
Abstract
Recently, unusual non-regulated ATP-dependent 6-phosphofructokinases (PFK) that belong to the PFK-B family have been described for the hyperthermophilic archaea Desulfurococcus amylolyticus and Aeropyrum pernix. Putative homologues were found in genomes of several archaea including the hyperthermophilic archaeon Methanocaldococcus jannaschii. In this organism, open reading frame MJ0406 had been annotated as a PFK-B sugar kinase. The gene encoding MJ0406 was cloned and functionally expressed in Escherichia coli. The purified recombinant enzyme is a homodimer with an apparent molecular mass of 68 kDa composed of 34 kDa subunits. With a temperature optimum of 85°C and a melting temperature of 90°C, the M. jannaschii nucleotide kinase represents one of the most thermoactive and thermostable members of the PFK-B family described so far. The recombinant enzyme was characterized as a functional nucleoside kinase rather than a 6-PFK. Inosine, guanosine, and cytidine were the most effective phosphoryl acceptors. Besides, adenosine, thymidine, uridin and xanthosine were less efficient. Extremely low activity was found with fructose-6-phosphate. Further, the substrate specificity of closely related PFK-Bs from D. amylolyticus and A. pernix were reanalysed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyperthermophilic prokaryotes are characterized by an optimal growth temperature higher than 80°C. They are considered to represent the phylogenetic most ancestral organisms (Stetter 1996). Recent comparative studies of the hexose degradation pathways in hyperthermophilic archaea and in the hyperthermophilic bacterium Thermotoga revealed that the classical Embden–Meyerhof (EM) pathway is operative only in Thermotoga, whereas in all archaea analysed so far, the EM pathway exists in modified versions. These modified pathways contain several unusual enzymes, e.g. non-regulatory 6-phosphofructokinases (PFKs) (for literature see Selig et al. 1997; Brunner et al. 1998; Hansen et al. 2002b; Ronimus and Morgan 2003; Verhees et al. 2003; Siebers and Schönheit 2005). 6-PFKs are present in all domains of life—Eukarya, Bacteria, and Archaea (for review see Ronimus and Morgan 2001). They catalyse the phosphorylation of fructose 6-phosphate (F-6-P) to fructose 1,6-bisphosphate with either ATP (ATP-PFK, EC 2.7.1.11), pyrophosphate (PP i -PFK, EC 2.7.1.90), or ADP (ADP-PFK, EC 2.7.1.146) as phosphoryl donor. Sequence and structural analyses group them into two convergent protein families (see SCOP http://www.scop.mrc-lmb.cam.ac.uk/scop/): PFK-A-family [SCOP 53785; Pfam PF00365] and the ribokinase superfamily [SCOP 53613], which include the PFK-B-family [Pfam, PF00294] and the ADP-PFK/GLK-family [Pfam, PF04587]. The PFK-A family comprises both ATP-dependent and PP i -dependent enzymes and is predominant in Eukarya and Bacteria. ATP-PFKs from this family including the enzyme from the hyperthermophile Thermotoga maritima represent key regulatory sites of sugar degradation via EM-pathway (Ronimus and Morgan 2001; Hansen et al. 2002a). In contrast, all archaeal PFKs described so far were characterized by a lack of allosteric response. Thus, they do not represent regulatory sites in the modified archaeal EM-pathways (for review see Ronimus and Morgan 2003; Verhees et al. 2003; Siebers and Schönheit 2005). The modified EM pathways of the euryarchaota Pyrococcus, Thermococcus, and Archaeoglobus fulgidus 7324 contain ADP-dependent 6-PFK (Kengen et al. 1994; Tuininga et al. 1999; Kengen et al. 2001; Labes and Schönheit 2001; Ronimus et al. 2001a; Hansen and Schönheit 2004), whereas in Methanocaldococcus jannaschii, a bifunctional ADP-dependent glucokinase/6-PFK was found, which might be involved in glycogen degradation (Verhees et al. 2001; Sakuraba et al. 2002). A pyrophosphate-dependent PFK was described for the crenarchaeon Thermoproteus (Siebers and Hensel 2001). We have recently characterized the non-regulatory ATP-PFKs from the crenarchaeota Desulfurococcus amylolyticus (DaPFK) and Aeropyrum pernix (ApPFK), which belong to the PFK-B family (Hansen and Schönheit 2000, 2001). The PFK-B protein family is a diverse family of ATP-dependent carbohydrate and pyrimidine kinases present in all domains. It comprises e.g. fructokinases, ribokinases, adenosine kinases, and the minor ATP-PFK from Escherichia coli (PFK-B or PFK2) (Bork et al. 1993). Several structures from the PFK-B family have been described so far: e.g. E. coli ribokinase, human and Toxoplasma gondii adenosine kinases, 2-keto-3-deoxy-gluconate (KDG) kinases from Thermus thermophilus and T. maritima, sheep pyridoxal kinase, and putative 1-PFK from T. maritima (Mathews et al. 1998; Sigrell et al. 1998; Schumacher et al. 2000; Li et al. 2002; Ohshima et al. 2004).
Five archaeal members of the PFK-B family have been biochemically characterized. These include fructokinase from Thermococcus litoralis, KDG kinases from T. tenax and Haloferax alicantei, as well as the DaPFK and the ApPFK (Holmes et al. 1997; Hansen and Schönheit 2000, 2001; Ronimus et al. 2001b; Qu et al. 2004; Siebers et al. 2004). Besides a lack of allosteric potential, the ApPFK exhibited a broad substrate specificity. However, structural analyses to understand these features have been hampered by the low expression level of this enzyme in A. pernix as well as for the recombinant enzyme in E. coli (Hansen and Schönheit 2001). A putative homolog of ApPFK, MJ0406, which had been annotated as a hypothetical PFK-B sugar kinase, has been found in the genome of M. jannaschii (Bult et al. 1996; Hansen and Schönheit 2001) indicating that M. jannaschii in addition to the ADP-PFK might contain an ATP-PFK as well.
Here, we describe cloning and heterologous expression of the open reading frame (ORF) MJ0406. The purified recombinant enzyme was characterized as a nucleoside kinase rather than an ATP-PFK.
Materials and methods
Identification of hypothetical ORFs
BLASTP searches were carried out to identify putative homologues of ORF MJ0406 (Hansen and Schönheit 2001). Additional archaeal sequences of the PFK-B family were retrieved from the PROSITE database and the Pfam database. Sequence alignments were constructed using the neighbour-joining method of Clustal X (Thompson et al. 1997).
Cloning and expression of ORF MJ0406
Open reading frame MJ0406 has previously been annotated as putative sugar kinase mjsk gene in the genome of M. jannaschii (Bult et al. 1996). This ORF was cloned and functionally overexpressed in E. coli as follows: The coding region of the gene was amplified by PCR with Pwo polymerase (PEQLAB, Erlangen, Germany) from genomic DNA of M. jannaschii as a template. Using the following primers 5′CTTAAAACATATGGGTGGTAAAATG3′ (forward) and 5′ATAGACATCCCTCGAGAAATTATAT3′ (reverse), restriction sites (underlined) for NdeI and XhoI, respectively, were introduced. After amplification and NdeI/Xho1 double-digestion, the PCR product was inserted by T4 DNA ligase (Roche Diagnostics, Mannheim, Germany) into a pET-17b (Novagen, Madison, WI, USA) expression vector linearized by NdeI/XhoI double-digestion. The resulting ORF MJ0406 carrying pET-17b-mjsk plasmid was introduced into E. coli JM109 and E. coli BL 21 (DE3) pLys S (Stratagene, La Jolla, CA, USA) via transformation. The inserted gene was confirmed by sequencing. Transformed E. coli BL 21 (DE3) pLys S cells were grown in 400 ml of Luria–Bertani medium containing 100 μg ml−1 carbenicillin and 34 μg ml−1 chloramphenicol at 37°C to an optical density at 600 nm of 0.8. Expression of the mjsk was initiated by induction with 1 mM IPTG. After 4 h of further growth (OD600 ∼ 3.2–3.6), the cells were harvested by centrifugation at 4°C, washed in 50 mM Tris/HCl, pH 7.0, containing 50 mM NaCl. Exponentially grown cells of M. jannaschii (100 mg) were disrupted by freezing in liquid nitrogen and subsequent thawing. RNA was isolated using the RNeasy isolation kit (Qiagen, Valencia, CA, USA) as specified by the manufacturer. RT-PCR was carried out by the Qiagen one Step RT-PCR kit using the PCR primers. RNA, which has not been incubated with reverse transcriptase, was used as negative control.
Purification of recombinant MjNK
All chromatographic steps were carried out at 4°C. Cell extracts were prepared by French press treatment (1.3×108 Pa) of cell suspensions in buffer A (50 mM Tris–HCl pH 8.4). After ultracentrifugation (100,000×g for 60 min), the solution was heat precipitated at 80°C for 45 min, centrifuged again (15,000×g for 30 min), three times dialysed against a 30-fold excess of 50 mM Tris–HCl pH 8.0 (buffer B), and applied to a DEAE- Sepharose column (60 ml) previously equilibrated with buffer B. The protein was eluted at a flow rate of 3 ml min−1 with 180 ml 50 mM piperazine pH 6.5 at 25°C (buffer C) and with a combination of fixed NaCl concentration and linear gradients from 0 M NaCl to 2 M NaCl in buffer B: 0–0.1 M (120 ml), 0.1–1 M (180 ml), 1–2 M (120 ml), and 2 M NaCl (120 ml). Fractions containing the highest activity (160 ml, 0.2–1 M NaCl) were pooled and diluted with 3 M ammonium sulphate in buffer B to a final ammonium sulphate concentration of 2 M. Subsequently, the solution was applied to a phenyl-Sepharose column (15 ml), previously equilibrated with 2 M ammonium sulphate in buffer B. The protein was desorbed by a linear ammonium sulphate gradient (2–0 M, 150 ml), followed by washing the column with 60 ml 50 mM Tris–HCl pH 8.0 and 60 ml water. Fractions containing the highest activity (75 ml, 1.2–0.2 M ammonium sulphate) were checked for purity with respect to protein and DNA impurities. Only almost pure MjNK (45 ml, 0.8–0.2 M ammonium sulphate) was used for further purification, concentrated by ultrafiltration (exclusion-size 10 kDa), and then applied to a Superdex 200 gel filtration column (120 ml) equilibrated with buffer D (150 mM NaCl, 50 mM Tris–HCl pH 7.5). From this step, pure MjNK was obtained (72–86 ml).
Analytical assays
The purity of the preparation was checked by SDS-PAGE in 12% gels followed by staining with Compassion Brilliant Blue R 250 according to standard procedures (Laemmli 1970). Protein concentrations were determined by the use of MjNK molar extinction coefficient at 280 nm (37,550 M−1 cm−1) as calculated by the ExPASy ProtParam Tool (http://us.expasy.org/tools/protparam.html). Molecular mass determination was performed by a Superdex 200 gel filtration column and carried out at ambient temperature in 50 mM Tris/HCl, 150 mM NaCl, pH 7.0 at a flow rate of 1 ml min−1.
Enzyme assays and determination of kinetic parameters
Since the enzyme activity was not sensitive to oxygen, all assays were performed under oxic conditions. The ATP-dependent MjNK activity with guanosine as substrate was measured at 50°C in a continuous assay and at 70°C in a discontinuous assay by coupling the ADP formation from ATP to the oxidation of NADH via pyruvate kinase and lactate dehydrogenase as described previously (Hansen and Schönheit 2000). The pH dependence of the enzyme was measured between 4.0 and 9.0 at 70°C using either piperazine, phosphate, MES, Tris/HCl, triethanolamine, or ethanolamine at a concentration of 100 mM each with inosine and guanosine (separate assays) as substrates. The cation specificities were examined at 70°C by exchanging Mg2+ (5 mM) for alternative divalent cations (Ni2+, Mn2+, Co2+, Ca2+, Zn2+, and Fe2+). In order to analyse the substrate specificity, the following substances were tested as putative substrates: fructose-6-phosphate, glucose-6-phosphate, mannose-6-phosphate, fructose, galactose, glucose, d-glucose, ribose, xylose, N-acetylglucosamine, adenosine, 2-deoxy-adenosine, cytidine, inosine, guanosine, thymidine, uridine, and xanthosine. For the determination of the temperature dependence of enzyme activity as well as the phosphoryl donor specificity of the enzyme, activity was determined in a continuous assay by coupling the formation of fructose-6-phosphate from fructose by MjNK to the reduction of NADP via phosphoglucose isomerase from Pyrococcus furiosus (Hansen et al. 2001) and glucose-6-phosphate dehydrogenase from T. maritima (Hansen et al. 2002c) as recently described for PaPGI/PMI (Hansen et al. 2004). The temperature dependence of the enzyme activity was measured between 70 and 95°C in 50 mM sodium phosphate buffer pH 7.0 using 2.5 mM ATP and 10 mM fructose. The phosphoryl donor specificity was tested by exchanging ATP (2 mM) for alternative phosphoryl donors (ITP, GTP, UTP, CTP, UDP, GDP, ADP, CDP, acetyl phosphate, and PP i ) at equimolar concentrations.
Thermal stability
Differential scanning calorimetry was carried out on a Microcal MCS calorimeter suitable for scanning rates up to 2 K min−1. The calorimeter was controlled by the MCS OBSERVER program (Microcal, Studio City, CA, USA). MjNK was dialysed against 20 mM potassium phosphate pH 7.0. Experiments were performed with a protein concentration of 2 mg ml−1, and both the buffer and sample were degassed for 30 min before being loaded into the calorimeter. To stabilize the baseline, six prescans were carried out before the sample scan. The scans were run from 20 to 120°C at 1°C min−1.
Results
In vivo transcription of mjnk in M. jannaschii
To test whether the mjnk gene is transcribed in vivo, RT-PCR experiments were performed to detect mRNA formation. Total RNA was extracted from M. jannaschii cells, grown lithoautotrophically on H2 and CO2 as energy and carbon source. Mjnk-specific RNA was amplified as cDNA by RT-PCR. mjnk-specific cDNA of the expected length (900 bp) was detected, indicating in vivo transcription of mjnk gene during lithoautotrophic growth.
Cloning, functional overexpression of ORF MJ0406, purification and molecular characterization of the recombinant protein
The ORF MJ0406 was previously identified as a homolog of the ApPFK (Hansen and Schönheit 2001) and had been annotated as a putative sugar kinase (Bult et al. 1996). ORF Mj0406 contains 909 bp coding for a polypeptide of 302 amino acids with a calculated molecular mass of 33.8 kDa. The coding function of this putative enzyme was analysed by its functional overexpression in E. coli and subsequent enzymatic analyses. The gene encoding for ORF MJ0460 was amplified by PCR, cloned into a pET-17b vector, and transformed into E. coli BL21 pLysS cells. After induction with IPTG, a polypeptide of 34 kDa was produced. The recombinant protein was purified from transformed E. coli about 15-fold to homogeneity by heat treatment at 80°C and subsequent chromatography on DEAE-Sepharose, Phenyl-Sepharose, and gel filtration. About 0.3 mg of purified protein was obtained from 1 g of transformed E. coli cells. The purified protein was electrophoretically homogeneous as judged by denaturing SDS-PAGE (Fig. 1), showing one subunit with an apparent molecular mass of 34 kDa. Since gel filtration of the native enzyme revealed a molecular mass of 68 kDa, a homodimeric α 2 structure has to be assumed (Table 1).

Thermal unfolding of MjNK as monitored by DSC
Temperature optimum and stability
The temperature dependence of MjNK was analysed between 40 and 95°C (not shown). At 40°C, the enzyme showed little activity, which, however, increased exponentially showing an apparent optimum at 85°C (Table 1). From the linear part of the Arrhenius plot between 40 and 80°C (not shown), activation energy of 57 kJ mol−1 was calculated. Thermal unfolding of the enzyme was monitored by DSC in 20 mM sodium phosphate buffer. The detected apparent T m-values were 90°C (Fig. 1). The DSC showed a second, but minor peak at 85°C, which might indicate subunit dissociation of the dimeric protein at this temperature.
Substrate specificity and catalytic properties
The purified enzyme catalysed the ATP-dependent phosphorylation of fructose-6-phosphate at an extremely low rate (0.01 U mg−1, 70°C). In order to analyse the substrate specificity, the ATP-dependent phosphorylation of several sugars, sugar-phosphates, and nucleosides was tested at 50 and 70°C (Table 2). Whereas all sugar-phosphates and sugars tested were phosphorylated by ATP to a very low extent, several nucleosides were used at significantly higher rates. Thus, the enzyme was defined as an ATP-dependent nucleoside kinase (MjNK) rather than an ATP-PFK. Among the nucleosides tested, highest catalytic efficiency was found for inosine, cytidine, and guanosine with apparent K m-values (50°C) of 21, 17, and 78 μM, respectively; and apparent V max-values (50°C) were about 18.8, 9, and 29.3 U mg−1, respectively (Table 2). Adenosine, xanthosine, 2-desoxy-adensosine, uridine, and thymidine were used at lower rates (Table 2). Rate dependence of all substrates analysed as well as of ATP followed Michaelis–Menten kinetics. The apparent K m-value for ATP measured with guanosine as phosphoryl acceptor was 0.25 mM (Table 1). Neither ADP, acetyl phosphate, nor PP i could replace ATP defining this archaeal MjNK as an ATP-dependent enzyme. Besides ATP (100%), ITP (41%) and GTP (40%) served as effective phosphoryl donors. Enzyme activity required the presence of divalent cations. Mg2+ (100%) could be efficiently replaced by Mn2+ (97%) and partially by Ni2+ (31%) or Co2+ (6%). The pH optimum of ATP-GLK was at pH 7.0; 50% of activity was found at pH 6.1 and 8.2.
Substrate specificities of DaPFK and ApPFK
Considering the broad substrate specificity of MjNK, the respective substrate specificities of its homologues DaPFK and ApPFK were reanalysed using several nucleosides at 5 mM each (Table 3). DaPFK used the tested nucleosides only to a very low extent. However, ApPFK phosphorylated inosine, xanthosine, and cytidine about to the same extent, but guanosine even at higher rates as fructose-6-phosphate.
Discussion
Molecular and catalytical properties
In this communication, we report cloning, heterologous expression, and characterization of a PFK-B from the euryarchaeon M. jannaschii. ORF MJ0406 was cloned due to its homology to the PFK-B ATP-PFKs from D. amylolyticus and A. pernix (Hansen and Schönheit 2000, 2001), indicating that this ORF might represent an ATP-dependent PFK as well, and thus would represent a second PFK in M. jannaschii in addition to the characterized ADP-PFK (Sakuraba et al. 2002). However, the recombinant protein exhibited some unique properties that separated it from the crenarchaeal ATP-PFKs from D. amylolyticus and A. pernix. First, the recombinant enzyme had a molecular mass of 68 kDa and was composed of a single 34 kDA subunit indicating a homodimeric structure. In comparison, DaPFK and ApPFK were characterized as homotetrameric enzymes (Hansen and Schönheit 2000, 2001). Both subunit size and oligomeric structure are typical properties of enzymes from the PFK-B family, most of them were described as homodimeric enzymes, e.g. T. litoralis fructokinase represents a homodimeric enzyme with 35 kDA subunits (Qu et al. 2004). In comparison, proteins from the PFK-A family and ADP-PFKs have subunits of about 34 and 50 kDA, respectively (Ronimus et al. 1999; Tuininga et al. 1999; Ding et al. 2000, 2001; Ronimus and Morgan 2001; Hansen et al. 2002a; Hansen and Schönheit 2004). The extremely high temperature optimum of activity at 85°C and thermostability of M. jannaschii MjNK is in accordance with its physiological function under hyperthermophilic growth conditions. In comparison, the temperature optima of DaPFK and ApPFK were at 90°C and of T. litoralis fructokinase at 80°C (Hansen and Schönheit 2000, 2001; Qu et al. 2004).
Most strikingly, MjNK showed the highest catalytic activity as a nucleoside kinase but only extremely low activity as PFK. Thus, MjNK probably plays a role in the nucleoside metabolism of M. jannaschii. The substrate specificity of MjNK is quite unusual with guanosine, inosine, and cytidine showing the highest catalytic activity. The guanosine kinases from Exiguobacterium acetylicum and E. coli were reported to phosphorylate guanosine and to some extent inosine (Mori et al. 1995; Usuda et al. 1997). Inosine differs from guanosine by lacking the amino group at C2 of the purine base, whereas cytidine contains a pyrimidine base. Though the PFK-B family includes pyrimidine kinases as well (see Pfam), the broad substrate range of the MjNK is, to our best knowledge, unique within the PFK-B family. This family includes pyrimidine kinases (see Pfam PF00294), e.g. the hydroxymethylpyrimidine kinase/phosphomethylpyrimidine kinase from E. coli (Mizote et al. 1999), but a PFK-B with cytidine kinase activity has not been described for a PFK-B kinase yet (Liacouras and Anderson 1975; Van Rompay et al. 2001). Further structural analyses will help to understand the substrate specificity of this enzyme. The recombinant enzyme could be produced in significant quantities, a prerequisite for crystallization and structural analysis, which are in progress. The extended reanalysation of DaPFK and ApPFK revealed that DaPFK represents a specific ATP-PFK, whereas the enzyme from A. pernix exhibits a broad substrate specificity with similar activities for F6P and various nucleosides. Therefore, this enzyme from A. pernix should be reassigned as ApPFK/NK. Consequently, DaPFK plays a role in glucose catabolism and MjNK in nucleotide metabolism, whereas ApPFK/NK might have a dual function in both glucose catabolism and nucleotide metabolism.
The distinct substrate specificity of hyperthermophilic MjNK cannot be explained on the sequence level yet. Like other PFK-Bs, the sequence of MjNK contains the two signature patterns of the PFK-B family (Hansen and Schönheit 2001). BLASTP searches of the non-redundant database with the sequence of the MjNK revealed more than 500 hits (March 2006), including a variety of several archaeal members of the PFK-B family (Table 4). The six best scores were obtained with putative methanogenic members of the PFK-B family (MMP0418, MM2358, ZP_00148642, ZP_00295129, MA373, MTH1544) indicating that these ORFs might encode for proteins with a substrate specificity similar to the one of MjNK. The MjNK shows the highest degree of sequence similarity to its putative homolog ORF MMP0418 from Methanococcus maripaludis (76% similarity). The sequence similarity of MjNK to ApPFK/NK is still significant (49%), but quite low to the E. acetylicum guanosine kinase (31%) or to the KDG kinase from T. tenax (20%).
Sequence comparison and phylogenetic affiliation
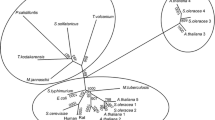
A multiple sequence alignment of the MjNK with characterized archaeal members of the PFK-B family (DaPFK, ApPFK/NK, fructokinase T. litoralis, KDG kinases from T. tenax, and H. alicantei) (Holmes et al. 1997; Hansen and Schönheit 2000, 2001; Ronimus et al. 2001b; Qu et al. 2004; Siebers et al. 2004) is given in Fig. 2. Presumably due to the diverse functions of the PFK-B proteins, only few residues are conserved, which are found in the two consensus patterns. The MjNK shows two deviations from these patterns. The characterization of the MjNK along with the reanalysation of the substrate specificity of ApPFK/NK allows tree constructions with respect to both phylogeny and function. For the majority of genomes from both archaeal hyperthermophilic species which have been sequenced so far, several proteins with different function have been annotated belonging to the PFK-B-family (e.g. see Table 4). Presumably, these paralogous sequences have evolved from an ancestral sequence by a combination of gene duplications and functional diversification. Indeed, the evolution of PFK-B appears to be complex as demonstrated in the phylogram in Fig. 3, which includes selected members of the PFK-B family as well as both characterized and putative archaeal PFK-B sequences. The PFK-B sequences cluster according to function and/or phylogeny. These overlapping evolutionary influences are probably also reflected by the low bootstrap values of the basal nodes. At least eleven groups could be discerned: (1) various bacterial PFK-B kinases, (2) bacterial and archaeal pyrimidine kinases, (3) mammalian ketohexokinaes, (4) bacterial and archaeal heptose-7-phosphate kinases, (5) archaeal and bacterial KDG-kinases, (6) fructokinases from all three domains, (7) archaeal PFK-Bs including the ApPFK/NK, (8) bacterial and archaeal ribokinases, (9) halobacterial PFK-Bs, (10) euryarchaeal and bacterial nucleoside kinases including the MjNK, and (11) eukaryal adenosine kinases.

Multiple sequence alignment of amino acid sequences of MjNK and the ApPFK/NK and its archaeal paralogous: Thermococcus litoralis fructokinase, Halobacterium alicantei KDG-kinase, Thermoproteus tenax KDG kinase (for accession numbers see Fig. 4). The alignment was generated by clustalX (Thompson et al. 1997). The two signature patterns of the PFK-B family: [AG]-G-X(0,1)-[GAP]-X-N-X-[STA]-X(2)-A-X-G-X-[GS]-X(9)-G and [DNSK]-[PSTV]-X-[SAG](2)-[GD]-D-X(3)-[SAGV]-[AG]-[LIVMFYA]-[LIVMSTAP] are printed bold and the respective deviations are highlighted

Phylogenetic relationships of PFK-B proteins. The tree was generated using the neighbour-joining method of clustalX (Thompson et al. 1997). Bootstrap values are based on 1,000 replicates and are given at each node (only values above 300 are shown). The accession numbers of the sequences as well as their annotation are given in brackets, characterized archaeal proteins (TxKDGK, HaKDGK, TlFrK, ApPFK/NK, MjNK) are marked bold. Abbreviations used for enzymes: GSK guanosine kinase, TK tagatose kinase, Fr1K fructose-1-kinase, PFK phosphofructokinase, THK hydroxyethylthiazole kinase, Pyk pyrimidine kinase, PmPK phosphomethylpyrimidine kinase/hydroxymethylpyrimidine kinase, HMPK hydroxymethylpyrimidine kinase, SK sugar kinase, KHK ketohexose kinase, HLDE heptose-7-phosphate kinase, KDGK 2-keto-3-deoxy-glucomnate kinase, Frk fructokinase, RK ribokinase, NK nucleoside kinase, AK adenosine kinase. Abbreviations used for species: Aa Aquifex aelicus, Af Archaeoglobus fulgidus, Ap Aeropyrum pernix, Acp Actinobacillus pleuromonia, Bcr Babesia canis rossi, Bs Bacillus subtilis, Cg Cricetulus griseus, Ec E. coli, Ea Exiguabacterium acetylicum, Gs Geobacillus stearothermophilus, Ha Halobacterium alicantei, Hl Halobacterium NRC 1, Hm Halobacterium marismortui, Ll Lactococcus lactis, m mouse, Ma Methanosarcina acetivorans, Mb Methanosarcina barkeri, Mj Methanocaldococcus jannaschii, Mk Methanopyrus kandleri, Mm Methanosarcina mazei, Mmp Methanococcus maripaludis, Mt Methanothermobacter thermoautotrophicus, MtM Methanothermobacter marburgensis, Pae Pyrobaculum aerophilum, Pa Pyrococcus abyssii, Pf Pyrococcus furiosus, Ph Pyrococcus horikoshii, Psa Pseudomonas aeruginosa, Pt Picrophilus torridus, Rl Rhizobium leguminosarum, Sa Staphylococcus aureus, Sat Salmonella typhimurium, Sato Salmonella thompson, Sm Streptococcus mutans, Ss Sulfolobus solfataricus, St Sulfolobus tokodai, Sotu Solanum tuberosum, Ta Thermoplasma acidophilum, Tg Toxoplasma gondii, Tk Thermococcus kodakarensis, Tl Thermococcus litoralis, Tm Thermotoga maritima, Tt Thermus thermophilus, Ttx Thermoproteus tenax, Tv Thermoplasma volcanium, Va Vibrio alginolyticus, Xp Xanthomonas campestris, zm Zea maize
Archaeal sequences were found among bacterial or bacterial and eukaryal sequences in at least five groups (pyrimidine kinases, heptose-7-phosphate kinases, KDG-kinases, fructokinases, and ribokinases) suggesting that these groups had resulted from a very early functional divergence in the evolution of the PFK-Bs, most likely before division of the domains. The separate clustering of the halobacterial PFK-Bs is presumably due to the unique physiology of these organisms with their high intracellular salt concentrations. Interestingly, the ApPFK/NK and the MjNK fall into separate groups despite their partial functional resemblance. The ApPFK/NK clusters with a variety of crenarchaeal and euryarchaeal sequences. However, the ApPFK/NK is the only protein in this group that has been functionally characterized yet. Though, the other archaeal sequences of this archaeal PFK-B group have been assigned as ribokinases or sugar kinases, further functional characterization of these archaeal PFK-B enzymes with respect to specificity are necessary to understand the evolution of this PFK-B group as well PFK-Bs in general. Further, the MjNK clusters with the adenosine kinase from the bacterium Mycoplasma tuberculosis (Long et al. 2003) and putative euryarchaeal sequences, indicating that these euryarchaeal sequences might encode for nucleoside kinases like the MjNK as well. Final proof will have to await further characterization of these enzymes.
References
Bork P, Sander C, Valencia A (1993) Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci 2:31–40
Brunner NA, Brinkmann H, Siebers B, Hensel R (1998) NAD+-dependent glyceraldehyde-3-phosphate dehydrogenase from Thermoproteus tenax. The first identified archaeal member of the aldehyde dehydrogenase superfamily is a glycolytic enzyme with unusual regulatory properties. J Biol Chem 273:6149–6156
Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, Fitzgerald LM, Clayton RA, Gocayne JD, Kerlavage AR, Dougherty BA, Tomb JF, Adams MD, Reich CI, Overbeek R, Kirkness EF, Weinstock KG, Merrick JM, Glodek A, Scott JL, Geoghagen NSM, Venter JC (1996) Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273:1058–1073
Ding Y-HR, Ronimus RS, Morgan HW (2000) Sequencing, cloning, and high-level expression of the pfp gene, encoding a PP i -dependent phosphofructokinase from the extremely thermophilic eubacterium Dictyoglomus thermophilum. J Bacteriol 182:4661–4666
Ding YH, Ronimus RS, Morgan HW (2001) Thermotoga maritima phosphofructokinases: expression and characterization of two unique enzymes. J Bacteriol 183:791–794
Hansen T, Musfeldt M, Schönheit P (2002a) ATP-dependent 6-phosphofructokinase from the hyperthermophilic bacterium Thermotoga maritima: characterization of an extremely thermophilic, allosterically regulated enzyme. Arch Microbiol 177:401–409
Hansen T, Oehlmann M, Schönheit P (2001) Novel type of glucose-6-phosphate isomerase in the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol 183:3428–3435
Hansen T, Reichstein B, Schmid R, Schönheit P (2002b) The first archaeal ATP-dependent glucokinase, from the hyperthermophilic crenarchaeon Aeropyrum pernix, represents a monomeric, extremely thermophilic ROK glucokinase with broad hexose specificity. J Bacteriol 184:5955–5965
Hansen T, Schlichting B, Schönheit P (2002c) Glucose-6-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: expression of the g6pd gene and characterization of an extremely thermophilic enzyme. FEMS Microbiol Lett 216:249–253
Hansen T, Schönheit P (2000) Purification and properties of the first-identified, archaeal, ATP-dependent 6-phosphofructokinase, an extremely thermophilic non-allosteric enzyme, from the hyperthermophile Desulfurococcus amylolyticus. Arch Microbiol 173:103–109
Hansen T, Schönheit P (2001) Sequence, expression, and characterization of the first archaeal ATP-dependent 6-phosphofructokinase, a non-allosteric enzyme related to the phosphofructokinase-B sugar kinase family, from the hyperthermophilic crenarchaeote Aeropyrum pernix. Arch Microbiol 177:62–69
Hansen T, Schönheit P (2004) ADP-dependent 6-phosphofructokinase, an extremely thermophilic, non-allosteric enzyme from the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus strain 7324. Extremophiles 8:29–35
Hansen T, Urbanke C, Schönheit P (2004) Bifunctional phosphoglucose/phosphomannose isomerase from the hyperthermophilic archaeon Pyrobaculum aerophilum. Extremophiles 8:507–512
Holmes ML, Scopes RK, Moritz RL, Simpson RJ, Englert C, Pfeifer F, Dyall-Smith ML (1997) Purification and analysis of an extremely halophilic beta-galactosidase from Haloferax alicantei. Biochim Biophys Acta 1337:276–286
Kengen SW, de Bok FA, van Loo ND, Dijkema C, Stams AJ, De Vos WM (1994) Evidence for the operation of a novel Embden-Meyerhof pathway that involves ADP-dependent kinases during sugar fermentation by Pyrococcus furiosus. J Biol Chem 269:17537–17541
Kengen SW, Tuininga JE, Verhees CH, Van der Oost J, Stams AJ, De Vos WM (2001) ADP-dependent glucokinase and phosphofructokinase from Pyrococcus furiosus. Methods Enzymol 331:41–53
Labes A, Schönheit P (2001) Sugar utilization in the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus strain 7324: starch degradation to acetate and CO2 via a modified Embden-Meyerhof pathway and acetyl-CoA synthetase (ADP-forming). Arch Microbiol 176:329–338
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Li MH, Kwok F, Chang WR, Lau CK, Zhang JP, Lo SC, Jiang T, Liang DC (2002) Crystal structure of brain pyridoxal kinase, a novel member of the ribokinase superfamily. J Biol Chem 277:46385–46390
Liacouras AS, Anderson EP (1975) Uridine-cytidine kinase. Purification from a murine neoplasm and characterization of the enzyme. Arch Biochem Biophys 168:66–73
Long MC, Escuyer V, Parker WB (2003) Identification and characterization of a unique adenosine kinase from Mycobacterium tuberculosis. J Bacteriol 185:6548–6555
Mathews II, Erion MD, Ealick SE (1998) Structure of human adenosine kinase at 1.5 Å resolution. Biochemistry 37:15607–15620
Mizote T, Tsuda M, Smith DD, Nakayama H, Nakazawa T (1999) Cloning and characterization of the thiD/J gene of Escherichia coli encoding a thiamin-synthesizing bifunctional enzyme, hydroxymethylpyrimidine kinase/phosphomethylpyrimidine kinase. Microbiology 145:495–501
Mori H, Iida A, Teshiba S, Fujio T (1995) Cloning of a guanosine-inosine kinase gene of Escherichia coli and characterization of the purified gene product. J Bacteriol 177:4921–4926
Ohshima N, Inagaki E, Yasuike K, Takio K, Tahirov TH (2004) Structure of Thermus thermophilus 2-Keto-3-deoxy-gluconate kinase: evidence for recognition of an open chain substrate. J Mol Biol 340:477–489
Qu Q, Lee SJ, Boos W (2004) Molecular and biochemical characterization of a fructose-6-phosphate-forming and ATP-dependent fructokinase of the hyperthermophilic archaeon Thermococcus litoralis. Extremophiles 8:301–308
Ronimus RS, de Heus E, Morgan HW (2001a) Sequencing, expression, characterisation and phylogeny of the ADP-dependent phosphofructokinase from the hyperthermophilic, euryarchaeal Thermococcus zilligii. Biochim Biophys Acta 1517:384–391
Ronimus RS, Kawarabayasi Y, Kikuchi H, Morgan HW (2001b) Cloning, expression and characterisation of a family B ATP-dependent phosphofructokinase activity from the hyperthermophilic crenarchaeon Aeropyrum pernix. FEMS Microbiol Lett 202:85–90
Ronimus RS, Koning J, Morgan HW (1999) Purification and characterization of an ADP-dependent phosphofructokinase from Thermococcus zilligii. Extremophiles 3:121–129
Ronimus RS, Morgan HW (2001) The biochemical properties and phylogenies of phosphofructokinases from extremophiles. Extremophiles 5:357–373
Ronimus RS, Morgan HW (2003) Distribution and phylogenies of enzymes of the Embden-Meyerhof-Parnas pathway from archaea and hyperthermophilic bacteria support a gluconeogenic origin of metabolism. Archaea 1:199–221
Sakuraba H, Yoshioka I, Koga S, Takahashi M, Kitahama Y, Satomura T, Kawakami R, Ohshima T (2002) ADP-dependent glucokinase/phosphofructokinase, a novel bifunctional enzyme from the hyperthermophilic archaeon Methanococcus jannaschii. J Biol Chem 277:12495–12498
Schumacher MA, Scott DM, Mathews II, Ealick SE, Roos DS, Ullman B, Brennan RG (2000) Crystal structures of Toxoplasma gondii adenosine kinase reveal a novel catalytic mechanism and prodrug binding. J Mol Biol 298:875–893
Selig M, Xavier KB, Santos H, Schönheit P (1997) Comparative analysis of Embden-Meyerhof and Entner-Doudoroff glycolytic pathways in hyperthermophilic archaea and the bacterium Thermotoga. Arch Microbiol 167:217–232
Siebers B, Hensel R (2001) Pyrophosphate-dependent phosphofructokinase from Thermoproteus tenax. Methods Enzymol 331:54–62
Siebers B, Tjaden B, Michalke K, Dörr C, Ahmed H, Zaparty M, Gordon P, Sensen CW, Zibat A, Klenk HP, Schuster SC, Hensel R (2004) Reconstruction of the central carbohydrate metabolism of Thermoproteus tenax by use of genomic and biochemical data. J Bacteriol 186:2179–2194
Siebers B, Schönheit P (2005) Unusual pathways and enzymes of central carbohydrate metabolism in archaea. Curr Opin Microbiol 8:695–705
Sigrell JA, Cameron AD, Jones TA, Mowbray SL (1998) Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 Å resolution: insights into a new family of kinase structures. Structure 6:183–193
Stetter KO (1996) Hyperthermophilic procaryotes. FEMS Microbiol Rev 18:149–158
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tuininga JE, Verhees CH, Van der Oost J, Kengen SW, Stams AJ, De Vos WM (1999) Molecular and biochemical characterization of the ADP-dependent phosphofructokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem 274:21023–21028
Usuda Y, Kawasaki H, Shimaoka M, Utagawa T (1997) Characterization of guanosine kinase from Brevibacterium acetylicum ATCC 953. Biochim Biophys Acta 1341:200–206
Van Rompay AR, Norda A, Linden K, Johansson M, Karlsson A (2001) Phosphorylation of uridine and cytidine nucleoside analogs by two human uridine-cytidine kinases. Mol Pharmacol 59:1181–1186
Verhees CH, Kengen SW, Tuininga JE, Schut GJ, Adams MW, De Vos WM, Van der Oost J (2003) The unique features of glycolytic pathways in archaea. Biochem J 375:231–246
Verhees CH, Tuininga JE, Kengen SW, Stams AJ, Van der Oost J, De Vos WM (2001) ADP-dependent phosphofructokinases in mesophilic and thermophilic methanogenic archaea. J Bacteriol 183:7145–7153
Acknowledgment
The expert technical assistance of K. Lutter-Mohr and M. Kusche is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Antranikian
Rights and permissions
About this article
Cite this article
Hansen, T., Arnfors, L., Ladenstein, R. et al. The phosphofructokinase-B (MJ0406) from Methanocaldococcus jannaschii represents a nucleoside kinase with a broad substrate specificity. Extremophiles 11, 105–114 (2007). https://doi.org/10.1007/s00792-006-0018-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-006-0018-1