
Abstract
Relaxin family peptides perform a variety of biological functions by binding and activating relaxin family peptide receptor 1–4 (RXFP1–4), four A-class G protein-coupled receptors. In the present work, we developed a novel ligand binding assay for RXFP3 and RXFP4 based on NanoLuc complementation technology (NanoBiT). A synthetic ligation version of the low-affinity small complementation tag (SmBiT) was efficiently ligated to the A-chain N terminus of recombinant chimeric agonist R3/I5 using recombinant circular sortase A. After the ligation product R3/I5-SmBiT was mixed with human RXFP3 or RXFP4 genetically fused with a secretory large NanoLuc fragment (sLgBiT) at the N terminus, NanoLuc complementation was induced by high-affinity ligand–receptor binding. Binding kinetics and affinities of R3/I5-SmBiT with sLgBiT-fused RXFP3 and RXFP4 were conveniently measured according to the complementation-induced bioluminescence. Using R3/I5-SmBiT and the sLgBiT-fused receptor as a complementation pair, binding potencies of various ligands with RXFP3 and RXFP4 were quantitatively measured without the cumbersome washing step. The novel NanoBiT-based ligand binding assay is convenient for use and suitable for automation, thus will facilitate interaction studies of RXFP3 and RXFP4 with ligands in future. This assay can also be applied to some other plasma membrane receptors for pharmacological characterization of ligands in future studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The relaxin family is a group of peptide hormones, including relaxin, relaxin-3, and insulin-like peptide 3–6 (INSL3–6) (Bathgate et al. 2013; Halls et al. 2015; Ivell et al. 2017; Ma et al. 2017; Patil et al. 2017). These peptides perform a variety of biological functions, such as regulating reproduction, food intake, stress responses, and glucose homeostasis. To date, four relaxin family peptide receptors, namely RXFP1–4, have been identified, and all are A-class G protein-coupled receptors (GPCRs). Relaxin and INSL3 are cognate agonists of the homologous RXFP1 and RXFP2, respectively (Hsu et al. 2002; Kumagai et al. 2002), while relaxin-3 and INSL5 are cognate agonists of the homologous RXFP3 and RXFP4, respectively (Liu et al. 2003b, 2005). Relaxin-3 can also activate RXFP1 and RXFP4 in vitro with high efficiency (Liu et al. 2003a; Sudo et al. 2003), but the receptors of INSL4 and INSL6 remain unknown.
In recent studies, we developed novel bioluminescent ligand binding assays for these relaxin family peptide receptors using NanoLuc-based tracers (Hu et al. 2016b; Liu and Guo 2016; Wang et al. 2017b; Wu et al. 2016; Zhang et al. 2013a). Using these binding assays, we studied the interaction mechanism of RXFP3 and RXFP4 with their respective ligands, and developed novel agonists and antagonists (Hu et al. 2016a, b, 2017; Liu et al. 2016; Wei et al. 2017). However, these ligand binding assays require a cumbersome washing step to remove unbound tracer before quantitation of bound tracer. To exclude the time-consuming washing step, in a recent study we developed a bioluminescence resonance energy transfer (BRET)-based ligand binding assay for RXFP3 using NanoLuc as an energy donor (Wang et al. 2017a). However, the BRET-based ligand binding assay also has some problems, such as low energy transfer efficiency and high background, especially for those receptors with poor plasma membrane location.
In one recent study, NanoLuc binary technology (abbreviated as NanoBiT) was developed based on NanoLuc complementation by splitting the brightest NanoLuc luciferase at C-terminal region (Dixon et al. 2016). High luciferase activity could be restored after the almost inactive large NanoLuc fragment (designated as LgBiT) was complemented with small complementation tags with either low affinity (such as SmBiT) or high affinity (such as HiBiT). The high-affinity HiBiT–LgBiT pair is suitable for quantitation assays in living cells or in solution (Oh-Hashi et al. 2017; Sasaki et al. 2018; Schwinn et al. 2017), while the low-affinity SmBiT–LgBiT pair is suitable for monitoring dynamic interactions in living cells in a real-time manner (Bodle et al. 2017; Dabo et al. 2017; Dixon et al. 2016; Dupuis et al. 2017; Mo et al. 2017). In the present work, we applied the low-affinity SmBiT–LgBiT pair to RXFP3 and RXFP4, and developed a novel ligand binding assay for both receptors. The novel NanoBiT-based ligand binding assay is quick, simple, and suitable for automation, and thus will facilitate interaction studies of both receptors with ligands in future. The novel assay can also be applied to some other plasma membrane receptors with at least one extracellular terminus for pharmacological characterization of ligands in future studies.
Materials and methods
Generation of expression constructs for secretory LgBiT-fused RXFP3 and RXFP4
The DNA sequence encoding the secretory LgBiT (designated as sLgBiT) was chemically synthesized (GeneWiz, Suzhou, China) according to the published DNA sequence of LgBiT and the cDNA sequence of the signal peptide of human interleukin-6. The resulting synthetic DNA fragment was cleaved with restriction enzymes NheI and KpnI, and ligated into pcDNA6 vector to generate the intermediate construct pcDNA6/sLgBiT. The coding sequence of human RXFP3 and human RXFP4 was either excised from our previous construct or PCR amplified, and inserted into pcDNA6/sLgBiT pretreated with KpnI and AgeI, resulting in the final pcDNA6/sLgBiT-RXFP3 and pcDNA6/sLgBiT-RXFP4 constructs encoding N-terminally sLgBiT-fused RXFP3 or RXFP4, respectively. The coding regions of sLgBiT-RXFP3 and sLgBiT-RXFP4 were confirmed by DNA sequencing, and their nucleotide and amino acid sequences are shown in supplementary Fig. S1.
Preparation of peptides
The ligation version of SmBiT (designated as SmBiT-srt) with a sortase recognition motif at the C terminus was chemically synthesized by solid-phase peptide synthesis using standard Fmoc methodology (GL Biochem, Shanghai, China). The ligation version of recombinant R3/I5 with four successive Gly residues at the A-chain N terminus (designated as R3/I5-4G) and other relaxin family peptides were prepared by overexpression in Escherichia coli and subsequent in vitro refolding and enzymatic maturation according to our previously described procedures (Luo et al. 2010; Zhang et al. 2013b). All peptides were purified to homogeneity by high-performance liquid chromatography (HPLC) using C18 reversed-phase columns (Zorbax 300SB-C18, 9.4 or 4.6 mm × 250 mm; Agilent Technologies, Santa Clara, CA, USA) and confirmed by mass spectrometry.
Preparation of a circular sortase A by split mini-intern-mediated peptide cyclization in E. coli
The gene encoding the split mini-intern derived from Synechocystis sp. PCC6803 DnaB was chemically synthesized (GeneWiz, Suzhou, China) according to the previously published sequence (Williams et al. 2002) and unique cleavage sites for EcoRI, SacI, HindIII, and NotI were introduced for insertion of target genes. After cleavage by restriction enzymes NdeI and XhoI, the synthetic mini-intern gene was ligated into a pET expression vector, resulting in the expression construct pET/intern. The coding region of the mini-intern was confirmed by DNA sequencing, and its nucleotide and amino acid sequences are shown in supplementary Fig. S2.
The coding sequence of the N-terminally truncated Staphylococcus aureus sortase A (residues 60-206) was PCR amplified using our previous pET/srtA construct as template (Wang et al. 2017b). After cleavage by restriction enzymes EcoRI and NotI, the sortase gene was inserted into the pET/intern construct pretreated with the same restriction enzymes, resulting in the pET/intern-srtA construct for overexpression of a circular sortase A in E. coli as described previously (Zhulenkovs et al. 2014). Nucleotide and amino acid sequences of the mini-intern-sortase A fusion protein are shown in supplementary Fig. S2. Thereafter, the pET/intern-srtA construct was transformed into E. coli strain BL21(DE3) and the circular sortase A was overexpressed following induction with 1.0 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 30°C overnight. Thereafter, E. coli cells were harvested by centrifugation (5000g, 10 min), resuspended in lysis buffer (20 mM sodium acetate, pH 4.5), and lysed by sonication. After centrifugation (10,000g, 30 min), the lysate supernatant was applied to a cation ion-exchange column (SP-Sephadex) and the fraction containing circular sortase A was eluted by 0.5 M sodium chloride (in 20 mM sodium acetate, pH 4.5). The eluted sortase fraction was dialysed against 20 mM Tris–HCl buffer (pH 8.5) and applied to an anion ion-exchange column (TSKgel DEAE-5PW, 7.5 mm × 75 mm; Sigma-Aldrich, St. Louis, MO, USA). The circular sortase fraction was eluted using a gradient of sodium chloride (in 20 mM Tris–HCl, pH 8.5) and confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Preparation of SmBiT-ligated R3/I5 using circular sortase A
Lyophilized R3/I5-4G and SmBiT-srt were dissolved in ligation buffer (100 mM Tris–HCl, 0.5 M urea, 5 mM CaCl2, pH 7.4) at a final concentration of ~ 0.2 and ~ 0.15 mM, respectively. To initiate ligation, recombinant circular sortase A was added to a final concentration of ~ 40 μM, and ligation was conducted at 30 °C overnight. Thereafter, the reaction mixture was acidified to pH 3–4 by trifluoroacetic acid, applied to a C18 reversed-phase column (Zorbax 300SB-C18, 4.6 mm × 250 mm; Agilent Technologies), and eluted by an acidic acetonitrile gradient. Eluted fractions were manually collected, lyophilized, and analyzed by tricine–SDS-PAGE and mass spectrometry.
NanoBiT-based ligand binding assays for RXFP3 and RXFP4
The expression constructs for sLgBiT-fused RXFP3 or RXFP4 were transiently transfected into human embryonic kidney (HEK) 293T cells. After transfection for ~ 36 h, cells were detached by gentle pipetting and suspended in serum-free Dulbecco’s modified Eagle’s medium (DMEM). For saturation and competition binding assays, living cells were directly used as a receptor source. For kinetic binding assays, the cell suspension was subjected to a short sonication (~ 30 s) and the cell homogenate was used as a receptor source. The cell suspension or homogenate was placed in a white opaque 96-well plate (25 μl/well, ~ 5 × 104 cells or equivalent/well). For binding kinetic assays, the cell homogenate was sequentially mixed with 1 μl of NanoLuc substrate and 25 μl of binding solution (serum-free DMEM plus 1% BSA) containing 20 nM of R3/I5-SmBiT, and bioluminescence data were collected for 20–30 min on a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA, USA) with an interval of 30 s. Thereafter, 1 μl of 50 μM R3/I5 stock solution was added and bioluminescence data were continuously collected for 50–65 min at an interval of 30 s. For saturation binding assays, the cell suspension was mixed with binding solution (25 μl/well) containing various concentrations of R3/I5-SmBiT. For competition binding assays, the cell suspension was mixed with binding solution (25 μl/well) containing a constant concentration of R3/I5-SmBiT and various concentrations of competitor. After incubation at 22°C for ~ 30 min, diluted substrate was added (5 μl/well) and bioluminescence was measured on a SpectraMax M5 plate reader (Molecular Devices) in a luminescence mode.
Results
Preparation of SmBiT-ligated R3/I5 and complementation with sLgBiT-fused RXFP3 and RXFP4
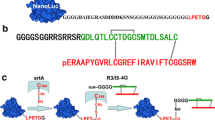
To develop a NanoBiT-based ligand binding assay for RXFP3 and RXFP4 (Fig. 1a), we attempted to attach the low-affinity SmBiT tag to the chimeric R3/I5 peptide, an efficient agonist for both receptors. We first genetically fused the SmBiT sequence to the A-chain N terminus of a single-chain R3/I5 precursor. Unfortunately, the resultant precursor could not be refolded in vitro after overexpression in E. coli, suggesting the fused SmBiT tag is detrimental to foldability of the precursor. Thus, we tried to attach a synthetic SmBiT tag to mature recombinant R3/I5 using sortase-catalyzed peptide ligation that was successfully employed in our previous study for rapid preparation of a bioluminescent tracer (Wang et al. 2017b). A sortase recognition motif was introduced to the C terminus of the SmBiT tag, and the resultant SmBiT-srt was conveniently prepared using solid-phase peptide synthesis (Fig. 1b). Four successive Gly residues and an Arg-rich solubilising tag were introduced to the A-chain N terminus of R3/I5, and the resultant R3/I5-4G was prepared through overexpression of a single-chain precursor in E. coli and subsequent in vitro refolding and enzymatic maturation according to our previous procedure (Fig. 1b). Sortase A can recognize the LPRTG motif of the synthetic SmBiT-srt, cleave the peptide bond between the Thr and Gly residues, and form a transient thioester bond between the carboxyl moiety of the Thr residue and the sulfhydryl moiety of its active site Cys residue (Fig. 1c). Thereafter, amine moiety of the first Gly residue of the recombinant R3/I5-4G can attack the transient thioester bond, release free sortase, and form a new peptide bond between the C-terminal Thr residue of the synthetic SmBiT-srt and the A-chain N-terminal Gly residue of R3/I5-4G (Fig. 1c). As a result, the synthetic SmBiT-srt was covalently attached to the A-chain N terminus of the recombinant R3/I5-4G via a peptide bond (Fig. 1b). For efficient catalysis of the peptide ligation, we prepared a circular sortase A via split mini-intern-mediated peptide chain cyclization in E. coli according to a previously reported procedure (Zhulenkovs et al. 2014). The circular sortase A is more stable than the linear form, and thus remains active in the presence of up to 2 M urea (Zhulenkovs et al. 2014). After the synthetic SmBiT-srt and the recombinant R3/I5-4G were ligated by the recombinant circular sortase A, two major peaks appeared on HPLC (Fig. 1d). Tricine–SDS-PAGE analysis (Fig. 1d, inner panel) revealed that the asymmetric peak 1 was a mixture of the circular sortase A and unreacted R3/I5-4G, while the symmetrical peak 2 was presumably the ligation product R3/I5-SmBiT because it displayed slightly lower mobility rate than R3/I5-4G. The identity of peak 2 was subsequently confirmed by mass spectrometry: its measured molecular mass (8824.0) was consistent with the theoretical value (8825.2) of the expected ligation product (Fig. S3). Thus, the novel SmBiT-based R3/I5 tracer could be conveniently prepared through sortase-catalyzed peptide ligation.

Development of the novel NanoBiT-based ligand binding assay for RXFP3 and RXFP4. a Schematic presentation of the NanoBiT-based ligand binding assay. b Amino acid sequences of synthetic SmBiT-srt, recombinant R3/I5-4G, and the ligation product R3/I5-SmBiT. The sequence of SmBiT is shown in pink, and the sortase recognition motif in blue. The A-chain of human INSL5 is shown in green, and the B-chain of human relaxin-3 in red. The disulfide linkages are shown as lines. c Mechanism for the sortase-catalyzed ligation of the synthetic SmBiT-srt and the recombinant R3/I5-4G. d Purification of R3/I5-SmBiT by HPLC after enzymatic ligation. Inner panel, tricine–SDS-PAGE analysis of peak 1 and peak 2. After electrophoresis, the gel was stained using Coomassie Brilliant Blue R250. e Bioluminescence measured using living HEK293T cells overexpressing sLgBiT-fused RXFP3 or RXFP4 under different conditions. The transfected cells were mixed with the indicated peptides and incubated at room temperature for ~ 30 min. After addition of NanoLuc substrate, bioluminescence was measured on a SpectraMax M5 plate reader under luminescence mode. Measured data are expressed as mean ± standard error (SE; n = 3) (color figure online)
To establish the NanoBiT-based ligand binding assay, the receptors RXFP3 and RXFP4 were genetically fused with a LgBiT harboring a human interleukin-6 signal peptide (designated as sLgBiT) for efficient extracellular translocation of the large LgBiT molecule. After the sLgBiT-fused RXFP3 and RXFP4 were transiently overexpressed in HEK293T cells, the measured bioluminescence was very low after addition of the NanoLuc substrate (Fig. 1e), confirming the low activity of LgBiT itself. After addition of SmBiT-srt (10 nM), the measured bioluminescence was not increased (Fig. 1e), suggesting that SmBiT itself could not complement LgBiT at low concentrations due to its low affinity with LgBiT. After addition of R3/I5-4G, the measured bioluminescence was again not increased as expected (Fig. 1e). However, strong bioluminescence was detected after addition of R3/I5-SmBiT (Fig. 1e), suggesting that receptor binding of R3/I5-SmBiT induced complementation of the ligand-tagged SmBiT with the receptor-fused LgBiT due to proximity effect (Fig. 1a). Moreover, complementation was abolished by competition with high concentration (1000 nM) of R3/I5-4G (Fig. 1e), confirming that the observed complementation effect was mediated by ligand–receptor binding. In summary, ligand–receptor binding-induced NanoLuc complementation was successfully achieved between R3/I5-SmBiT and sLgBiT-fused RXFP3 and RXFP4.
Binding kinetics of R3/I5-SmBiT with sLgBiT-fused RXFP3 and RXFP4
As discussed above, binding of R3/I5-SmBiT with sLgBiT-RXFP3 and sLgBiT-RXFP4 restored high NanoLuc activity. According to the complementation-induced bioluminescence, we determined binding kinetics of R3/I5-SmBiT with the sLgBiT-fused receptors. In the kinetic assays, we used cell homogenate instead of living cells because the sLgBiT-fused receptors would be internalized after binding the tracer, and would, therefore, not be displaced by the competitor in the dissociation phase. After addition of R3/I5-SmBiT (final concentration of 10 nM) into the cell homogenate containing overexpressed sLgBiT-RXFP3, the measured bioluminescence increased quickly, reaching a plateau within 20 min with a calculated half-life of ~ 2.6 min (Fig. 2a). After addition of R3/I5 to a high concentration (1 μM), the measured bioluminescence decreased slowly due to displacement of the receptor-bound R3/I5-SmBiT by the competitor, with a half-life of ~ 19 min (Fig. 2a). The data were fitted well by the association/dissociation functions, implying that the R3/I5-tagged SmBiT could complement the RXFP3-fused LgBiT rapidly after ligand–receptor binding.

Binding kinetics of R3/I5-SmBiT with sLgBiT-RXFP3 (a) and sLgBiT-RXFP4 (b). NanoLuc substrate and R3/I5-SmBiT were added to the homogenate of HEK293T cells transiently overexpressing sLgBiT-fused receptor and bioluminescence was measured on a SpectraMax M5 plate reader with an interval of 30 s. For dissociation measurement, competitor R3/I5 was added and bioluminescence was continuously measured with an interval of 30 s. Association data were fitted to Y = Bmax(1 − e−kX), and dissociation data were fitted to Y = Bmaxe−kX, using SigmaPlot 10.0 software
After R3/I5-SmBiT (final concentration of 10 nM) was added to the cell homogenate containing overexpressed sLgBiT-RXFP4, the measured bioluminescence increased slowly, reaching a plateau within 30 min with a half-life of ~ 8 min (Fig. 2b). The association data for sLgBiT-RXFP4 displayed a short delay, and did not fit well to the association function. This might be caused by a slow complementation of the R3/I5-tagged SmBiT with the RXFP4-fused sLgBiT after ligand–receptor binding, because the extracellular N-terminal domain of RXFP4 is much shorter than that of RXFP3. After addition of a high concentration (1 μM) of competitor, bioluminescence decreased slowly, with a half-life of ~ 58 min (Fig. 2b). In summary, binding of the SmBiT-based R3/I5 tracer to the sLgBiT-fused receptors could be conveniently monitored using the NanoLuc complementation approach.
Binding affinities of R3/I5-SmBiT with sLgBiT-fused RXFP3 and RXFP4
According to the complementation-induced bioluminescence, we measured receptor binding affinities of R3/I5-SmBiT through saturation binding assays using living HEK293T cells transiently overexpressing sLgBiT-RXFP3 or sLgBiT-RXFP4 as a receptor source. As the concentration of R3/I5-SmBiT was increased, measured bioluminescence increased in a typical saturation manner (Fig. 3). The measured data were fitted well to one-site binding model, with a calculated dissociation constant (Kd) of 0.70 ± 0.04 nM (n = 3) for RXFP3 and 2.77 ± 0.22 nM (n = 3) for RXFP4. These Kd values measured by the NanoLuc complementation approach are slightly lower than previous values measured using the NanoLuc-based bioluminescent tracer or the BRET approach (Wang et al. 2017a, b), since binding between the ligand-tagged SmBiT and the receptor-fused sLgBiT also contributes to the total binding affinity. Moreover, nonspecific binding measured by the complementation approach was very low, which would benefit its use in competition binding assays.

Saturation binding of R3/I5-SmBiT with sLgBiT-RXFP3 (a) and sLgBiT-RXFP4 (b). Various concentrations of R3/I5-SmBiT were mixed with living HEK293T cells transiently overexpressing sLgBiT-RXFP3 or sLgBiT-RXFP4. After incubation at 22 °C for ~ 30 min, bioluminescence was measured on a SpectraMax M5 plate reader after addition of NanoLuc substrate. Nonspecific binding data were obtained by competition with 1.0 μM of R/I5. The measured data are expressed as mean ± SE (n = 3) and were fitted to Y = BmaxX/(X + Kd) + NsX for total binding, Y = BmaxX/(X + Kd) for specific binding, and Y = NsX for nonspecific binding, using SigmaPlot 10.0 software
NanoBiT-based competition binding assays for characterization of various ligands
Since R3/I5-SmBiT could bind the sLgBiT-fused receptors in a reversible manner with high affinity, we established competition ligand binding assays using this complementation pair for characterization of various ligands for RXFP3 and RXFP4. Using R3/I5-SmBiT as a tracer and living HEK293T cells transiently overexpressing sLgBiT-fused RXFP3 or RXFP4 as a receptor source, sigmoidal competition curves were obtained for different ligands (Fig. 4). The pIC50 values, indicating the receptor binding potencies of different ligands, were calculated from these binding curves (Table 1). The measured pIC50 value for R3/I5-4G was similar to that of wild-type R3/I5 for both RXFP3 and RXFP4, hence they displayed similar binding potency towards RXFP3 and towards RXFP4. However, INSL5 displayed ~ 50-fold lower binding potency towards RXFP3, but displayed only ~ threefold lower binding potency towards RXFP4, compared with wild-type R3/I5. As shown in Table 1, the present data measured by the NanoBiT-based binding assay were consistent with our previous results measured by conventional receptor binding assays using NanoLuc-based bioluminescent tracer (Wang et al. 2017b). Thus, the NanoBiT-based binding assay could sensitively discriminate the binding potency of various ligands for RXFP3 and RXFP4 and, therefore, represents a novel ligand binding assay for characterization of various ligands.

NanoBiT-based competition binding assays for RXFP3 (a) and RXFP4 (b). Nonspecific data were obtained by competition with 1.0 μM of R3/I5. Measured data are expressed as mean ± SE (n = 3) and fitted to sigmoidal curves using SigmaPlot 10.0 software. The calculated pIC50 values are listed in Table 1
Discussion
In the present study, we developed a novel NanoBiT-based ligand binding assay for G protein-coupled receptors RXFP3 and RXFP4. The novel assay involves two simple steps without the cumbersome and time-consuming washing procedure, as follows: (1) SmBiT-based tracer and competitor were mixed with cells overexpressing LgBiT-fused receptor and incubated for a short time (~ 30 min); (2) NanoLuc substrate was added and bioluminescence measured on a plate reader in a luminescence mode. Thus, the NanoBiT-based ligand binding assay is convenient for use and suitable for automation.
In future studies, the NanoBiT-based ligand binding assay can be applied to some other plasma membrane receptors with at least one extracellular terminus for genetic fusion with LgBiT. To establish the NanoBiT-based binding assay, the large LgBiT should be genetically fused to the extracellular terminus of the receptor with an appropriate linker, ensuring that the fused LgBiT has an extracellular location and good flexibility. For example, a secretory LgBiT can be fused at the N terminus of a GPCR, since all GPCRs have an extracellular N terminus and an intracellular C terminus. On the other hand, the small SmBiT tag should be attached to an appropriate position of the ligand with an appropriate linker, ensuring that the tag has a good flexibility, but no serious detrimental effect on ligand–receptor binding. For protein or peptide ligands, the SmBiT tag can either be genetically fused at one terminus, or chemically/enzymatically attached to an appropriate site. Given its small size (13 amino acids, ~ 1.3 kDa, similar to some fluorescent dyes), SmBiT might also be attached to nonpeptidic hormones through an appropriate approach. Essentially, if significant NanoLuc complementation is induced by ligand–receptor binding between the LgBiT-fused receptor and the SmBiT-tagged ligand, a NanoBiT-based ligand binding assay could be developed using this complementation pair.
References
Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ (2013) Relaxin family peptides and their receptors. Physiol Rev 93:405–480
Bodle CR, Hayes MP, O’Brien JB, Roman DL (2017) Development of a bimolecular luminescence complementation assay for RGS: G protein interactions in cells. Anal Biochem 522:10–17
Dabo S, Maillard P, Collados Rodriguez M, Hansen MD, Mazouz S, Bigot DJ, Tible M, Janvier G, Helynck O, Cassonnet P, Jacob Y, Bellalou J, Gatignol A, Patel RC, Hugon J, Munier-Lehmann H, Meurs EF (2017) Inhibition of the inflammatory response to stress by targeting interaction between PKR and its cellular activator PACT. Sci Rep 7:16129
Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, Machleidt T, Kirkland TA, Wood MG, Eggers CT, Encell LP, Wood KV (2016) NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol 11:400–408
Dupuis N, Laschet C, Franssen D, Szpakowska M, Gilissen J, Geubelle P, Soni A, Parent AS, Pirotte B, Chevigné A, Twizere JC, Hanson J (2017) Activation of the orphan G protein-coupled receptor GPR27 by surrogate ligands promotes β-arrestin 2 recruitment. Mol Pharmacol 91:595–608
Halls ML, Bathgate RA, Sutton SW, Dschietzig TB, Summers RJ (2015) International Union of Basic and Clinical Pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1–4, the receptors for relaxin family peptides. Pharmacol Rev 67:389–440
Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD, Hsueh AJ (2002) Activation of orphan receptors by the hormone relaxin. Science 295:671–674
Hu MJ, Shao XX, Wang JH, Wei D, Guo YQ, Liu YL, Xu ZG, Guo ZY (2016a) Mechanism for insulin-like peptide 5 distinguishing the homologous relaxin family peptide receptor 3 and 4. Sci Rep 6:29648
Hu MJ, Shao XX, Wang JH, Wei D, Liu YL, Xu ZG, Guo ZY (2016b) Identification of hydrophobic interactions between relaxin-3 and its receptor RXFP3: implication for a conformational change in the B-chain C-terminus during receptor binding. Amino Acids 48:2227–2236
Hu MJ, Wei D, Shao XX, Wang JH, Liu YL, Xu ZG, Guo ZY (2017) Interaction mechanism of insulin-like peptide 5 with relaxin family peptide receptor 4. Arch Biochem Biophys 619:27–34
Ivell R, Agoulnik AI, Anand-Ivell R (2017) Relaxin-like peptides in male reproduction - a human perspective. Br J Pharmacol 174:990–1001
Kumagai J, Hsu SY, Matsumi H, Roh JS, Fu P, Wade JD, Bathgate RA, Hsueh AJ (2002) INSL3/Leydig insulin-like peptide activates the LGR8 receptor important in testis descent. J Biol Chem 277:31283–31286
Liu YL, Guo ZY (2016) Novel bioluminescent binding assays for interaction studies of protein/peptide hormones with their receptors. Amino Acids 48:1151–1160
Liu C, Chen J, Sutton S, Roland B, Kuei C, Farmer N, Sillard R, Lovenberg TW (2003a) Identification of relaxin-3/INSL7 as a ligand for GPCR142. J Biol Chem 278:50765–50770
Liu C, Eriste E, Sutton S, Chen J, Roland B, Kuei C, Farmer N, Jörnvall H, Sillard R, Lovenberg TW (2003b) Identification of relaxin-3/INSL7 as an endogenous ligand for the orphan G-protein-coupled receptor GPCR135. J Biol Chem 278:50754–50764
Liu C, Kuei C, Sutton S, Chen J, Bonaventure P, Wu J, Nepomuceno D, Kamme F, Tran DT, Zhu J, Wilkinson T, Bathgate R, Eriste E, Sillard R, Lovenberg TW (2005) INSL5 is a high affinity specific agonist for GPCR142 (GPR100). J Biol Chem 280:292–300
Liu Y, Zhang L, Shao XX, Hu MJ, Liu YL, Xu ZG, Guo ZY (2016) A negatively charged transmembrane aspartate residue controls activation of the relaxin-3 receptor RXFP3. Arch Biochem Biophys 604:113–120
Luo X, Bathgate RA, Zhang WJ, Liu YL, Shao XX, Wade JD, Guo ZY (2010) Design and recombinant expression of insulin-like peptide 5 precursors and the preparation of mature human INSL5. Amino Acids 39:1343–1352
Ma S, Smith CM, Blasiak A, Gundlach AL (2017) Distribution, physiology and pharmacology of relaxin-3/RXFP3 systems in brain. Br J Pharmacol 174:1034–1048
Mo X, Qi Q, Ivanov AA, Niu Q, Luo Y, Havel J, Goetze R, Bell S, Moreno CS, Cooper LA, Johns MA, Khuri FR, Du Y, Fu H (2017) AKT1, LKB1, and YAP1 revealed as MYC interactors with NanoLuc-based protein-fragment complementation assay. Mol Pharmacol 91:339–347
Oh-Hashi K, Furuta E, Fujimura K, Hirata Y (2017) Application of a novel HiBiT peptide tag for monitoring ATF4 protein expression in Neuro2a cells. Biochem Biophys Rep 12:40–45
Patil NA, Rosengren KJ, Separovic F, Wade JD, Bathgate RAD, Hossain MA (2017) Relaxin family peptides: structure-activity relationship studies. Br J Pharmacol 174:950–961
Sasaki M, Anindita PD, Phongphaew W, Carr M, Kobayashi S, Orba Y, Sawa H (2018) Development of a rapid and quantitative method for the analysis of viral entry and release using a NanoLuc luciferase complementation assay. Virus Res 243:69–74
Schwinn MK, Machleidt T, Zimmerman K, Eggers CT, Dixon AS, Hurst R, Hall MP, Encell LP, Binkowski BF, Wood KV (2017) CRISPR-mediated tagging of endogenous proteins with a luminescent peptide. ACS Chem Biol. https://doi.org/10.1021/acschembio.7b00549
Sudo S, Kumagai J, Nishi S, Layfield S, Ferraro T, Bathgate RA, Hsueh AJ (2003) H3 relaxin is a specific ligand for LGR7 and activates the receptor by interacting with both the ectodomain and the exoloop 2. J Biol Chem 278:7855–7862
Wang JH, Shao XX, Hu MJ, Wei D, Liu YL, Xu ZG, Guo ZY (2017a) A novel BRET-based binding assay for interaction studies of relaxin family peptide receptor 3 with its ligands. Amino Acids 49:895–903
Wang JH, Shao XX, Hu MJ, Wei D, Nie WH, Liu YL, Xu ZG, Guo ZY (2017b) Rapid preparation of bioluminescent tracers for relaxin family peptides using sortase-catalysed ligation. Amino Acids 49:1611–1617
Wei D, Hu MJ, Shao XX, Wang JH, Nie WH, Liu YL, Xu ZG, Guo ZY (2017) Development of a selective agonist for relaxin family peptide receptor 3. Sci Rep 7:3230
Williams NK, Prosselkov P, Liepinsh E, Line I, Sharipo A, Littler DR, Curmi PM, Otting G, Dixon NE (2002) In vivo protein cyclization promoted by a circularly permuted Synechocystis sp. PCC6803 DnaB mini-intein. J Biol Chem 277:7790–7798
Wu QP, Zhang L, Shao XX, Wang JH, Gao Y, Xu ZG, Liu YL, Guo ZY (2016) Application of the novel bioluminescent ligand–receptor binding assay to relaxin-RXFP1 system for interaction studies. Amino Acids 48:1099–1107
Zhang L, Song G, Xu T, Wu QP, Shao XX, Liu YL, Xu ZG, Guo ZY (2013a) A novel ultrasensitive bioluminescent receptor-binding assay of INSL3 through chemical conjugation with nanoluciferase. Biochimie 95:2454–2459
Zhang WJ, Jiang Q, Wang XY, Song G, Shao XX, Guo ZY (2013b) A convenient method for europium-labeling of a recombinant chimeric relaxin family peptide R3/I5 for receptor-binding assays. J Pept Sci 19:350–354
Zhulenkovs D, Jaudzems K, Zajakina A, Leonchiks A (2014) Enzymatic activity of circular sortase A under denaturing conditions: an advanced tool for protein ligation. Biochem Eng J 82:200–209
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31670773, 31470767).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: F. Albericio.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hu, MJ., Shao, XX., Li, HZ. et al. Development of a novel ligand binding assay for relaxin family peptide receptor 3 and 4 using NanoLuc complementation. Amino Acids 50, 1111–1119 (2018). https://doi.org/10.1007/s00726-018-2588-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-018-2588-5