration) sodium sulphide was added by means of a syringe to counteract potential oxygen intake. Within 4 h after sampling, aliquots of 500 μL per sample were fixed for 1 h with 3 % (final concentration) paraformaldehyde for subsequent FISH analyses as described by Ravenschlag et al. (2000) and stored at −20 °C. The original samples were stored at 4 °C.
Determination of chemical parameters
Aliquots of approx. 120 mL of the original samples were centrifuged at 3,800×g for 40 min. The supernatants were filtered (pore size of the filters <0.45 μm) and analysed for chemical parameters applying the following methods/instruments: optical emission spectroscopy with inductively coupled plasma (Optima 7000 DV OES/ICP, PerkinElmer, Inc.) (Na, K, Mg, Ca, Fe, S), ultraviolet–visible spectrophotometry (Lambda 25 UV–VIS, PerkinElmer, Inc.) (NH4 +, Fe2+, NO2−), ion chromatography (Metrohm AG, Germany) (Cl−, NO3 −, SO3 2−, SO4 2−, PO4 3−) and TOC analyser (highTOC, Elementar Analysensysteme GmbH, Germany) (DOC, TIC).
Isolation of DNA
Total community DNA was isolated as described by Zhou et al. (1996), based on lysis with high-salt extraction buffer and extended heating in the presence of sodium dodecyl sulphate (SDS). The samples were centrifuged at 3,939×g for 1 h and 2–3 g of the pellet was resuspended in DNA extraction buffer, containing 1 % hexadecylmethylammonium bromide (CTAB). Prior to DNA isolation, three freeze-and-thaw cycles in liquid nitrogen and a water bath at 65 °C were conducted. The precipitated DNA was dissolved in 120 μL sterile water and analysed spectrophotometrically at 260/280 nm and by agarose gel electrophoresis (0.4 % agarose). DNA solutions derived from samples with high proportions of sediments appeared intensively orange or yellowish, indicating the coextraction of humic substances which interfere with UV-photometric measurement of DNA concentration. Thus, quantification of DNA extracted from these samples was merely done by agarose gel electrophoresis.
In addition, we applied the PowerMax™ Soil DNA Isolation Kit (MO BIO Laboratories, Inc., Carlsbad, USA) as an alternative method for isolation of genomic DNA from all samples. Microorganisms were lysed by a combination of SDS and further disruption agents, and mechanical force using beads. Approx. 2 g of sediment was used for DNA extraction. The DNA was eluted in 2 mL sterile water and quantified as described above. Pooled DNA from both methods was used for metagenomic pyrosequencing or PCR amplification.
Denaturing gradient gel electrophoresis
PCR amplification and DGGE were performed as described by Muyzer et al. (1998). We routinely added 3 μg/μL (final concentration) bovine serum albumin to the PCR reaction mixture to prevent inhibition of the DNA polymerase by humic substances. The sequences of the primers used for amplification of 16S rRNA gene-fragments were as follows: Arc334F: 5′-CGCCCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGCCCGACGGGGYGCAGCAGGCGCGA-3′, Arc915R: 5′-GTGCTCCCCCGCCAATTCCT-3′ (Archaea); 314F: 5′-CGCCCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGCCCGCCTACGGGAGGCAGCAG-3′, 907R: 5′-CCGTCAATTCMTTTGAGTTT-3′ (Bacteria). Taq DNA polymerase (Fermentas GmbH, St. Leon-Rot, Germany) was used in PCR. The touchdown PCR protocol by Muyzer (annealing temperatures ranging from 65 to 55 °C) was applied for both primer pairs. We used the Dcode™ System (Bio-Rad Laboratories GmbH, Munich, Germany) for DGGE. Electrophoresis was carried out using a 6 % polyacrylamide gel with a denaturing gradient of 20–80 %, at 55 °C, 200 V for 5 h (Bacteria-DGGE), and a denaturing gradient of 30–80 % at 60 °C, 150 V for 2 h (Archaea-DGGE). After electrophoresis, the gels were stained in Roti®-Gel Stain (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) and photographed on a UV transilluminator.
Amplification of 16S rRNA gene fragments and pyrosequencing
DNA preparations from both isolation approaches were used as template for individual PCR amplification of 16S rRNA gene fragments. The PCR products derived from either DNA preparation were combined for sequencing in equal amounts. The sequences of the bacterial and archaeal V2/V3 region-specific primers used in respective PCRs are as follows: Bacteria, V2f: 5′-AGTGGCGGACGGGTGAGTAA-3′; V3r, 5′-CCGCGGCTGCTGGCAC-3′ [(Will et al. 2010) V3r modified]; Archaea, Arc113f: 5′-ACKGCTSAGTAACACGTGG-3′; Arc520r, 5′-TACCGCGGCKGCTGGCA-3′ [Arc113f, (Baker and Cowan 2004); Arc520r, this study]. The template-specific primer sequences were complemented with a GS FLX Titanium primer sequence including the sequence adaptor, a four-base library key sequence, and a MID (multiplex identifier) sequence at the 5′ end of the specific primer. The Amplicon Fusion Primers were commercially synthesized (Fermentas GmbH, St. Leon-Rot, Germany).
The PCR mixtures (final volume 50 μL) contained 10 μL fivefold Phusion HF buffer (New England Biolabs GmbH, Frankfurt am Main, Germany), 300 μg BSA, 0.2 mM final concentration of each dNTP, 0.4 μM final concentration of each primer, 0.5 U Phusion High-Fidelity DNA polymerase (New England Biolabs GmbH, Frankfurt am Main, Germany), and 30–70 ng DNA. For Archaea-specific PCR the following steps were conducted: initial denaturation at 98 °C for 30 s and 28 cycles of denaturation at 98 °C for 10 s, primer annealing for 20 s using a temperature gradient ranging from 68 °C to 55 °C (1 °C touchdown every two cycles) and extension at 72 °C for 15 s, followed by a final extension period at 72 °C for 10 min. For Bacteria-specific PCR the cycling scheme deviated from the one described above by the annealing temperature gradient which was from 68 to 58 °C over 22 cycles and six additional cycles at a constant annealing temperature of 58 °C. Subsequently, the PCR products were purified by employing the GeneJet™ Gel Extraction Kit (Fermentas GmbH, St. Leon-Rot, Germany) and quantified spectrophotometrically.
The isolated metagenomic DNA was used to create a 454-shotgun library following the GS Rapid library protocol (Roche). The library was sequenced with the Genome Sequencer FLX (Roche, Mannheim, Germany) using Titanium chemistry. Sequencing was performed by the Göttingen Genomics Laboratory. In total, 2,044,797 shotgun reads were generated by two complete sequencing runs.
The partial 16S rRNA gene amplicons were sequenced using the unidirectional sequencing protocol (Roche) and Titanium chemistry. One medium lane of a Titanium Picotiter plate was used for sequencing, resulting in 125,702 shotgun sequences.
The sequences have been deposited at NCBI database under the accession number: SRA059339.
Analysis of pyrosequencing-derived data
All pyrosequencing reads obtained from partial 16S rRNA gene amplicons were reassigned to samples AI and AIV based on the unique MIDs. Different scripts of the QIIME software pipeline (Caporaso et al. 2010) and additional programs were applied to perform pyrosequencing data preprocessing and downstream analyses. Removal of sequences <200 bp, and sequences containing more than two primer mismatches or long homopolymers (>8 bp) was performed by applying the script “split_libraries.py”. Pyrosequencing noise was removed by using the scripts “denoise_wrapper.py” and “inflate_denoiser.py”. Subsequently, the programs cutadapt (Martin 2011) and UCHIME (Edgar et al. 2011) (reference database: Greengenes gold database) were applied to remove remaining primer sequences and chimeras, respectively.
Operational taxonomic units (OTUs) were identified at genetic distances of 3 and 20 %, representing the species and phylum level, respectively, according to Schloss and Handelsman (2005) by applying the QIIME script “pick_otus.py” (Schloss and Handelsman 2005). In addition, taxonomic classification of OTUs as well as calculation of rarefaction curves, the Shannon index and the Chao1 index were performed using QIIME (Shanon and Weaver 1949; Chao and Bunge 2002). With respect to the script “assign_taxonomy.py”, related to taxonomic classification of OTUs, preprocessed sequences were compared to the SILVA ribosomal RNA database (release 108) using BLASTN (Pruesse et al. 2007). Furthermore, a customized script to remove all OTUs from the OTU table that have been classified as chloroplasts was applied.
Metagenomic 16SrRNA gene sequences with a minimum length of 100 bp were analysed in the same way by comparing them to the SILVA ribosomal RNA database (release 108) using BLASTN (Pruesse et al. 2007).
Fluorescence in situ hybridization
An 80 μL aliquot of the fixed sample was diluted 1:10 in PBS/ethanol and treated by sonication using Branson Sonifier 450 (Branson Ultrasonics Corporation, Danbury, USA) at a setting of 1 min, duty cycle 20 and output control 2, three times with 60 s lapse between each sonication step while cooled on ice. Subsequently, the sample was resuspended in 400 μL PBS/ethanol. In situ hybridization was conducted following the protocol by Snaidr et al. (1997). An aliquot of 2.5 μL was applied to a poly-l-lysine-coated microscopic slide. After dehydration, hybridization of the respective 5′-cyanine 3-labelled oligonucleotide probe to the ribosomal sequence in the target cells was carried out at 46 °C for 100–120 min. The slide was washed and stained with 2 μg/mL 4′,6′-diamidino-2-phenylindol (DAPI) for 5 min on ice. Cells were counted using a fluorescent microscope (Zeiss Axioplan) and means were calculated by counting 10–20 randomly chosen fields for each analysis, which corresponded to at least 1,000 Dapi-stained cells.
The FISH probes applied in this study were Eub338 (5′-GCTGCCTCCCGTAGGAGT-3′, specificity: domain Bacteria, 35 % formamide in hybridization buffer) (Amann et al. 1990), Arc915 (5′-GTGCTCCCCCGCCAATTCCT-3′, specificity: domain Archaea, 35 % formamide in hybridization buffer) (Stahl and Amann 1991), Cal450 (5′- CTCCCCGTCCAAAGAGGT-3′, specificity: genus Caldicellulosiruptor, 30 % formamide in hybridization buffer) (O-Thong et al. 2007), SHyd540 (5′-TCGCGCAACGTTCGGGACC-3′, specificity: genus Sulfurihydrogenibium, 60 % formamide in hybridization buffer), and Fer660 (5′-GTTCCGTCTGCCTCTGCC-3′, specificity: genus Fervidobacterium, 20 % formamide in hybridization buffer). Probes were commercially synthesized as 5′-indocyanine 3-labelled oligonucleotides (Fermentas GmbH, St. Leon-Rot, Germany). Specificity for the newly designed oligonucleotide probes SHyd540 and Fer660 was verified by hybridization to control strains that possess one mismatch position within the ribosomal target sequence (SHyd540: Rhodothermus marinus (DSMZ 4252); Fer660: Fervidobacterium gondwanense (DSMZ 13020) and F. nodosum (DSMZ 5306)). Under the given stringency discrimination of one mismatch was possible.
Results
Sampling site and sample characteristics
Samples were collected in September 2010 in the Valley of Furnas from nine sites with a wide variety of physicochemical characteristics. In situ temperatures ranged from 51 to 92 °C, whereas pH values varied between 8 and 2.5 (Table 1). Two sampling sites (AI and AIV) were selected for in-depth analysis, including the determination of several chemical parameters (Table 2). Site AI had a temperature of 51 °C and an acidic pH of 3. The greyish muddy hot spring exhibited oxidized conditions with high concentrations of sulphate (12.5 mM) and iron, which was mainly present as Fe3+ (1,400 μM). Site AIV, with a temperature of 92 °C and a pH of 8, exhibited reduced conditions, indicated by the proportion of Fe2+ to Fe3+, which can be taken as a proxy for redox conditions (Mathur et al. 2007). The concentrations of sulphate and iron were low (100 and 20 μM) and iron was mainly present as Fe2+. Organic carbon content was high with 0.006 % (60 mg/L) at site AI and 0.03 % (300 mg/L) at site AIV.
Community fingerprinting using DGGE
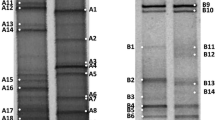
Prokaryotic diversity was estimated using denaturing gradient gel electrophoresis (DGGE) based on 16S rRNA gene fragments amplified with Bacteria- and Archaea-specific primers. Results are shown in Figs. 2 and 3. Taking the number of bands in the DGGE-profile as diversity indicator, the highest bacterial diversity was detected at sites with temperatures between 55 and 85 °C and pH values between 7 and 8. Analysis of the DGGE-profiles revealed a prominent effect of pH. In particular, the combination of acidic pH and high temperature (pH 2.5–3 and 84 °C) led to a very low bacterial diversity (Fig. 2, lane 2) while at moderate temperature of 51 °C and pH 3 more bacterial bands were detected (Fig. 2, lane 1). Analysis of archaeal 16S rRNA genes using DGGE revealed an overall lower number of bands (Fig. 3). Sites AIII and AIV exhibit an almost identical band-pattern despite the difference in temperature (85 vs. 92 °C).

DGGE analysis of 16S rRNA gene fragments amplified with Bacteria-specific primers. PCR products were analysed on a denaturing gradient from 20 to 80 %. The samples were grouped according to sampling site (A, B, C) and increasing temperature. The lanes can be attributed to the following sites: 1 AI (51 °C, pH 3), 2 AII (84 °C, pH 2.5–3), 3 AIII (85 °C, pH 8), 4 AIV (92 °C, pH 8), 5 BI (60 °C, pH 6), 6 BII (65 °C, pH 7), 7 BIII (70 °C, pH 6), 8 CI (55 °C, pH 8), 9 CII (76 °C, pH 8)

DGGE analysis of 16S rRNA gene fragments amplified with Archaea-specific primers. PCR products were analysed on a denaturing gradient from 30 to 80 %. The samples were grouped according to sampling site (A, B, C) and increasing temperature. The lanes can be attributed to the following sites: 1 AI (51 °C, pH 3), 2 AII (84 °C, pH 2.5–3), 3 AIII (85 °C, pH 8), 4 AIV (92 °C, pH 8), 5 BI (60 °C, pH 6), 6 BII (65 °C, pH 7), 7 BIII (70 °C, pH 6), 8 CI (55 °C, pH 8), 9 CII (76 °C, pH 8)
Based on site-specific characteristics and the DGGE results we selected two sites (AI and AIV) for further analysis. Both sites are connected, with the outflow of AIV running into AI. Furthermore, they exhibit medium diversity and particularly challenging conditions with regard to pH (AI: pH 3) or temperature (AIV: 92 °C).
Community composition determined by fluorescence in situ hybridization
To quantify the relative contributions of Bacteria and Archaea to the communities of sites AI and AIV, we applied FISH using domain-specific probes (Fig. 4). The acidic hydrothermal spring AI was dominated by Bacteria (68 %). Archaea could not be detected at all and 32 % of the cells could only be stained with DAPI. At the high-temperature site AIV (92 °C) 75 % of all cells could be detected by FISH. The relative contribution of the specific domains was almost even with 35 % Archaea and 40 % Bacteria. Total cell number determined by DAPI-staining was 4.6 × 107/ml for AI and 2.6 × 108/ml for AIV.

Domain-level composition of microbial communities AI and AIV as determined by FISH. For Bacteria EUB333 was used and for Archaea probe Arch915. Relative abundance is based on the total cell number determined by DAPI-staining. a Depicts the results for site AI and b for site AIV
Phylogenetic analyses based on 16S rRNA gene sequences
To evaluate prokaryotic diversity of the selected sites, partial 16S rRNA genes were amplified from metagenomic DNA and sequenced by pyrosequencing. Based on former studies, we decided to amplify the V2/V3 region for both, Bacteria and Archaea, comprising 435 bp (Nacke et al. 2011; Yu et al. 2008; Will et al. 2010; Kysela et al. 2005). While primers for Bacteria were already available, new primers to amplify the corresponding region in the archaeal 16S rRNA gene had to be designed. The forward primer was based on the data of Baker and Cowan with slight modifications (Baker and Cowan 2004) while the reverse primer was designed de-novo. A total number of 125,702 sequences were generated. After preprocessing including quality filtering and denoising, 93,576 sequences with an average length of 390 bases could be obtained for further analyses. The number of total reads of every sample and remaining numbers of reads after quality filtering and denoising are depicted in Table 3. Amplification was specific, yielding the expected domain-specific sequences apart from archaeal amplification of DNA from site AIV.
Furthermore, metagenomic DNA derived from site AIV was analysed using direct pyrosequencing without prior amplification. Of the data generated 0.1 % represented partial 16S rRNA genes. The 16S rRNA gene fragments f ≥ 100 bp were extracted from the dataset. The resulting 725 partial 16S rRNA gene sequences were analysed by comparing them to the SILVA ribosomal RNA database using BLASTN.
Comparison of the sequencing approaches
Combined results of the two sequencing approaches are shown in Tables 4, 5 and 6. The acidic spring AI was dominated by Proteobacteria (>80 %) and Firmicutes (10 %) (Table 4) while site AIV was dominated by bacteria belonging to 4 different phyla: Thermotogae, Firmicutes, Dictyoglomi, and Aquificae (Table 5). Each of these phylum-groups mainly consisted of one dominating genus. The acidic spring AI was dominated by Proteobacteria known for their acidophilic properties: the heterotrophic genus Acidicaldus (38 %) and the chemolithoautotrophic Acidithiobacillus (43 %). In addition, almost 8 % of the sequences was related to Anoxybacillus (Table 4). Within site AIV (92 °C, pH 8) members of Dictyoglomus, Caldicellulosiruptor, and Fervidobacterium constituted up to 61 % of the bacterial community according to data resulting from non-amplified pyrosequencing of metagenomic DNA, whereas analysis of the amplified V2/V3 16S rRNA gene region indicated an even higher abundance of these heterotrophic genera (88 %) (Table 5). Data from direct metagenome sequencing also revealed high abundance of two genera from the phylum Aquificae, with the chemolithoautotrophic Sulfurihydrogenibium being the dominant genus (22 %).

Rarefaction analysis of V2/V3 sequencing results for Bacteria and Archaea. Rarefaction curves were calculated on the basis of 22,100 sequences for Bacteria (a) and based on 35,200 sequences for Archaea (b). The curves indicate the observed number of OTUs within the two samples AI (51 °C, pH 3) and AIV (92 °C, pH 8). OTUs are shown at the level of 3 and 20 % genetic distance
The archaeal community of the hot spring AIV (92 °C) was almost exclusively composed of Crenarchaeota while in the acidic spring AI only Euryarchaeota were detected. Table 6 depicts the detailed composition. The archaeal community of site AI mainly harboured Thermoplasma (89 %), while clone group BLSdp215 represented the second largest group (9 % of all sequences). Site AIV was dominated by Desulfurococcaceae (75 %) with the genera Sulfophobococcus, Desulfurococcus being most abundant within this family. Furthermore, the genus Pyrobaculum of the Thermoprotaceae made up 25 % of the sequences.
Microbial diversity indices
Hot springs are usually referred to as low-diversity habitats. Diversity indices are well established measures to quantify diversity on a parametric (Shanon) or non-parametric (Chao1) basis. While the Shanon index takes species richness and species evenness into account, the Chao index is based on species richness, with the obtained value giving the number of expected operational taxonomic units (OTUs). To calculate rarefaction curves, richness, and diversity indices, OTUs at genetic distances of 3 and 20 % were determined. With respect to the Bacteria, OTU-based analyses were performed at the same level of surveying effort by using 22,100 randomly selected sequences per sample to enable accurate OTU-based comparisons between site AI and AIV.
Comparison of the rarefaction analyses with the number of OTUs determined by Chao1 richness estimator revealed that 76.2–90.6 % (20 % genetic distance) and 54.5–77.7 % (3 % genetic distance) of the taxonomic richness were covered by the surveying effort (Fig. 5; Table 7). Thus, we did not survey the full extent of taxonomic diversity at these genetic distances, but especially at the phylum level a substantial fraction of the estimated richness was assessed by the surveying effort. The bacterial diversity at sites AI and AIV exhibits an interesting difference: on the phylum level the acidic spring is more diverse than the slightly alkaline high-temperature site AIV, with 50 versus 30 OTUs (Table 7), whereas on the species level the opposite pattern was discovered (AI: 178 OTUs, AIV: 236 OTUs).
Using Chao1 richness estimator, the maximum number of OTUs for the acidic spring bacterial community predicted at 3 % genetic distance was 229 belonging to 59 predicted phyla. With respect to site AIV (92 °C, pH8), 371 bacterial OTUs on species level and 33 OTUs on the phylum level were predicted by Chao1 richness estimator. The Shanon index (H′), determined for both habitats, indicated a slightly lower diversity in site AI than in site AIV (3 % genetic distance: AI: 1.8, AIV: 2.3; 20 % genetic distance: AI: 0.8, AIV: 1.0).
Archaeal diversity could only be estimated for site AI and was very low (H’: 0.54 at 3 % and 0.09 at 20 % genetic distance) as expected due to the high dominance of Thermoplasma (Table 6). At 3 % sequence divergence a maximum number of 99 OTUs were predicted while 21 OTUs were predicted at 20 % sequence divergence using Chao1 richness estimator.
Evaluation of the pyrosequencing-based analyses by group-specific fluorescence in situ hybridization
At site AIV relative abundance of Sulfurihydrogenibium and Fervidobacterium differed depending on the sequencing approach (Table 5). While sequencing of non-amplified metagenomic DNA indicated high abundance of Aquificae (approx. 28 %), in particular Sulfurihydrogenibium, the sequencing of partial 16S rRNA gene amplicons hardly detected any. On the other hand, amplicon sequencing indicated dominance of Fervidobacterium (approx. 47 %). We applied FISH to quantify and further analyse these specific groups including also Caldicellulosiruptor, which represented approx. 21 % of the bacterial community according to data derived from direct sequencing and pyrosequencing of the V2/V3 16S rRNA gene region. Dictyoglomus, the fourth dominant genus in sample AIV could not be evaluated because no specific probe could be designed.
Two probes had to be designed de-novo. The probes specific for Sulfurihydrogenibium and Fervidobacterium were developed on the basis of signature sequences described by Harmsen et al. (1997), which had been specific for Hydrogenobacter and Thermotoga. An exchange of one and two bases, respectively, allowed to change the specificity to the genera in question. Primer specificity was confirmed in-silico using ProbeMatch of the RDP Database Project and hybridization conditions were experimentally verified using controls with one mismatch. At the stringency conditions applied, no signal could be detected in cells with 1 mismatch to the probe. A comparison of all three approaches to quantify dominant bacterial genera is shown in Fig. 6. Quantification by FISH underlined the high abundance of Caldicellulosiruptor, revealing that this genus represented 28 % of all Bacteria (detected with EUB-specific probe) as compared to 19 and 21 % detected by sequencing of partial 16S rRNA gene amplicons and direct sequencing of metagenomic DNA, respectively. Furthermore, FISH indicated that Fervidobacterium accounted for 22 % of the bacterial community and thus had been overestimated by analysis of the V2/V3 16S rRNA region (47 %) and underestimated according to data derived from direct sequencing of metagenomic DNA (14 %). While Sulfurihydrogenibium was almost undetected by sequencing of the V2/V3 16S rRNA gene region (0.4 %), the genus was overestimated by direct sequencing of metagenomic DNA (17 %) since FISH revealed it to account for 9 % of the Bacteria. Altogether, almost 60 % of the bacterial cells could be assigned to the three genera Caldicellulosiruptor, Fervidobacterium, and Sulfurihydrogenibium using FISH.

Comparative quantification using FISH, V2/V3 and metagenome sequencing. The relative abundance relates to % of bacterial cells for FISH, % of bacterial rRNA gene sequences for V2/V3 and metagenome sequences
Discussion
Prokaryotic diversity
Hot springs are considered low-diversity habitats due to the high physicochemical constraints and the high degree of geographical isolation (Papke et al. 2003). The effect of temperature on prokaryotic diversity has been studied and results are contradictory. Yim et al. (2006) did not find a linear correlation between prokaryotic diversity and temperature in mats and streamers at temperatures between 52 and 69 °C in alkaline geothermal springs from Tibet. On the other hand, Miller et al. suggested that bacterial diversity along a temperature gradient from 39 to 72 °C in two alkaline hot springs from Yellowstone National Park is primarily controlled by temperature in photosynthetically active microbial mats which has also been shown by Hiraishi et al. in hot springs in Japan (Yim et al. 2006; Miller et al. 2009; Hiraishi et al. 1999). Furthermore, Skirnisdottir et al. (2000) found the lowest bacterial diversity at the highest temperature (80 °C) and highest sulphide concentration when comparing hot springs in Iceland with low and high concentration of sulphide. Based on DGGE analysis of nine sites, we found high bacterial diversity over a range of temperature between 55 and 85 °C as long as the pH was neutral to slightly alkaline. With respect to Archaea, the temperature range was slightly shifted upwards from 70 to 92 °C. In the hydrothermal springs investigated here, lowest bacterial diversity was detected at acidic pH and high temperature.
If phylogenetic distance is taken into account, sites AI and AIV display a distinctive difference. OTU-based analyses showed that bacterial diversity was more phylum rich in site AI compared to site AIV. However, site AIV was more species rich than site AI. Similar observations have been published previously (Roesch et al. 2007; Yim et al. 2006). Yim et al. suggested that extreme stress might inhibit the survival of large numbers of closely related guild taxa in microbial population from hot spring sediments and streamers leading to higher phylum numbers (Yim et al. 2006). Roesch et al. (2007) found that agricultural soil was phylum poor and species rich, whereas undisturbed forest soil showed the opposite pattern. However, the authors could not find a clear explanation for this. The factors causing the differences between the two habitats AI and AIV investigated here remain elusive. It is possible that coping with low pH is a property more widespread in the phylogenetic tree on phylum level than coping with temperatures above 90 °C. On the other hand it could also mean, that a habitat with temperature above 90 °C allows faster speciation than the low pH habitat. As long as there is no additional data available to show that this is not a coincidental observation, we can only speculate. Further comparative studies will be needed to evaluate this and to establish, whether it also holds for archaeal populations.
Diversity indices help to compare habitats. However, only few data are available on diversity indices from hot springs. For circumneutral springs at temperatures between 73 and 86 °C Costa et al. (2009) calculated Shanon indices between 2.02 and 2.37 at 3 % genetic distance for Bacteria and between 1.18 and 2.7 for Archaea. At a genetic distance of 20 %, the values were between 1.82 and 2.29 for Bacteria and between 0.95 and 2.16 for Archaea. For slightly alkaline springs along a temperature gradient from 52 to 69 °C Lau et al. (2009) calculated slightly higher Shanon indices between 3 and 4 at 3 % genetic distance for Bacteria. In acidic springs at temperatures between 60 and 75 °C Mathur et al. (2007) calculated Shanon indices for bacteria at 3 % genetic distance between 0.83 and 3.28 at springs with high sulphide and low iron concentration and between 2.1 and 3.6 in springs with high iron and low sulphide concentration. Shanon indices for bacteria calculated for sites AI and AIV fall within the same range. The acidic spring AI, which contained high iron and low sulphide concentrations, exhibited a Shanon index of 1.8 for Bacteria and 0.54 for Archaea at 3 % sequence divergence and values of 0.83 and 0.09 at 20 % sequence divergence. The slightly alkaline high-temperature (92 °C) spring AIV shows a Shanon index of 2.29 at 3 % and 1.04 at 20 % sequence divergence for Bacteria. These values are, as expected, lower than in terrestrial soils, which are considered the most complex microbial ecosystems on Earth, with Shanon indices of 5–7 at 3 % sequence divergence and 2.5–4.5 at 20 % sequence divergence (Will et al. 2010; Nacke et al. 2011). Shanon indices calculated for another extreme environment, glacier ice (3.4 and 1.7 for 3 and 20 % sequence divergence, respectively) (Simon et al. 2009), were in the same range as in our study.
Dominant metabolic pathways in the hot springs of Furnas
Inferring physiology from 16S rRNA gene phylogeny is inherently biased (Jaspers and Overmann 2004), in particular if the closest relatives are sequences from uncultured organisms. Keeping this in mind we suggest putative physiology on the basis of the closest cultivated 16S rRNA gene relatives for our habitat, in particular for AIV because a large proportion of the partial sequences is more than 99 % identical to sequences from cultivated organisms. For Fervidobacterium 91 % of the sequences were more than 99 % identical to F. islandicum, for Dictyoglomus 88 % of the sequences were more than 99 % identical to D. thermophilum, and for Caldicellulosiruptor 85 % of the sequences were more than 99 % identical to C. lactoaceticus. Altogether only the rare members of the sequence pool amounting to 11 % were <97 % identical to cultivated or related to uncultivated species. The situation is different for AI and here in particular for Acidicaldus. 94 % of the Acidicaldus-specific sequences were related to uncultivated organisms. However, 99 % of the Acidithiobacillus-sequences were 99 % identical to Acidithiobacillus caldus. This is in contrast to the majority of other environmental studies, in which the largest proportion of sequences does not affiliate with cultivated species (Meyer-Dombard et al. 2005; Hugenholtz et al. 1998; Ivanova et al. 2011; Pagaling et al. 2012).
Although differing for two groups (Sulfurihydrogenibium and Fervidobacterium), both 16S rRNA gene sequence-based approaches indicated the dominance of heterotrophic bacterial genera in both springs. This is in contrast to many other studies in hot spring-environments where Aquificales have been repeatedly found to dominate 16S rRNA gene-libraries, e.g. from Yellowstone National Park, Iceland, New Zealand, and Japan (Hall et al. 2008; Meyer-Dombard et al. 2005; Blank et al. 2002; Jackson et al. 2001; Skirnisdottir et al. 2000; Hetzer et al. 2007; Yamamoto et al. 1998). These findings led to the hypothesis that chemolithotrophic physiology probably based on the oxidation of H2 or reduced sulphur compounds is the major metabolic pathway in hot springs. Skirnisdottir et al. compared results from different studies on hot spring microbial mats and sediments and found that, depending on the chemical characteristics of the spring, different subgroups of Aquificales dominated (Yamamoto et al. 1998; Reysenbach et al. 2000; Hugenholtz et al. 1998). In high-sulphide and sometimes iron-rich habitats groups, J and S were dominating and the related sequences can today be attributed to Sulfurihydrogenibium. The high abundance (21 %) of Sulfurihydrogenibium-related sequences within the metagenome from site AIV fitted into the picture. However, evaluation with FISH revealed that Sulfurihydrogenibium had been overestimated and was only accounting for 9 % of the bacterial cells.
The comparatively low abundance of chemolithoautotrophic organisms at site AIV might be related to the high concentration of dissolved organic carbon (DOC) of 284 mg/L (0.028 %). Only little data are available on the DOC content of hot springs. Yamamoto et al. measured a DOC content of 0.41–0.72 mg/L within the microbial sulphur-turf mat, while Hetzer et al. determined 0.7 mg/L in Champagne Pool, New Zealand, and Hall et al. described a DOC content of 10 mg/L in Coffee Pot hot spring (YNP) (Yamamoto et al. 1998; Hetzer et al. 2007; Hall et al. 2008). All sites showed clear dominance of Aquificales. The more than 20-fold or even 400-fold higher concentration of DOC in the Furnas spring could well be a reason for the abundant occurence of heterotrophic bacteria. Regarding autotrophic processes at site AIV, however, also Archaea have to be taken into account, since FISH detected 35 % of the cells to be of archaeal origin. Metagenomic rRNA data suggest that the heterotrophic genera Sulfophobococcus spp. and Desulfurococcus spp. account for approx. 74 % of the sequences while the remaining 26 % may well be related to chemolithotrophic genera. Further studies will show, if high abundance of heterotrophic microorganisms is a common feature for springs with higher organic carbon content.
The dominant genera in site AIV (Caldicellulosiruptor, Dictyoglomus, and Fervidobacterium) have also been detected in in situ enrichment cultures in hot springs from Kamchatka (Kublanov et al. 2009). While Caldicellulosiruptor sp. and Dictyoglomus sp. grew on polysaccharides like cellulose and chitin, Fervidobacterium sp. was enriched on proteinaceous substrates. Although dominated by one OTU, all groups show additional sequence diversity in particular Dictyoglomus with 44 OTUs at 3 % divergence. For Fervidobacterium and Caldicellulosiruptor the number of specific OTUs is 18 and 14, respectively. These results suggest that the hot springs of Furnas are a valuable source to retrieve new polymer-degrading organisms.
Conclusions
The microbial community of two hot springs of Furnas Valley was characterized on the basis of different rRNA gene-based methods. The combination of different primer-dependent and independent methods revealed major differences, emphasizing the importance of combining different methodological approaches. A large dataset of short rRNA gene sequences and metagenome analysis revealed that the habitats were dominated by three to four genera, both for the bacterial and the archaeal population. Archaeal diversity was particularly limited in the acidic spring AI (pH 3, 51 °C), in which Thermoplasma represented almost 90 %. However, overall abundance of Archaea in this acidic environment was low as revealed by FISH, while the hot slightly alkaline spring AIV (pH 8, 92 °C) had almost even abundances of Archaea and Bacteria. Unlike other hydrothermal habitats, the high-temperature slightly alkaline low-sulphate spring AIV was dominated by heterotrophic bacteria, probably due to a high content of dissolved organic carbon, suggesting that not all hydrothermal habitats are dominated by chemolithoautotrophs and that a wide range of chemical characteristics needs to be taken into account when analysing hot springs. This supports the efforts of the Earth Microbiome Project which states the importance of careful and comprehensive metadata collection for each and in particular for metagenomic environmental studies in order to improve comparability, so that general patterns can be identified on the basis of different studies (Knight et al. 2012; Yilmaz et al. 2011).
Our results suggest a natural enrichment of heterotrophic, polymer-degrading genera in the Furnas springs which make them particularly promising for the search of novel thermostable enzymes for application in biotechnology and biorefinery of the second generation.
References
Albuquerque L, Rainey FA, Nobre MF, da Costa MS (2008) Elioraea tepidiphila gen. nov., sp. nov., a slightly thermophilic member of the Alphaproteobacteria. Int J Syst Evol Microbiol 58:773–778
Albuquerque L, Rainey FA, Nobre MF, da Costa MS (2010) Meiothermus granaticius sp. nov., a new slightly thermophilic red-pigmented species from the Azores. Syst Appl Microbiol 33:243–246
Albuquerque L, Rainey FA, Nobre MF, da Costa MS (2012) Hydrotalea sandarakina sp. nov., isolated from a hot spring runoff and emended description of the genus Hydrotalea and the species Hydrotalea flava. Int J Syst Evol Microbiol 62:1603–1608
Allen ET, Day AL (1935) Hot springs of Yellowstone National Park. Carnegie Institution of Washington Publication no. 466
Amann RI, Krumholz L, Stahl DA (1990) Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J Bacteriol 172:762–770
Baker GC, Cowan DA (2004) 16 S rDNA primers and the unbiased assessment of thermophile diversity. Biochem Soc Trans 32:218–221
Barth TFW (1950) Volcanic geology, hot springs and geysers of Iceland. Carnegie Institution of Washington Publication no. 587
Blank CE, Cady SL, Pace NR (2002) Microbial composition of near-boiling silica-depositing thermal springs throughout Yellowstone National Park. Appl Environ Microbiol 68:5123–5135
Brock TD, Brock ML (1967) The hot springs of Furnas Valley, Azores. Int Revue ges Hydrobiol 52:545–558
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Chao A, Bunge J (2002) Estimating the number of species in a stochastic abundance model. Biometrics 58:531–539
Costa KC, Navarro JB, Shock EL, Zhang CL, Soukup D, Hedlund BP (2009) Microbiology and geochemistry of great boiling and mud hot springs in the United States Great Basin. Extremophiles 13:447–459
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Franca L, Rainey FA, Nobre MF, da Costa MS (2006) Tepidicella xavieri gen. nov., sp. nov., a betaproteobacterium isolated from a hot spring runoff. Int J Syst Evol Microbiol 56:907–912
Friedrich AB, Antranikian G (1996) Keratin Degradation by Fervidobacterium pennavorans, a novel thermophilic anaerobic species of the order Thermotogales. Appl Environ Microbiol 62:2875–2882
Hall JR, Mitchell KR, Jackson-Weaver O, Kooser AS, Cron BR, Crossey LJ, Takacs-Vesbach CD (2008) Molecular characterization of the diversity and distribution of a thermal spring microbial community by using rRNA and metabolic genes. Appl Environ Microbiol 74:4910–4922
Hamamura N, Meneghin J, Reysenbach AL (2012) Comparative community gene expression analysis of Aquificales-dominated geothermal springs. Environ Microbiol
Harmsen H, Prieur D, Jeanthon C (1997) Group-specific 16S rRNA-targeted oligonucleotide probes to identify thermophilic bacteria in marine hydrothermal vents. Appl Environ Microbiol 63:4061–4068
Hetzer A, Morgan HW, McDonald IR, Daughney CJ (2007) Microbial life in Champagne Pool, a geothermal spring in Waiotapu, New Zealand. Extremophiles 11:605–614
Hiraishi A, Umezawa T, Yamamoto H, Kato K, Maki Y (1999) Changes in quinone profiles of hot spring microbial mats with a thermal gradient. Appl Environ Microbiol 65:198–205
Hugenholtz P, Pitulle C, Hershberger KL, Pace NR (1998) Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180:366–376
Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML (2008) Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet 4:e1000255
Inskeep WP, Rusch DB, Jay ZJ, Herrgard MJ, Kozubal MA, Richardson TH, Macur RE, Hamamura N, Jennings R, Fouke BW, Reysenbach AL, Roberto F, Young M, Schwartz A, Boyd ES, Badger JH, Mathur EJ, Ortmann AC, Bateson M, Geesey G, Frazier M (2010) Metagenomes from high-temperature chemotrophic systems reveal geochemical controls on microbial community structure and function. PLoS ONE 5:e9773
Ivanova I, Atanassov I, Lyutskanova D, Stoilova-Disheva M, Dimitrova D, Tomova I, Derekova A, Radeva G, Buchvarova V, Kambourova M (2011) High Archaea diversity in Varvara hot spring, Bulgaria. J Basic Microbiol 51:163–172
Jackson CR, Langner HW, Donahoe-Christiansen J, Inskeep WP, McDermott TR (2001) Molecular analysis of microbial community structure in an arsenite-oxidizing acidic thermal spring. Environ Microbiol 3:532–542
Jaspers E, Overmann J (2004) Ecological significance of microdiversity: identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl Environ Microbiol 70:4831–4839
Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, Hugenholtz P, van der Lelie D, Meyer F, Stevens R, Bailey MJ, Gordon JI, Kowalchuk GA, Gilbert JA (2012) Unlocking the potential of metagenomics through replicated experimental design. Nat Biotechnol 30:513–520
Kublanov IV, Perevalova AA, Slobodkina GB, Lebedinsky AV, Bidzhieva SK, Kolganova TV, Kaliberda EN, Rumsh LD, Haertle T, Bonch-Osmolovskaya EA (2009) Biodiversity of thermophilic prokaryotes with hydrolytic activities in hot springs of Uzon Caldera, Kamchatka (Russia). Appl Environ Microbiol 75:286–291
Kysela DT, Palacios C, Sogin ML (2005) Serial analysis of V6 ribosomal sequence tags (SARST-V6): a method for efficient, high-throughput analysis of microbial community composition. Environ Microbiol 7:356–364
Lau MC, Aitchison JC, Pointing SB (2009) Bacterial community composition in thermophilic microbial mats from five hot springs in central Tibet. Extremophiles 13:139–149
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17:10–12
Mathur J, Bizzoco RW, Ellis DG, Lipson DA, Poole AW, Levine R, Kelley ST (2007) Effects of abiotic factors on the phylogenetic diversity of bacterial communities in acidic thermal springs. Appl Environ Microbiol 73:2612–2623
Meyer-Dombard DR, Shock EL, Amend JP (2005) Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA. Geobiology 3:211–227
Miller SR, Strong AL, Jones KL, Ungerer MC (2009) Bar-coded pyrosequencing reveals shared bacterial community properties along the temperature gradients of two alkaline hot springs in Yellowstone National Park. Appl Environ Microbiol 75:4565–4572
Muyzer G, Brinkhoff T, Nübel U, Santegoeds C, Schäfer H, Wawer C (1998) Denaturing gradient gel electrophoresis (DGGE) in microbial ecology. In: Akkermans ADL, van Elsas JD, de Bruijn FJ (eds) Molecular microbial ecology manual. Kluwer Academic Publishers, London, pp 3.4.4.1–3.4.4.27
Nacke H, Thurmer A, Wollherr A, Will C, Hodac L, Herold N, Schoning I, Schrumpf M, Daniel R (2011) Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS ONE 6:e17000
O-Thong S, Prasertsan P, Karakashev D, Angelidaki I (2007) Specific detection of Thermoanaerobacterium spp., Thermoanaerobacterium thermosaccharolyticum and Caldicellulosiruptor spp. in thermophilic biohydrogen reactor using fluorescent in situ hybridization (FISH). Int J Hydrogen Energy 33:6082–6091
Pagaling E, Grant WD, Cowan DA, Jones BE, Ma Y, Ventosa A, Heaphy S (2012) Bacterial and archaeal diversity in two hot spring microbial mats from the geothermal region of Tengchong, China. Extremophiles 16:607–618
Papke RT, Ramsing NB, Bateson MM, Ward DM (2003) Geographical isolation in hot spring cyanobacteria. Environ Microbiol 5:650–659
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196
Ravenschlag K, Sahm K, Knoblauch C, Jorgensen BB, Amann R (2000) Community structure, cellular rRNA content, and activity of sulfate-reducing bacteria in marine arctic sediments. Appl Environ Microbiol 66:3592–3602
Reysenbach AL, Wickham GS, Pace NR (1994) Phylogenetic analysis of the hyperthermophilic pink filament community in Octopus Spring, Yellowstone National Park. Appl Environ Microbiol 60:2113–2119
Reysenbach AL, Ehringer M, Hershberger K (2000) Microbial diversity at 83 °C in Calcite Springs, Yellowstone National Park: another environment where the Aquificales and “Korarchaeota” coexist. Extremophiles 4:61–67
Riessen S, Antranikian G (2001) Isolation of Thermoanaerobacter keratinophilus sp. nov., a novel thermophilic, anaerobic bacterium with keratinolytic activity. Extremophiles 5:399–408
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD, Daroub SH, Camargo FA, Farmerie WG, Triplett EW (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Shanon WE, Weaver W (1949) The mathematical theory of communication. MD Comput 14:306–317
Simon C, Wiezer A, Strittmatter AW, Daniel R (2009) Phylogenetic diversity and metabolic potential revealed in a glacier ice metagenome. Appl Environ Microbiol 75:7519–7526
Skirnisdottir S, Hreggvidsson GO, Hjorleifsdottir S, Marteinsson VT, Petursdottir SK, Holst O, Kristjansson JK (2000) Influence of sulfide and temperature on species composition and community structure of hot spring microbial mats. Appl Environ Microbiol 66:2835–2841
Snaidr J, Amann R, Huber I, Ludwig W, Schleifer KH (1997) Phylogenetic analysis and in situ identification of bacteria in activated sludge. Appl Environ Microbiol 63:2884–28896
Spear JR, Walker JJ, McCollom TM, Pace NR (2005) Hydrogen and bioenergetics in the Yellowstone geothermal ecosystem. Proc Natl Acad Sci USA 102:2555–2560
Stahl DA, Amann RI (1991) Development and application of nucleic acid probes. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, Chichester, pp 205–248
Swingley WD, Meyer-Dombard DR, Shock EL, Alsop EB, Falenski HD, Havig JR, Raymond J (2012) Coordinating environmental genomics and geochemistry reveals metabolic transitions in a hot spring ecosystem. PLoS ONE 7:e38108
Will C, Thürmer A, Wollherr A, Nacke H, Herold N, Schrumpf M, Gutknecht J, Wubet T, Buscot F, Daniel R (2010) Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol 76:6751–6759
Yamamoto H, Hiraishi A, Kato K, Chiura HX, Maki Y, Shimizu A (1998) Phylogenetic evidence for the existence of novel thermophilic bacteria in hot spring sulfur-turf microbial mats in Japan. Appl Environ Microbiol 64:1680–1687
Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, Gilbert JA, Karsch-Mizrachi I, Johnston A, Cochrane G, Vaughan R, Hunter C, Park J, Morrison N, Rocca-Serra P, Sterk P, Arumugam M, Bailey M, Baumgartner L, Birren BW, Blaser MJ, Bonazzi V, Booth T, Bork P, Bushman FD, Buttigieg PL, Chain PS, Charlson E, Costello EK, Huot-Creasy H, Dawyndt P, DeSantis T, Fierer N, Fuhrman JA, Gallery RE, Gevers D, Gibbs RA, San Gil I, Gonzalez A, Gordon JI, Guralnick R, Hankeln W, Highlander S, Hugenholtz P, Jansson J, Kau AL, Kelley ST, Kennedy J, Knights D, Koren O, Kuczynski J, Kyrpides N, Larsen R, Lauber CL, Legg T, Ley RE, Lozupone CA, Ludwig W, Lyons D, Maguire E, Methe BA, Meyer F, Muegge B, Nakielny S, Nelson KE, Nemergut D, Neufeld JD, Newbold LK, Oliver AE, Pace NR, Palanisamy G, Peplies J, Petrosino J, Proctor L, Pruesse E, Quast C, Raes J, Ratnasingham S, Ravel J, Relman DA, Assunta-Sansone S, Schloss PD, Schriml L, Sinha R, Smith MI, Sodergren E, Spo A, Stombaugh J, Tiedje JM, Ward DV, Weinstock GM, Wendel D, White O, Whiteley A, Wilke A, Wortman JR, Yatsunenko T, Glockner FO (2011) Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol 29:415–420
Yim LC, Hongmei J, Aitchison JC, Pointing SB (2006) Highly diverse community structure in a remote central Tibetan geothermal spring does not display monotonic variation to thermal stress. FEMS Microbiol Ecol 57:80–91
Youssef N, Sheik CS, Krumholz LR, Najar FZ, Roe BA, Elshahed MS (2009) Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl Environ Microbiol 75:5227–5236
Yu Z, Garcia-Gonzalez R, Schanbacher FL, Morrison M (2008) Evaluations of different hypervariable regions of archaeal 16S rRNA genes in profiling of methanogens by Archaea-specific PCR and denaturing gradient gel electrophoresis. Appl Environ Microbiol 74:889–893
Zhou J, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:316–322
Acknowledgments
The authors gratefully acknowledge Milton da Costa for supporting their sampling on a field trip to the Valley of Furnas.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. da Costa.
Rights and permissions
About this article
Cite this article
Sahm, K., John, P., Nacke, H. et al. High abundance of heterotrophic prokaryotes in hydrothermal springs of the Azores as revealed by a network of 16S rRNA gene-based methods. Extremophiles 17, 649–662 (2013). https://doi.org/10.1007/s00792-013-0548-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-013-0548-2