
Abstract
The hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus strain 7324 has been shown to degrade starch via glucose using a modified Embden-Meyerhof pathway. In this pathway phosphorylation of fructose-6-phosphate to fructose-1,6 bisphosphate is catalyzed by an ADP-dependent 6-phosphofructokinase (ADP-PFK), which was purified 1,800-fold to homogeneity. The enzyme is composed of 50 kDa subunits and is eluted from gel filtration as both a homotetramer and a homodimer. It had a temperature optimum at 85°C and showed significant thermostability up to 95°C. Kinetic constants were determined for both reaction directions at pH 6.6 and 80°C. Rate dependence for all substrates followed Michaelis Menten kinetics. The apparent K m for ADP and fructose-6-phosphate (forward reaction) was 0.6 mM and 2.2 mM, respectively; the apparent V max was 1,200 U/mg. ADP-PFK catalyzed in vitro the reverse reaction, with apparent K m for fructose-1,6-bisophosphate and AMP of 5.7 and 1.4 mM, respectively, and a V max value of 85 U/mg. The enzyme did not use ATP, PPi, or acetyl phosphate as phosphoryl donor and was highly specific for fructose-6-phosphate as substrate. The A. fulgidus ADP-PFK did not phosphorylate glucose and thus differs from the bifunctional ADP-PFK/GLK from Methanococcus jannaschii. Divalent cations were required for catalytic activity; Mg2+, which was most effective, could be partially replaced by Mn2+, Ni2+, and Co2+. Enzyme activity was not allosterically regulated by classical effectors of bacterial and eukaryal ATP-PFKs, such as ADP, AMP, phosphoenolpyruvate, or citrate. N-terminal amino acid sequence showed high similarity to known ADP-PFKs. In the genome of Archaeoglobus fulgidus strain VC 16, which is closely related to strain 7324, no homologous gene for ADP-PFK could be identified.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyperthermophilic prokaryotes, with an optimal growth temperature higher than 80°C, are considered to represent the phylogenetically most ancestral organisms (Stetter 1996). Recent comparative studies of the hexose degradation pathways in hyperthermophilic archaea and in the hyperthermophilic bacterium Thermotoga revealed that the classical Embden-Meyerhof (EM) pathway is operative only in Thermotoga, whereas in all archaea analyzed so far, the EM pathway exists in modified versions. The modified EM pathways of the euryarchaeota Pyrococcus and Thermococcus contain, e.g., novel ADP-dependent 6-phosphofructokinases (ADP-PFK). In the crenarchaeota Desulfurococcus, Aeropyrum, and Thermoproteus, non-regulatory ATP-PFKs or pyrophosphate-dependent PFKs (PPi-PFKs) were found.
6-Phosphofructokinases catalyze the phosphorylation of fructose 6-phosphate (F-6-P) to fructose 1,6-bisphosphate with ATP (ATP-PFK, EC 2.7.1.11), pyrophosphate (PPi-PFK, EC 2.7.1.90), or ADP (ADP-PFK, EC 2.7.1.146) as phosphoryl donor. The enzymes are present in all domains of life—eukarya, bacteria, and archaea (for reviews, see Ronimus and Morgan 2001; Uyeda 1979). Sequence and structural analysis group them into two convergent protein families (see Pfam http://www.sanger.ac.uk/Software/Pfam/) (Bateman et al. 2001): the PFK-A family (PF00365) and the ribokinase superfamily, including the PFK-B family (PF00294) and the ADP-PFK/GLK family (PF04587). The PFK-A family comprises both ATP-dependent and PPi-dependent enzymes. ATP-PFKs from this family have been purified and characterized from a variety of eukarya and bacteria including the hyperthermophile Thermotoga maritima (Hansen et al. 2002a). They show allosteric regulation by compounds of the intermediary metabolism (for review, see Uyeda 1979). PPi-PFKs have been isolated from eukarya, from bacteria, and from the archaeon Thermoproteus tenax (Siebers et al. 1998). These enzymes catalyze a reversible reaction and are not allosterically regulated.
The PFK-B family is a diverse family of ATP-dependent carbohydrate and pyrimidine kinases that includes the minor ATP-PFK from Escherichia coli (PFK-B or PFK2) and is present in all domains of life. In addition to the E. coli PFK-B, only the homologues from the crenarchaeota Desulfurococcus amylolyticus and Aeropyrum pernix (Daldal 1983; Hansen and Schönheit 2000, 2001) have been characterized. The lack of allosteric regulation differentiates these ATP-PFKs from ATP-PFKs from the PFK-A family.
ADP-PFKs were found only in the euryarchaeota; they have been characterized from Thermococcales and from glycogen-forming methanogens, including the hyperthermophile Methanococcus jannaschii (Kengen et al. 1995; Koga et al. 2000; Ronimus et al. 1999; Selig et al. 1997; Tuininga et al. 1999; Verhees et al. 2001). The latter organism has been reported to contain a bifunctional ADP-dependent sugar kinase, showing both ADP-GLK and ADP-PFK activity (Sakuraba et al. 2002). Like the other archaeal PFKs, euryarchaeal ADP-PFKs are characterized by the absence of allosteric regulation. So far crystal structures are not available from ADP-PFKs but are available from closely related Thermococcus litoralis and Pyrococcus horikoshii ADP-GLKs (Ito et al. 2001; Tsuge et al. 2002). According to a similar fold, the ADP-GLKs were grouped in the ribokinase superfamily.
Recently, the sugar metabolism of the hyperthermophilic, sulfate-reducing euryarchaeon Archaeoglobus fulgidus has been studied. It was found that A. fulgidus strain 7324 (Beeder et al. 1994) could grow on starch and sulfate. Extracts of starch-grown cells contained all activities of a modified Embden-Meyerhof pathway similar to those of Thermococcales, including activities of ADP-PFK (Labes and Schönheit 2001). In the completely sequenced genome of A. fulgidus type strain VC16 (Klenk et al. 1997), no open reading frame with significant similarity to the ADP-PFKs from Pyrococcus and Thermococcus or ADP-PFK/GLK from Methanococcus could be identified. This might indicate that ADP-PFK in Archaeoglobus species is significantly different from those of Thermococcales and Methanococcus.
In this communication we report the purification and molecular and catalytic properties of ADP-PFK from Archaeoglobus fulgidus strain 7324. The enzyme was characterized as an extremely thermophilic, homodimeric/homotetrameric protein of 50 kDa subunits, showing high specificity to both ADP and fructose-6-phosphate. Using this N-terminal sequence, no gene encoding a homologous ADP-PFK could be identified in the genome of the closely related A. fulgidus strain VC16.
Materials and methods
Growth of the organism and preparation of cell free extracts
Archaeoglobus fulgidus 7324 (DSM 3109) (Beeder et al. 1994) was grown anaerobically at 80°C in a 100-l Biostat fermentor on a medium containing starch (1 g/l) and yeast extract (5 g/l) as carbon and energy source as previously described (Labes and Schönheit 2001). Cells were harvested at the late exponential growth phase. The following steps were carried out under anoxic conditions: Cell extracts were prepared from 50 g of frozen cells (wet mass), which were suspended in 100 ml 50 mM Tris/HCl, pH 7.0 (25°C) containing 1 mM dithiothreithol, 2 mM EDTA, and 20 mM NaCl. Upon thawing and passing three times through a French pressure cell at 8 MPa, the cells were almost completely disrupted. Cell debris and unbroken cells were removed by ultracentrifugation for 90 min at 100,000 g at 4°C.
Purification of ADP-dependent 6-phosphofructokinase
All chromatographic steps were carried out at 4°C. The 100,000 g supernatant was diluted with 20 mM Tris pH 9.0 containing 2 mM EDTA and 1 mM DTE (buffer A) to a final volume of 1,425 ml and was applied to a HiLoad Q-Sepharose column (10 cm×2.6 cm) equilibrated with buffer A. Protein was desorbed at a flow rate of 2 ml/min with fixed concentration as well as linear gradients from 0 to 1 M NaCl in buffer A: 0–0.01 M (5 ml), 0.01–0.1 M (120 ml), 0.1 M (10 ml), 0.1–0.25 M (60 ml), 0.25 M (10 ml), 0.25–1 M (60 ml), and 1 M (80 ml). Fractions containing the highest ADP-PFK activity (17 ml, 0.16–0.18 M NaCl) were pooled, diluted to a final NaCl concentration of 30 mM with 50 mM Tris/HCl pH 7.0 containing 1 mM DTE and 2 mM MgCl2 (buffer B), and applied to an ADP-Agarose column (7 cm×1 cm) at a flow rate of 0.75 ml/min equilibrated with buffer B. After washing the column with 15 ml buffer B, protein was desorbed at a flow rate of 0.2 ml/min with fixed concentration as well as linear gradients from 0 to 100 mM of both F-6-P and ADP in buffer B: 0–2 mM (6 ml), 2–25 mM (12 ml), 25–100 mM (8 ml), and 100 mM (3 ml). Fractions with the highest ADP-PFK activity (2.8 ml, 14–20 mM F-6-P/ADP) were pooled, diluted twofold with 50 mM MES pH 5.7 (25°C) containing 1 mM DTE (buffer C), adjusted to pH 5.7 with 1 M acetic acid, and loaded on an Uno S 1 column (1 ml) previously equilibrated with buffer. Protein was eluted at a flow rate of 1 ml/min using linear NaCl gradients from 0 to 0.5 M (10 ml) and 0.5–2 M (6 ml) in buffer C. Fractions containing the highest ADP-PFK-activity (2.5 ml) were recovered from 0.4–0.6 M NaCl. Pure protein fractions were obtained after gel filtration on Superdex Tm200 using 150 mM NaCl, 50 mM Tris/HCl, 1 mM DTE as elution buffer. The eluate was stored at −20°C. Under these conditions activity remained about constant.
Analytical assays
The purity of the preparations was checked by SDS-PAGE in 14% polyacrylamide gels followed by staining with Coomassie Brilliant Blue R 250 according to standard procedures (Laemmli 1970). Protein concentrations were determined by the method of Bradford (1976) with BSA as standard. Gel-filtration chromatography was carried out at ambient temperature on a Superdex 200 (50 mM Tris/HCl, 150 mM NaCl, pH 7.0, 1 ml/min). Blotting onto a poly(vinylidene diflouride) membrane and N-terminal microsequencing on a model 473A sequencer (Applied Biosystems) were carried out as described in Meyer et al. (1996).
Enzyme assays and determination of kinetic parameters
Since the enzyme activity was not sensitive to oxygen, all assays were carried out under oxic conditions. The ADP-dependent PFK activity (F-6-P + ADP ⇌ F-1,6-BP + AMP) was determined in both directions. The forward reaction was measured by coupling the ADP-dependent formation of F-1,6-BP to the oxidation of NADH via F-1,6-BP aldolase, triosephosphate isomerase, and glycerol-3-phosphate-dehydrogenase, whereas the reverse reaction was investigated by coupling the AMP-dependent formation of F-6-P to the reduction of NADP+ via glucose-6-phosphate isomerase (GPI) and glucose 6-phosphate dehydrogenase. ADP-PFK activity was measured at 50°C in a continuous assay and above 50°C in a discontinuous assay as previously described (Hansen and Schönheit 2000), except for the standard assay mixtures containing (1) 50 mM Tris/HCl (pH 6.6 at the respective temperature), 5 mM F-6-P, 2 mM ADP, and MgCl2:ADP at an optimized ratio of 2.5:1 (forward reaction) or (2) 25 mM F-1,6-BP, 2 mM ADP, and MgCl2:ADP at an optimized ratio of 2.5:1 (reverse reaction). Initial velocities were investigated in at least six parallel assays stopped at different time intervals. One unit (U) of ADP-PFK activity is defined as the conversion of either 1 μmol F-6-P to F-1,6-BP (forward reaction) or the formation of 1 μmol F-6-P from F-1,6-BP (reverse reaction). The coupling enzymes in all assays were routinely tested to ensure that they were not rate limiting.
pH dependence, substrate specificity, cation specificity, and effectors
The pH dependence of the enzyme was measured between 3.9 and 9.0 at 80°C using succinate, acetate, piperazine, phosphate, MES, Tris/HCl, triethanolamine, or ethanolamine at a concentration of 50 mM each. The cation and nucleotide specificity were examined using the standard discontinuous test system at 80°C by exchanging either Mg2+ (5 mM) or ADP (2 mM) for alternative divalent cations (Ni2+, Mn2+, Co2+, Ca2+, Zn2+, and Fe2+) or alternative phosphoryl donors (ATP, ITP, GTP, UTP, CTP, UDP, CDP, GDP, acetyl phosphate, and PPi) at equimolar concentrations. For the test of substrate specificity for sugars, F-6-P was exchanged for fructose 1-phosphate and glucose. Potential glucose-6-phosphate formation measured in a continuous assay with the following concentrations: 5 mM glucose, 2 mM ADP, 5 mM MgCl2, 0.5 mM NADP, 1 U glucose-6-phosphate dehydrogenase from Thermotoga maritima (Hansen et al. 2002b), 50 mM Tris/HCl pH 6.6 (80°C ). The following classical effectors of ATP-PFKs from bacterial and eukaryotic sources were tested at 80°C using the following concentrations: PEP (0.2 mM, 2 mM), AMP (0.2 mM, 1 mM, 10 mM), and citrate (0.2 mM, 10 mM). In addition the following metabolites were tested at 80°C at concentrations of both 0.1 and 10 mM for any potential effect on ADP-PFK activity at maximal and half-maximal velocity: ATP, acetate, alanin, formate, isocitrate, glyceraldehyde-3-phosphate, 3-phosphoglycerate, cis-aconitate, malate, oxaloacetate, pyruvate, and succinate.
Temperature dependence and thermal stability
The temperature dependence of the enzyme activity was measured between 25°C and 95°C in 50 mM sodium phosphate buffer, pH 6.6. The activity was measured in the direction of F-1,6-BP formation using standard concentrations of F-6-P (5 mM), ADP (2 mM), and MgCl2 (5 mM), which ensured specific activities close to V max. The thermostability of the purified enzyme (1 μg in 40 μl sodium phosphate buffer pH 7.0) as well as the effects of potential stabilizing additives [1 M NaCl, 1 M KCl, 1 M (NH4)2SO4)] were tested in sealed vials that were incubated at temperatures between 70°C and 100°C for 2–120 min. The vials were then cooled on ice for 10 min and the remaining enzyme activity was tested at 50°C and compared with the controls (unheated sample).
Source of material
All commercially available chemicals used were of reagent grade and were obtained from Merck (Darmstadt, Germany), Fluka (Buchs, Switzerland), or Sigma (Deisenhofen, Germany). Yeast extract and peptone were from Difco (Stuttgart, Germany). Enzymes, coenzymes, and other chemicals were from Roche Diagnostics (Mannheim, Germany) or Sigma (Deisenhofen, Germany). ATP, ADP, fructose 6-phosphate, and PEP, each at highest available purity, were purchased from Roche Diagnostics (Mannheim, Germany). Gases were from Linde (Hamburg, Germany). Archaeoglobus fulgidus 7324 (DSM 8774) was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). All FPLC material and columns used were from Pharmacia (Freiburg, Germany), Sigma, and Biorad (Munich, Germany).
Results
Purification of ADP-PFK
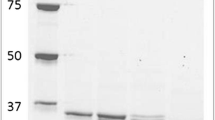
ADP-PFK was purified aerobically from cell extracts of Archaeoglobus fulgidus 7324 by four purification steps: anion exchange chromatography on Q-Sepharose, affinity chromatography on ADP-agarose, cation exchange chromatography on Uno-S1, and gel filtration on Sephadex. By this procedure the enzyme was purified about 1,800-fold to a specific activity of 400 U/mg (50°C) with a yield of 11% (Table 1). The purified protein was electrophoretically homogeneous as judged by denaturing SDS-PAGE (not shown). Thus, ADP-PFK represents about 0.06% of the cellular protein of A. fulgidus 7324.
Molecular properties
Native ADP-PFK eluted from gel filtration columns with an apparent molecular mass of both 100 kDa (80%) and 200 kDa (20%) (200 μg applied). SDS-PAGE revealed only one single subunit with an apparent molecular mass of 50 kDa (not shown), indicating the presence of both a homodimeric (α2) and a homotetrameric (α4) structure under the condition tested (100 μg enzyme). However, only a dimeric fraction could be detected after gel filtration applying a lower protein concentration (25 μg enzyme). The specific activity of the dimeric as well as the tetrameric ADP-PFK fraction was identical. The N-terminal amino acid sequence of the 50 kDa subunit (Fig. 1) showed a high degree of identity (43%) to the N-termini of ADP-PFKs from Pyrocccus (43%), Thermococcus (43%), and Methanococcus (32%) (Ronimus et al. 2001; Tuininga et al. 1999; Verhees et al. 2001).

Alignment of N-terminal amino acid sequences of ADP-PFK from Archaeoglobus fulgidus 7324 with N-terminal regions of ADP-PFKs from from Thermococcus zilligii (Q9HH12) and Pyrococcus furiosus (Q9V2Z7) and ADP-PFK/GLK from Methanococcus jannaschii (Q58999)
Catalytic properties
Kinetic constants of purified ADP-PFK were determined in both reaction directions (ADP + F-6-P ⇌ F-1,6-BP + AMP) (Table 2). The rate dependence of the enzyme on the ADP, F-6-P (Fig. 2), AMP, and F-1,6-BP concentrations followed Michaelis-Menten kinetics. Substrate inhibition was observed for ADP and AMP at concentrations above 1.5 mM and 3 mM, respectively. The forward reaction was measured at both 50°C and 80°C. At 80°C the apparent K m values for F-6-P and ADP were 2.2 mM and 0.6 mM, respectively; apparent V max values were about 1,200 U/mg. At 50°C the apparent K m value for F-6-P (4.7 mM) was about 2.5-fold higher, whereas the apparent K m value for ADP (0.3 mM) was twofold lower, indicating a lower affinity for the sugar substrate and a higher affinity for the phosphoryl donor at lower temperatures. ADP could not be replaced by ATP or PPi, defining this archaeal PFK as an ADP-dependent enzyme (Table 2). The enzyme was highly specific for both the phosphoryl donor and the sugar substrate. ADP (100%, 1,200 U/mg at 80°C) could hardly be replaced by GDP (8%), UDP (3%), and CDP (3%). All other substances tested could not serve as phosphoryl donors. ADP-PFK did not accept any other sugar tested as phosphoryl acceptors, including glucose (see Materials and Methods). Thus, the enzyme is a true ADP-PFK and therefore differs from bifunctional ADP-PFK/GLK from Methanoccus jannaschii (Sakuraba et al. 2002). Enzyme activity required divalent cations; Mg2+ (100%, 1,200 U/mg at 80°C), which was most effective, could partially be replaced by Mn2+ (62%), Ni2+ (33%), and Co2+ (30%), whereas Fe2+ (16%), Zn2+ (6%), Cu2+ (4%), and Ca2+ (2%) were less effective. The pH optimum of ADP-PFK was at pH 6.6; 50% of activity was found at pH 6.1 and at pH 7.9. Neither the classical effectors of bacterial and eukaryal ATP-PFKs—ADP, AMP, citrate, and phosphoenolpyruvate—nor several metabolites showed any significant effect on the ADP-PFK activity under the various conditions tested (for details, see Materials and methods).

Rate dependence of the ADP-PFK from A. fulgidus 7324 on the fructose-6-phosphate concentration (A) and the ADP concentration (B) at 80°C
The enzyme also catalyzed the reverse reaction in vitro, i.e., AMP-dependent F-1,6-BP conversion to ADP and F-6-P. At 80°C an apparent K m value for F-1,6-BP (5.7 mM) and V max (85 U/mg) were determined; however, the catalytic efficiency (k cat/ K m) was about 37-fold lower when compared to the forward reaction (12 versus 392 s–1mM–1).
Temperature optimum and stability
The temperature dependence of ADP-PFK is shown in Fig. 3. At 40°C the enzyme showed little activity, which, however, increased exponentially and showed an optimum of 90°C. From the linear part of the Arrhenius plot between 40°C and 70°C, an activation energy of 57 kJ/mol was calculated. The temperature stability of ADP-PFK was tested between 80°C and 100°C in 50 mM sodium phosphate pH 7 by incubating the enzyme up to 120 min, followed by measuring residual activity at 50°C (Fig. 4). After 120 min incubation at 80°C, the enzyme did not lose activity. At 100°C, an almost complete loss of activity was observed after 60 min. Addition of KCl and, to a lower extent, of NaCl (each 1 M) effectively stabilized ADP-PFK against heat inactivation at 100°C; almost 50% and 30% residual activity was retained after 90 min in the presence of KCl (Fig. 4) and NaCl, respectively.

Effect of temperature on the specific activity of the ADP-PFK from A. fulgidus 7324. The assay mixture contained 0.5 μg enzyme, 50 mM sodium phosphate buffer, pH 6.6 (at the indicated temperature), 5 mM fructose-6-phosphate, 2 mM ADP, and 10 mM MgCl2

Thermostability of ADP-PFK from A. fulgidus. 1 μg of enzyme was incubated in 40 μl sodium phosphate pH 7.0 between 80°C and 100°C in the presence or absence of 1 M salts. 80°C (▼), 90°C (■), 95°C (●), 100°C (▲), 100°C containing 1 M KCl (◆). 100% activity corresponded to a specific activity of 400 U/mg at 50°C
Discussion
Table 2 summarizes molecular and kinetic properties of ADP-PFKs from Archaeoglobus fulgidus in comparison to ADP-PFKs from Pyrococcus furiosus, Thermococcus zilligii, and the bifunctional ADP-PFK/GLK from Methanococcus jannaschii (Ronimus et al. 1999, 2001; Sakuraba et al. 2002; Tuininga et al. 1999; Verhees et al. 2001).
The N-terminal sequence of the A. fulgidus enzyme was more similar to the ADP-PFKs from T. zilligii and P. furiosus (12 identical aa out of 28 aa) than to the bifunctional ADP-PFK/GLK from M. jannaschii (9 identical aa). Using N-terminal amino acid sequence of ADP-PFK from A. fulgidus strain 7324, no open reading frame coding for a homologous protein could be identified in the genome of A. fulgidus strain VC16 (Klenk et al. 1997), which is closely related to the A. fulgidus strain 7324 analyzed in this study. This indicates that A. fulgidus strain VC16 did not contain homologous ADP-PFK. In accordance it was found that A. fulgidus VC 16—in contrast to strain 7324—could not grow on starch and sulfate and did not exhibit ADP-PFK activity in crude extracts (Labes and Schönheit 2001). The absence of ADP-PFK in A. fulgidus strain VC16 might indicate that this strain has lost its corresponding gene; alternatively, A. fulgidus strain 7324 might have taken up this gene from a Thermococcus-like organism by lateral gene transfer.
All ADP-PFKs have subunits of about 50–54 kDa. The bifunctional ADP-PFK/GLK from M. jannaschii was described as a monomer, whereas the A. fulgidus ADP-PFK eluted from gel filtration predominantly as a dimer and to a certain extent as a tetramer at increased protein concentration. A similar observation has been described for the ADP-PFK from T. zilligii, which was reported as a dimer when purified from T. zilligii (Ronimus et al. 1999) but has been shown to be a tetramer, as reported for the recombinant P. furiosus ADP-PFK, when heterologously expressed in E. coli (Ronimus et al. 2001; Tuininga et al. 1999).
ADP-PFK of A. fulgidus showed a temperature optimum of 85°C, which is in accordance with the optimal growth temperature of strain 7324 (about 80°C). In addition, the enzyme from A. fulgidus exhibited high thermostability up to 95°C. In comparison, the bifunctional enzyme from M. jannaschii was less thermostable; 20% of activity was lost upon a 10-min incubation at 90°C (Sakuraba et al. 2002).
The ADP-PFK from A. fulgidus was highly specific for both fructose-6-phosphate as phosphoryl acceptor and ADP as phosphoryl donor. Apparent K m values for both substrates were in the same range as reported for ADP-PFKs from P. furiosus and T. zilligii (Ronimus et al. 1999; Tuininga et al. 1999). Besides fructose-6-phosphate, glucose could not serve as phosphoryl acceptor by the A. fulgidus ADP-PFK. Thus, the A. fulgidus enzyme is a true ADP-PFK and differs from the bifunctional ADP-PFK/GLK from M. jannaschii.
Kinetic parameters, specificities, and potential allosteric effects of the ADP-PFK from A. fulgidus were assayed at 80°C, i.e., near both the growth temperature optimum of A. fulgidus 7324 and the temperature optimum of the enzyme (85°C). In contrast to the homologues from P. furiosus, T. zilligii and M. jannaschii, these data have been determined at 50°C, i.e., 30–50°C below the growth temperature of the respective organisms. It is important to note that the assay temperature at the physiologically relevant temperature could be crucial in demonstrating allosteric properties of a hyperthermophilic enzyme, as has been reported for the Thermotoga maritima ATP-PFK (PFK-A family) (Hansen et al. 2002a). When assayed at 80°C, the A. fulgidus ADP-PFK was not subject to allosteric control (see Materials and Methods), a property also reported for the ADP-PFKs from P. furiosus, T. zilligii, and M. jannaschii (Ronimus et al. 1999; Tuininga et al. 1999; Verhees et al. 2001). This confirms the present view that archaeal PFKs, being ADP-, PPi- or ATP-dependent—in contrast to the bacterial and eukaryal ATP-PFKs (PFK-A)—are generally not the sites of allosteric control in the modified EM pathways (Hansen and Schönheit 2000, 2001; Siebers et al. 1998).
When comparing the kinetic properties of the A. fulgidus ADP-PFK with purified hyperthermophilic PFKs at physiologically relevant temperatures, it is worth noticing that the catalytic efficiency in terms of k cat /K m of the A. fulgidus enzyme (392 s–1mM–1) was in the same range as the physiologically relevant ATP-PFK (PFK-A) from the hyperthermophilic bacterium T. maritima (287 s–1mM–1[s 0.5/K m]) (Hansen et al. 2002a); however, the catalytic efficiency of the A. fulgidus ADP-PFK was significantly higher (20–35-fold) when compared to the crenarchaeotal ATP-PFKs (PFK-B) of Desulfurococcus amylolyticus (19 s–1mM–1) and Aeropyrum pernix (11 s–1mM–1). In contrast to the A. fulgidus ADP-PFK and the T. maritima ATP-PFK, which were very specific for F-6-P, the A. pernix ATP-PFK exhibited a broad specificity for sugars, presumably at the cost of catalytic efficiency (Hansen and Schönheit 2001).
References
Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL (2001) The Pfam protein families database. Nucleic Acids Res 28:263–266
Beeder J, Nilsen RK, Rosnes JT, Torsvik T, Lien T (1994) Archaeoglobus fulgidus isolated from hot North Sea oil field waters. Appl Environ Microbiol 60:1227–1231
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Daldal F (1983) Molecular cloning of the gene for phosphofructokinase-2 of Escherichia coli and the nature of a mutation, pfkB1, causing a high level of the enzyme. J Mol Biol 168:285–305
Hansen T, Schönheit P (2000) Purification and properties of the first-identified, archaeal, ATP-dependent 6-phosphofructokinase, an extremely thermophilic non-allosteric enzyme, from the hyperthermophile Desulfurococcus amylolyticus. Arch Microbiol 173:103–109
Hansen T, Schönheit P (2001) Sequence, expression, and characterization of the first archaeal ATP-dependent 6-phosphofructokinase, a non-allosteric enzyme related to the phosphofructokinase-B sugar kinase family, from the hyperthermophilic crenarchaeote Aeropyrum pernix. Arch Microbiol 177:62–69
Hansen T, Musfeldt M, Schönheit P (2002a) ATP-dependent 6-phosphofructokinase from the hyperthermophilic bacterium Thermotoga maritima: characterization of an extremely thermophilic, allosterically regulated enzyme. Arch Microbiol 177:401–409
Hansen T, Schlichting B, Schönheit P (2002b) Glucose-6-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: expression of the g6pd gene and characterization of an extremely thermophilic enzyme. FEMS Microbiol Lett 216:249–253
Ito S, Fushinobu S, Yoshioka I, Koga S, Matsuzawa H, Wakagi T (2001) Structural basis for the ADP-specificity of a novel glucokinase from a hyperthermophilic archaeon. Structure (Camb) 9:205–214
Kengen SW, Tuininga JE, de Bok FA, Stams AJ, De Vos WM (1995) Purification and characterization of a novel ADP-dependent glucokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem 270:30453–30457
Klenk HP et al. (1997) The complete genome sequence of the hyperthermophilic, sulphate- reducing archaeon Archaeoglobus fulgidus. Nature 390:364–370
Koga S, Yoshioka I, Sakuraba H, Takahashi M, Sakasegawa S, Shimizu S, Ohshima T (2000) Biochemical characterization, cloning, and sequencing of ADP-dependent (AMP-forming) glucokinase from two hyperthermophilic archaea, Pyrococcus furiosus and Thermococcus litoralis. J Biochem (Tokyo) 128:1079–1085
Labes A, Schönheit P (2001) Sugar utilization in the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus strain 7324: starch degradation to acetate and CO2 via a modified Embden-Meyerhof pathway and acetyl-CoA synthetase (ADP-forming). Arch Microbiol 176:329–338
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Meyer C, Schmid R, Scriba PC, Wehling M (1996) Purification and partial sequencing of high-affinity progesterone- binding site(s) from porcine liver membranes. Eur J Biochem 239:726–731
Ronimus RS, Morgan HW (2001) The biochemical properties and phylogenies of phosphofructokinases from extremophiles. Extremophiles 5:357–373
Ronimus RS, Koning J, Morgan HW (1999) Purification and characterization of an ADP-dependent phosphofructokinase from Thermococcus zilligii. Extremophiles 3:121–129
Ronimus RS, de Heus E, Morgan HW (2001) Sequencing, expression, characterisation and phylogeny of the ADP-dependent phosphofructokinase from the hyperthermophilic, euryarchaeal Thermococcus zilligii. Biochim Biophys Acta 1517:384–391
Sakuraba H, et al (2002) ADP-dependent glucokinase/phosphofructokinase, a novel bifunctional enzyme from the hyperthermophilic archaeon Methanococcus jannaschii. J Biol Chem 277:12495–12498
Selig M, Xavier KB, Santos H, Schönheit P (1997) Comparative analysis of Embden-Meyerhof and Entner-Doudoroff glycolytic pathways in hyperthermophilic archaea and the bacterium Thermotoga. Arch Microbiol 167:217–232
Siebers B, Klenk HP, Hensel R (1998) PPi-dependent phosphofructokinase from Thermoproteus tenax, an archaeal descendant of an ancient line in phosphofructokinase evolution. J Bacteriol 180:2137–2143
Stetter KO (1996) Hyperthermophilic procaryotes. FEMS Microbiol Rev 18:149–158
Tsuge H, Sakuraba H, Kobe T, Kujime A, Katunuma N, Ohshima T (2002) Crystal structure of the ADP-dependent glucokinase from Pyrococcus horikoshii at 2.0-A resolution: A large conformational change in ADP-dependent glucokinase. Protein Sci 11:2456–2463
Tuininga JE, Verhees CH, Van der Oost J, Kengen SW, Stams AJ, De Vos WM (1999) Molecular and biochemical characterization of the ADP-dependent phosphofructokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem 274:21023–21028
Uyeda K (1979) Phosphofructokinases. Adv Enzymol 48:193–244
Verhees CH, Tuininga JE, Kengen SW, Stams AJ, van der OJ, De Vos WM (2001) ADP-dependent phosphofructokinases in mesophilic and thermophilic methanogenic archaea. J Bacteriol 183:7145–7153
Acknowledgements
The authors thank Dr. R. Schmid (Mikrobiologie, Universität Osnabrück) for N-terminal amino acid sequencing and H. Preidel for mass culturing Archaeoglobus fulgidus strain 7324. Some preliminary experiments of the purification and characterization of A. fulgidus ADP-PFK were carried out by Bettina Schlichting in an advanced student course. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SCHO 316/8–1) and the Fonds der Chemischen Industrie.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Antranikian
Rights and permissions
About this article
Cite this article
Hansen, T., Schönheit, P. ADP-dependent 6-phosphofructokinase, an extremely thermophilic, non-allosteric enzyme from the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus strain 7324. Extremophiles 8, 29–35 (2004). https://doi.org/10.1007/s00792-003-0356-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-003-0356-1