
Abstract
Objective
In this study, we investigated the occurrence of papillomavirus (PV) infection in non-human primates (NHPs) in northeastern Argentina. We also explored their evolutionary history and evaluated the co-speciation hypothesis in the context of primate evolution.
Methods
We obtained DNA samples from 57 individuals belonging to wild and captive populations of Alouatta caraya, Sapajus nigritus, and Sapajus cay. We assessed PV infection by PCR amplification with the CUT primer system and sequencing of 337 bp (112 amino acids) of the L1 gene. The viral sequences were analyzed by phylogenetic and Bayesian coalescence methods to estimate the time to the most common recent ancestor (tMRCA) using BEAST, v1.4.8 software. We evaluated viral/host tree congruence with TreeMap v3.0.
Results
We identified two novel putative PV sequences of the genus Gammapapillomavirus in Sapajus spp. and Alouatta caraya (SPV1 and AcPV1, respectively). The tMRCA of SPV1 was estimated to be 11,941,682 years before present (ybp), and that of AcPV1 was 46,638,071 ybp, both before the coalescence times of their hosts (6.4 million years ago [MYA] and 6.8 MYA, respectively). Based on the comparison of primate and viral phylogenies, we found that the PV tree was no more congruent with the host tree than a random tree would be (P > 0.05), thus allowing us to reject the model of virus-host coevolution.
Conclusion
This study presents the first evidence of PV infection in platyrrhine species from Argentina, expands the range of described hosts for these viruses, and suggests new scenarios for their origin and dispersal.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Papillomaviruses (PVs) are members of a highly diverse family of viruses (Papillomaviridae) that were initially described in mammals but have since been found in birds, turtles, snakes, and fish and probably infect all vertebrates [1]. PVs have a circular double-stranded DNA genome with a size close to 8 kb, and their taxonomy is based on the percentage of nucleotide sequence identity in the L1 gene and the complete genome [2]. Thus far, a total of 429 viral types have been identified. These include 211 types found in non-human animals (86 species) and 218 found exclusively in humans (HPV), which are the most intensively studied hosts [1,2,3]. Because there is a larger body of research focused on the study of PVs of clinical significance for humans (members of the genera Alpha-, Beta-, Gamma-, Mu-, and Nupapillomavirus), the described host range is probably not a true reflection of the biology of these viruses [4,5,6].
PVs have evolved in close relationship with their hosts, leading to the hypothesis of co-speciation [7, 8]. However, multiple incongruences between their evolutionary history and those of their hosts have been demonstrated previously [9,10,11,12]. For this reason, additional evolutionary forces such as cross-species infection, recombination, and gene duplication are likely to have influenced PV evolution [13,14,15,16].
PVs infect animals in an anatomically site-specific fashion (epithelia and mucosa), with genital infections being associated with cervical dysplasia and carcinomas in humans, macaques, and baboons [17,18,19]. Moreover, at least 30 PVs types/putative types have been detected in the genital mucosa of non-human primate (NHP) species, such as Macaca fascicularis (MfPV1-11 and MmPV2-7), Macaca mulatta (MmPV1-7), Macaca fuscata (MfuPV1-2), Pan paniscus (PpPV1), Pan troglodytes (PtPV1), Colobus guereza (CgPV1-2), and Papio hamadryas anubis (PhPV1) [1, 3, 5, 19,20,21,22,23,24]. However, information about PV infection in neotropical NHP species is still scarce, with only four PVs identified in Saimiri sciureus (SscPV1-3) and Alouatta guariba (AgPV1) [23, 25, 26] and two putative types in Ateles geoffroyi (strain SMAA1) and Callicebus cupreus (PV006) [4, 27].
In Argentina, there are five species of NHPs. These include Alouatta caraya (black and gold howler monkeys), Alouatta guariba clamitans (brown howler monkeys), Aotus azarae (owl monkeys), Sapajus nigritus (black capuchins), and Sapajus cay (brown-capped capuchins) [28]. They inhabit the remaining forest in Argentina, and their populations are facing severe threats due to habitat alteration for agriculture (soy and rice crops, deforestation, etc.) and illegal hunting [28]. Moreover, the expansion of human populations and human activities within primate habitats has resulted in a high potential for pathogen exchange [29].
This research investigated the occurrence of PV infection in the New World monkey species Alouatta caraya, Sapajus nigritus, and Sapajus cay in northeastern Argentina by using PCR-CUT primer system and sequencing of 337 bp (112 amino acids) of the L1 gene. We conducted phylogenetic and coalescence analysis with partial viral (L1) and host (CYTB) sequences in order to describe their evolutionary history and evaluate the cospeciation hypothesis in the context of primate evolution. The results of this study expand our knowledge about viral infections in those species and contribute to the understanding of PV evolution in primate species more broadly.
Material and methods
Biological samples and bioethics
For this study, we collected biological samples (feces, genital swabs and oral swabs) from 57 NHP individuals (19 from captive and 38 from wild populations) in Argentina. These included members of the species Alouatta caraya (family Atelidae) and Sapajus nigritus, S. cay, and unidentified monkeys from the genus Sapajus (Family Cebidae). The latter belong to a captive population living in the Ecological Park El Puma in Misiones Province (see below).
This study complied with the Code of Best Practices for Field Primatology (International Primatological Society, 2014) [30]. It was conducted with the approval of authorities from Corrientes and Chaco, the Administración de Parques Nacionales in Argentina (number NEA350), and the Ministerio de Ecología y Recursos Renovables de la Provincia de Misiones (number DISP 121 EXP 9910-00076/12). The animal capture and identification techniques were designed to be less invasive to preserve the welfare of the animals and relieve potential stress.
Sampling sites and methods
Wild populations: Sampling sites were located at the Biological Station of Corrientes, Argentine Museum of Natural Sciences (EBCo-MACN) and State Park San Cayetano Corrientes [27°30′ S, 58°41′ W]; Paraje Santa Rita, Chaco [26° 01′ 32.46″ S, 59° 58′ 33.49″ W]; and the Iguazú National Park, Misiones [25°40′S, 54°30′W].
Fifteen fecal samples were collected at the Iguazú National Park at Misiones during 2012–13. The park is part of the Upper Paraná Atlantic Forest at the southwestern edge of the South American Atlantic Forest complex. Researchers were trained for a period of three months at the site in order to be able to individually recognize every member of the troop by their physical features (facial color pattern, body size, and shape of tufts). Researchers followed S. nigritus troops and waited until an identified monkey defecated. Immediately after defecation, approximately 5 g of feces per individual was taken from the forest floor and then transferred to a sterile 50-ml polypropylene tube. Samples were kept cool (2–8°C) until reaching the lab and stored at -20°C until DNA extraction.
A total of 31 sample swabs from the genital (n = 23) and oral (n = 8) cavities of A. caraya were obtained through capture and anesthesia in EBCo-MACN at the Biological Station of Corrientes and State Park San Cayetano in Corrientes and Paraje Santa Rita in Chaco. The research group has conducted long-term studies with these troops over the past 15 years [28, 29, 31]. Individuals were immobilized with medetomidine hydrochloride combined with ketamine hydrochloride, administered via a dart driven by compressed air. A trained veterinarian collected the samples by swabbing the oral or genital area and then transferring them to a 15-ml sterile tube with 1X phosphate buffered saline (PBS). During this procedure, the body temperature of the NHPs was maintained by covering the animals with blankets and placing warm water bottles next to their bodies. After sampling, each animal was transferred to the exact site of capture and observed until it fully recovered. The samples were maintained at a cold temperature until they were taken to the lab, whereupon they were frozen at -20°C until subjected to DNA extraction.
Captive populations: Eighteen archival samples of DNA extracted from oral and genital samples were available at the laboratory (LaBiMAp-FCEQyN-UNaM) as a part of previously unpublished studies from our group conducted at the ecological reserve “El Puma” (Candelaria, Misiones) [27°27′36.5″ S, 55°48′00.5″ W]. This site houses rescued animals kept as pets and receives confiscated animals from illegal trafficking in Misiones Province and nearby areas. The Institution is open to the public and abides by local regulations.
Briefly, animals are kept in outdoor cages of wire mesh with a soil floor and roof. These cages have crossbars, ropes, platforms, dry tree trunks, and some refuges to provide environmental and behavioral enrichment to the animals. Different species occupy different cages. The primates are grouped according to their physical characteristics (see below), age, size of the resident troop (approximately 15 individuals per cage). They are fed with a balanced and varied diet consisting of seasonal fruits, vegetables and seeds, and red and white meats. Water is freely available. The wellbeing of the animals is monitoring daily by trained personnel from the park.
The sampling was conducted as follows. The animals were captured using a net and received an injection of anesthetic (0.1-0.3 ml of Zelazol or ketamine, 25 mg/kg). Once the animal was asleep, a trained veterinarian collected the samples by swabbing the oral or genital area and then transferring them to a 15-ml sterile tube with 1X PBS. These samples were kept cold in an ice bucket and later transferred to the lab for DNA extraction. After sampling, each animal received a vitamin supplement and was observed until it fully recovered and returned to the home colony.
At the time of sampling, the species assignment was based on the phenotypic diagnosis of the individual. Sapajus nigritus (black capuchin) is characterized by a very dark brown or grey, even blackish, pelage, with no (or very vague) dorsal stripe, and the face is white and contrasts with the color of the body. By contrast, Sapajus cay (yellow-bearded capuchin) is marked by a yellow to white head with black crown and sideburns, which contrast with the light-colored body; the presence of a prominent dark dorsal stripe; limbs mainly dark to blackish, with upper arms not lighter than the body; and a yellowish or reddish underside, often overlaid with black, as described previously [32].
Following these criteria, most samples at this site were recorded as belonging to S. nigritus and S. cay. However, not all species were unambiguously identified by the zookeepers and/or veterinarians and were therefore denoted as Sapajus sp. Details about the species and study sites are shown in Fig. 1.

(A) Study sites in Misiones, Corrientes, and Chaco provinces, Northeastern Argentina. Locations: 1. Paraje Santa Rita, Chaco [26° 01′ 32.46″ S, 59° 58′ 33.49″’ W]: A. caraya; 2. Biological Station of Corrientes, Argentine Museum of Natural Sciences and State Park San Cayetano, Corrientes [27°30′ S, 58°41′ W]: A. caraya; 3. Ecological Reserve “El Puma”, Misiones [27°27′S, 55°48′W]: A. caraya, Sapajus sp., S. nigritus and S. caraya; 4. Iguazú National Park, Misiones [25°40′S, 54°30′W]: S. nigritus. (B) Non-human primates sampled: A. caraya (left), S. cay (center), and S. nigritus (right). Pictures from https://cma.sarem.org.ar/
Sample processing and PV identification
DNA was extracted from feces using a PowerFecal® DNA Isolation Kit (12830-50 MO BIO Laboratories, Inc., Carlsbad, United States) according to the manufacturer’s instructions. DNA from genital and oral swabs was extracted using the ADN PuriPrep-S kit (K1205-250, Inbio HighWay®, Buenos Aires, Argentina). We determined the DNA concentration using a QubitTM fluorometer (Thermo Fisher Scientific, Waltham, USA).
The general strategy for PV DNA identification involved the use of an improved version of the original CUT primer system with a “hanging droplet PCR” amplification strategy, as described previously [33, 34]. The CUT primer system is a cocktail of five degenerate primers (four forward and one reverse) targeting 370 bp of the L1 gene (position 6,052–6,424 of the HPV10 genome). This approach allowed us to detect a broad spectrum of HPVs (mucosal/genital and cutaneous) within all genera [33].
More specifically, a primary 20 μl-reaction mixture was placed in a 0.2-ml tube and covered with one drop of mineral oil. After the addition of a 5-μl sample, the mixture had a final volume of 25 μl, containing 0.4 μM CUT1Fw (5′-TRCCiGAYCCiAATAARTTTG-3′), 0.4 μM CUT1EFw (5′-TRCCiGAYCCiAATAGATTTG-3′), 0.266 μM CUT1BRv (5′-TCiACCATRTCiCCRTCYT-3′), 0.266 μM CUT1CRv (5′-TCiCACATRTCiCCRTCYTG-3′) and 0.266 μM CUT1DRv (5′-TCiSCCATRTCiCCRTCYTG-3′), 200 μM each dNTP, 3.5 mM MgCl2, 1.5x PCR buffer with (NH4)2SO4, and 1 U of Taq DNA polymerase (Thermo Fisher Scientific). Before closing the tube, a 25-μl droplet containing the same reaction mixture (but with 400 μM of each dNTP and 2.5 U of Taq DNA polymerase) was placed in the center of the inside of the reaction tube cap.
Using a thermocycler programmed for block temperature without a heated lid, the mixture was heated for 2 min at 94°C, followed by 20 cycles of a step-down protocol (4 cycles of 30 s at 94°C, 30 s at 52°C, and 40 s at 72°C; 4 cycles of 30 s at 94°C, 30 s at 51°C, and 40 s at 72°C; 4 cycles of 30 s at 94°C, 30 s at 50°C, and 40 s at 72°C; 4 cycles of 30 s at 94°C, 30 s at 49°C, and 40 s at 72°C; and 4 cycles of 30 s at 94°C, 30 s at 48°C, and 40 s at 72°C). After the first round of amplification, we incorporated the “hanging droplet” into the reaction mixture (final volume, 50 μl) by centrifugation for 1 min at 11,000 × g, and a second round of 40 cycles (30 s at 94°C and 2 s at 60°C, followed by a ramp of 0.2 °C/s to 50°C, 50°C for 10 s, and 40 s at 72°C) was performed.
Amplicons derived from the modified CUT primer systems (~ 370 bp) were purified using a spin column (Nucleospin II, Thermo Fisher Scientific) and eluted in 30 μl of elution buffer. All purified amplicons were cloned using a pGEM®-T Easy Cloning Kit (Promega). Clones containing target inserts were identified by PCR amplification using M13 Fw/Rv primers. We sequenced at least three recombinant clones from each sample. All samples were subjected to Sanger sequencing by a commercial service (Macrogen, Inc., Seoul, South Korea). The resulting sequences were compared to available PV sequences in the GenBank database using the BLAST algorithm “Somewhat similar sequences (blastn)”. A new putative PV type was defined when the L1 fragment sequence showed less than 90% sequence identity to any of the previously known PV types [2]. The resulting sequences have been deposited in the GenBank database under accession numbers MT450752 (AcPV1) and MT450753 (SPV1).
Cytochrome B amplification of non-human primate DNA
In order to confirm the NHP origin of the PV-positive samples, we determined the mitochondrial DNA (mtDNA) cytochrome b (CYTB) sequence in each sample. All PCR amplifications were conducted using the following conditions: 75 μl of a mixture containing 10-50 ng of template DNA, 50 mM KCl, 20 mM Tris–HCl (pH 8.4), 1.5 mM MgCl2, 200 μM each dNTP, 1 μM each primer (FW, 5′-CCATCCAACATCTCAGCATGATGAAA-3′; RV, 5′-CCCCTCAGAATGATATTTGTCCTCA-3′) and 2 U of Taq DNA polymerase (Invitrogen, Thermo Fisher Scientific). We included negative (no-template) controls in all amplifications. Ten μl of each PCR product was subjected to electrophoresis in a 2% agarose gel and visualized using SYBR™ Safe DNA Gel Stain (S33102, Invitrogen, Thermo Fisher Scientific) to confirm amplification success. A single band of 359 bp was indicative of a positive amplification of the L1 gene. Positive amplicons were purified using an ADN PuriPrep-GP Kit (K1206-100 Inbio HighWay) and then sequenced by the Sanger method, using the original primers (Macrogen, Inc). The two sequences belonging to the positive PV samples were deposited in the GenBank database [43] under the accession numbers MT451931 (Alouatta caraya) and MT451932 (Sapajus sp).
Phylogenetic analysis
A phylogeny was constructed using published reference sequences of primate PVs for the genera Alpha-, Beta-, and Gammapapillomavirus (n = 266) available at Papillomavirus Episteme [3] and GenBank and those obtained in this study (n = 2). Details of the dataset are provided in Supplementary Table S1. The dataset was aligned using MUSCLE v3.8.31 [35], and the best-fit model of nucleotide substitution was selected by the Bayesian information criterion using the FindModel procedure [36]. A phylogenetic tree was constructed by the maximum-likelihood method using IQTree 1.6.8 for Linux [37], which automatically sets the substitution model according to the results of ModelFinder (in this case, GTR+Γ+I). The branch support was evaluated by ultrafast bootstrapping with 1,000 pseudo-replicates [38]. The tree was visualized and prepared for publication using FigTree V1.4.4 [39].
Molecular dating of gammapapillomaviruses
To estimate the tMRCA (time to the most recent common ancestor) of novel strains within the genus Gammapapillomavirus, we used a dataset of 126 sequences that were 337 nucleotides in length (the L1 CUT PCR fragment). Details about accession numbers, PV types, and host species are provided in Supplementary Table S2. An initial phylogenetic reconstruction was carried out by the maximum-likelihood method and ultrafast bootstrapping as branch support, both implemented in IQTree v 1.6.8 [37]. Coalescence dates were then obtained using the Bayesian Markov chain Monte Carlo (MCMC) method in BEAST v1.10.4 [40]. We used the following priors for this analysis: general time-reversible substitution model with gamma distribution and invariant sites (GTR+G+I); a relaxed (uncorrelated lognormal) molecular clock; a Bayesian skyline plot (BSP) demographic growth [40]; and a substitution rate of 1.84 × 10-8 s/s/y (substitutions per site per year) [8].
The MCMC was run for 5 × 106 generations, sampling every 5,000th generation in order to achieve an effective sample size (ESS) > 200. We analyzed all BEAST run logs using the TRACER program version 1.7 [41] after discarding 2% of the run-length as burn-in. We constructed a maximum-clade-credibility tree (MCCT) with the TreeAnnotator tool after discarding 2% of the sampling [40]. We further visualized the MCCT summarizing the posterior information of topologies and the median branch lengths from the trees sampled with FigTree V1.4.4 [39]. The tMRCA values were expressed in years before present with a high posterior probability density of 95% (HPD 95% range).
Cophylogeny analysis
To evaluate the hypothesis that members of the genus Gammapapillomavirus evolved in association with their host, we used TreeMap v3 [42]. This program evaluates the significance of any congruence between viral-host trees through randomization, using a Markov model to reconstruct random associated trees. The null hypothesis was that the parasite tree is no more congruent with the host tree than a random tree would be [43]. A practical limit to the size of tanglegrams that are manageable by older versions of TreeMap is less than 50 samples, as large numbers may have unreasonable calculation times or memory requirements [43]. Hence, we reduced our data set to a single representative PV sequence per monophyletic lineage occurring in a single host species (n = 30). A mirror primate host phylogeny was constructed using complete mitochondrial genomes of Homo, Pan, Gorilla, Macaca, Alouatta, Ateles, and Cebus, using sequences available in GenBank. Details about the accession numbers and host species are shown in Supplementary Table S2.
A phylogenetic tree was constructed by the maximum-likelihood method using the PhyML platform [44]. Both phylogenies were visualized as a tanglegram and subjected to reconciliation analysis through cophylogeny mapping (25 random maps and 25 generations) [43]. The maximum number of codivergence events (CEs = 28) was later used to run the statistical test with 50 randomized phylogenies. The latter gave us a p-value and 95% confidence interval, with the null hypothesis being rejected if p < 0.05.
Results
NHP population characteristics
We analyzed 64 samples from 57 individuals from the species S. nigritus, S. cay, and A. caraya. Of these individuals, 47.7% were female and 52.3% were male. The characteristics of the NHP study samples are shown in Table 1.
PV detection typing
Of the 64 samples analyzed, 62 were negative and two were positive for PV. One of the positive samples was detected in an oral swab sample of a wild female A. caraya from Corrientes, while the other was found in an oral swab of a captive female Sapajus sp. from Misiones. We provisionally named these partial sequences AcPV1 (strain CUT-Pr145) and SPV1 (strain CUT-Pr035), respectively. The translation of 337 bp (112 amino acids) of the L1 gene confirmed the amplification of papillomavirus L1 gene for both sequences.
Pairwise comparison by the nucleotide BLAST algorithm indicated that AcPV1 shared 71.9% nucleotide sequence identity with HPV126 (species Gammapapillomavirus 11) and 71.6% nucleotide sequence identity with non-human putative PV type MfAA11 (species Gammapapillomavirus 4) identified in Macaca fascicularis. By contrast, the putative lineage SPV1 shared 98.1% nucleotide sequence identity with HPV-msk022 (Gamma-unclassified) and 70.2% nucleotide sequence identity with putative non-human PV type MfAA13 (species Gammapapillomavirus 4) identified in Macaca fascicularis. Thus, BLAST analysis identified these new partial sequences as putative novel types and variants, respectively. Yet, to fully fulfill these taxonomic criteria, complete genomes sequences are needed. No sequences from non-primate hosts were retrieved in the BLAST search.
Phylogenetic analysis
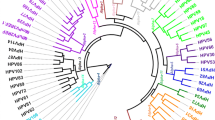
In Fig. 2, we show the phylogenetic relationships between the new putative PV sequences AcPV1 and SPV1 and other known primate alpha, beta-, and gammapapillomaviruses. The tree topology was consistent in revealing three supported monophyletic clusters for the genera Alpha-, Beta- and Gammapapillomavirus. The AcPV1 and SPV1 sequences from Argentina were placed in the cluster of gammapapillomaviruses. Therefore, the phylogenetic analysis confirmed that both PV sequences were members of this genus.

Phylogenetic classification of novel putative PV sequences AcPV1 and SPV1 within the family Papillomaviridae. The evolutionary history of the family Papillomaviridae (genera Alpha-, Beta-, and Gammapapillomavirus) was inferred by the maximum-likelihood method. The analysis involved 266 nucleotide sequences, each 337 bp in length. Viruses from this study belonging to the genus Gammapapillomavirus are indicated by black dots.
In Fig. 3, we show the phylogenetic relationships and molecular dating of the novel putative PV sequences AcPV1 and SPV1 in relation to other known gammapapillomavirus types. In our dataset, all gammapapillomaviruses coalesced to a tMRCA of 51,367,435 years ago (HPD 95% = 33,282,867-72,480,552 years), with the emergence of SPV1 occurring during the last 11,941,682 years (HPD 95% = 6,751,416–18,042,367 years) and AcPV during the last 46,638,071 years (HPD 95% = 26,810,762–61,367,537 years). Since a number of clusters were not strongly supported across the tree, we viewed these dates as provisional.

Phylogenetic analysis and molecular dating of primate gammapapillomaviruses. The evolutionary history was inferred using the Bayesian method. A maximum-clade-credibility tree is shown. The analysis involved 126 nucleotide sequences. The final dataset included a total of 337 positions. The x-axis indicates years ago. The posterior probability values are shown at the nodes of the tree. Novel putative PV types identified in this study are shown in red.
Cophylogeny analysis
A tanglegram is shown in Supplementary Fig. S1. Reconciliation analysis through cophylogeny mapping indicated that we could not reject the null hypothesis that the PV tree was no more congruent with the host tree than a random tree would be, with a P-value of 0.340 (range, 0.212-0.484).
Discussion
In this study, we investigated the occurrence of PV infections in NHPs in northern Argentina and explored the possible scenarios for the evolution and dispersal of these viruses in primate lineages. We identified two putatively novel PV sequences. One was found in the oral mucosa of a wild Alouatta caraya female from Corrientes Province (called AcPV1), and the other was identified in a captive Sapajus sp. female (SPV1) from Misiones Province. Genetic and phylogenetic analysis of these PVs sequences allowed us to assign them to the genus Gammapapillomavirus. Prior to this study, PV infections had been reported in a number of other platyrrhine species, including Saimiri sciureus, Alouatta guariba, Ateles geoffroyi, Callicebus cupreus, and Callithrix penicillata [4, 23, 25,26,27, 45]. Thus, our findings expand the range of described hosts for these viruses.
The diversity of cutaneous gammapapillomaviruses is known to be high. Several hundred partial PCR sequences and more than 100 complete reference genome sequences have been described in the last 10 years (3, 15, 33, 34, 46, 47]. The evolutionary basis for this genetic diversity is presently unclear. It has been suggested that UV-light-induced damage may contribute to a higher mutation rate in the sun-exposed PVs in the skin in the case of beta- and gammapapillomaviruses [46].
Interestingly, the gammapapillomaviruses were initially reported as belonging to the group of cutaneous PVs because they were predominantly isolated from the cutaneous epithelium of human skin [15, 34, 46]. However, studies have shown that the oral cavity contains a broad spectrum of gammapapillomaviruses, which enlarges their proposed tropism [6, 47, 48]. Recently, Chen et al. studied PV infection in different body parts of macaques and found frequencies of 55.6% in genital swabs, 35.9% in oral swabs, and 29.9% in perianal swabs [6]. Moreover, they found a significant difference in the distribution of members of different PV genera at different body sites, with alphapapillomavirus infections being more frequent in genital samples (86.2%) and gammapapillomavirus infections more frequent in the oral cavity (90.3%) [6]. Our identification of AcPV1 and SPV1 in samples of desquamated cells of the oral mucosa is consistent with these findings.
On the other hand, cross-body-site infection has also been reported as being common among macaques [6]. Thus, an alternative explanation for our results is that primate skin-to-mouth contact could be responsible for the transmission of skin PV types to the oral cavity. In the case of NHPs, grooming is a widespread activity that involves looking for and eating parasites in the fur of peers [49]. In this context, the oral mucosa could represent a satellite niche produced by grooming. This is not a minor issue, since tissue tropism has been indicated as one of the main determinants for the evolution of PVs [13, 14, 23]. Thus, future studies addressing the tropism of gammapapillomaviruses will be crucial for understanding virus niche adaptation.
The gammapapillomavirus tree topology indicates the absence of a monophyletic pattern for viruses that infect the same species, with those infecting humans being the most striking example. In support of this observation, our statistical analysis rejected the host/hosted coevolution model. This finding is consistent with previous reports for gammapapillomaviruses [27] and members of other PV genera. Among the alphapapillomaviruses, viruses from papions (PCPV), rhesus (RhPV-1) and colobus (CCPV) are closely related to the human oncogenic types of the species Alphapapillomavirus 9 and therefore do not occupy a basal position in the phylogeny [9,10,11]. Increasing evidence therefore supports the view that the first step in PV evolution was niche adaptation to tissue tropism. The conflicts between the pathogen and host phylogenies may therefore have a reasonable explanation [11, 13, 23].
Regarding the evolutionary history of PVs, molecular dating of the gammapapillomaviruses has estimated their origin to be 51 million years ago (MYA). Previous studies based on complete PV genome sequences have generated dates ranging from 33 MYA to 45 MYA [9, 30, 56] and 50 to 60 MYA [14, 50]. This extent of variation can be attributed to the diversity (the number of species) and length (base pairs) of sequences involved in these different datasets. Nevertheless, all studies agree that the ancestral virus dates back to the Eocene (56-34 MYA).
Current views of primate taxonomy agree that the extant genera originated from a common ancestor during the Cretaceous/Paleocene boundary roughly 80–90 MYA, with the expansion of the major extant lineages Strepsirrhini, Tarsiiformes, and Simiiformes occurring during the Eocene [51]. The importance of these data for our study concerns the Simiiformes group, which comprises Platyrrhini (New World monkeys) and Catarrhini (Old World monkeys and humans) and has an estimated tMRCA of 43 (36–50) MYA [51]. Our molecular dating of the gammapapillomavirus types produces an estimate that falls within the time frame of the evolution of the order primates.
Yet, the emergence of SPV1 during the last 11.9 MYA and of AcaPV at 46.6 MYA is not consistent with the evolutionary history of their primate host species. The biogeographic history of capuchins suggests a late Miocene geographic isolation of the gracile (Cebus) and robust (Sapajus) forms at 6.7 MYA [52]. The divergence time between Alouatta species has also been estimated at 6.6–6.8 MYA [53]. Given this evidence, it is clear that the origin of the PVs occurred before the speciation of their respective hosts.
Finally, it is important to mention the potential role of cross-species transmission in our findings. For example, it is known that bovine deltapapillomavirus infection causes tumors in horses, cape mountain zebras, giraffes, sable antelopes, and buffaloes [54]. Unfortunately, there is relatively little information about humans as sources of PVs in NHPs. Recent studies have revealed that a zookeeper transiently tested positive for a chimpanzee PV [4], while a cat was infected with human HPV9 (possible by a cat owner, who was not tested) [55]. These examples raise the possibility of viral transfer between human and non-human species. In our study, the Sapajus sp. sample came from a captive animal, but we were unable to include the zookeepers’ samples in this study.
The discovery of novel PVs, particularly in hosts in which PV infection had not been reported previously is significant, as it increases our knowledge about PV evolution and diversification. However, one of the limitations of this study is the use of a small DNA fragment for taxonomic, phylogenetic, and molecular dating inferences. Unfortunately, we were unsuccessful in retrieving larger genes from our samples to expand this analysis. For this reason, other approaches such as enrichment of circular DNA by rolling-circle amplification, and/or next-generation sequencing may be needed for the characterization of these novel viruses in the future [56].
Conclusions
This is the first report of PV infection of platyrrhine species from Argentina. It expands the range of the described hosts for these viruses, and is consistent with recent models for PV-primate origin and emergence. We believe that additional analysis will confirm the phylogenetic status of these newly identified PVs and that similar kinds of studies of other NHP species should be conducted to enlarge our understanding of PV infection and evolution.
References
Rector A, Van Ranst M (2013) Animal papillomaviruses. Virology. https://doi.org/10.1016/j.virol.2013.05.007
de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H (2004) Classification of papillomaviruses. Virology. https://doi.org/10.1016/j.virol.2004.03.033
Van Doorslaer K, Li Z, Xirasagar S, Maes P, Kaminsky D, Liou D et al (2017) The papillomavirus episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw879
Antonsson A, Hansson BG (2002) Healthy skin of many animal species harbors papillomaviruses which are closely related to their human counterparts. J Virol. https://doi.org/10.1128/JVI.76.24.12537-12542.2002
Chen Z, van Doorslaer K, DeSalle R, Wood CE, Kaplan JR, Wagner JD et al (2009) Genomic diversity and interspecies host infection of alpha12 Macaca fascicularis papillomaviruses (MfPVs). Virology. https://doi.org/10.1016/j.virol.2009.07.012
Chen Z, Long T, Wong PY, Ho WCS, Burk RD, Chan PKS (2019) Non-human primate papillomaviruses share similar evolutionary histories and niche adaptation as the human counterparts. Front Microbiol. https://doi.org/10.3389/fmicb.2019.02093
Chan SY, Bernard HU, Ratterree M, Birkebak TA, Faras AJ, Ostrow RS (1997) Genomic diversity and evolution of papillomaviruses in rhesus monkeys. J Virol 71(7):4938–4943
Rector A, Lemey P, Tachezy R, Mostmans S, Ghim SJ, Van Doorslaer K et al (2007) Ancient papillomavirus-host co-speciation in Felidae. Genome Biol. https://doi.org/10.1186/gb-2007-8-4-r57
Gottschling M, Stamatakis A, Nindl I, Stockfleth E, Alonso A, Bravo IG (2007) Multiple evolutionary mechanisms drive papillomavirus diversification. Mol Biol Evol. https://doi.org/10.1093/molbev/msm039
Gottschling M, Göker M, Stamatakis A, Bininda-Emonds OR, Nindl I, Bravo IG (2011) Quantifying the phylodynamic forces driving papillomavirus evolution. Mol Biol Evol. https://doi.org/10.1093/molbev/msr030
Shah SD, Doorbar J, Goldstein RA (2010) Analysis of host-parasite incongruence in papillomavirus evolution using importance sampling. Mol Biol Evol. https://doi.org/10.1093/molbev/msq015
García-Pérez R, Ibáñez C, Godínez JM, Aréchiga N, Garin I, Pérez-Suárez G et al (2014) Novel papillomaviruses in free-ranging Iberian bats: no virus-host co-evolution, no strict host specificity, and hints for recombination. Genome Biol Evol. https://doi.org/10.1093/gbe/evt211
Bravo IG, de Sanjosé S, Gottschling M (2010) The clinical importance of understanding the evolution of papillomaviruses. Trends Microbiol. https://doi.org/10.1016/j.tim.2010.07.008
Van Doorslaer K (2013) Evolution of the papillomaviridae. Virology. https://doi.org/10.1016/j.virol.2013.05.012
Bolatti EM, Chouhy D, Casal PE, Pérez GR, Stella EJ, Sanchez A et al (2016) Characterization of novel human papillomavirus types 157, 158 and 205 from healthy skin and recombination analysis in genus γ-Papillomavirus. Infect Genet Evol. https://doi.org/10.1016/j.meegid.2016.04.018
Murahwa AT, Tshabalala M, Williamson AL (2020) Recombination between high-risk human papillomaviruses and non-human primate papillomaviruses: evidence of ancient host switching among alphapapillomaviruses. J Mol Evol. https://doi.org/10.1007/s00239-020-09946-0
Ostrow RS, McGlennen RC, Shaver MK, Kloster BE, Houser D, Faras AJ (1990) A rhesus monkey model for sexual transmission of a papillomavirus isolated from a squamous cell carcinoma. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.87.20.8170
Wood CE, Chen Z, Cline JM, Miller BE, Burk RD (2007) Characterization and experimental transmission of an oncogenic papillomavirus in female macaques. J Virol. https://doi.org/10.1128/JVI.00233-07
Bergin IL, Bell JD, Chen Z, Zochowski MK, Chai D, Schmidt K et al (2013) Novel genital alphapapillomaviruses in baboons (Papio hamadryas anubis) with cervical dysplasia. Vet Pathol. https://doi.org/10.1177/0300985812439725
Van Ranst M, Fuse A, Fiten P, Beuken E, Pfister H, Burk RD et al (1992) Human papillomavirus type 13 and pygmy chimpanzee papillomavirus type 1: comparison of the genome organizations. Virology. https://doi.org/10.1016/0042-6822(92)90896-W
Joh J, Hopper K, Van Doorslaer K, Sundberg JP, Jenson AB, Ghim SJ (2009) Macaca fascicularis papillomavirus type 1: a non-human primate betapapillomavirus causing rapidly progressive hand and foot papillomatosis. J Gen Virol. https://doi.org/10.1099/vir.0.006544-0
Wood CE, Tannehill-Gregg SH, Chen Z, Kv D, Nelson DR, Cline JM et al (2011) Novel betapapillomavirus associated with hand and foot papillomas in a cynomolgus macaque. Vet Pathol. https://doi.org/10.1177/0300985810383875
Chen Z, Wood CE, Abee CR, Burk RD (2018) Complete Genome sequences of three novel Saimiri sciureus papillomavirus types isolated from the cervicovaginal region of squirrel monkeys. Genome Announc. https://doi.org/10.1128/genomeA.01400-17
Long T, Wong PY, Ho WCS, Burk RD, Chan PKS, Chen Z (2018) Complete genome sequences of six novel Macaca mulatta papillomavirus types isolated from genital sites of Rhesus Monkeys in Hong Kong SAR, China. Microbiol Resour Announc. https://doi.org/10.1128/MRA.01414-18
Chen Z, DeSalle R, Schiffman M, Herrero R, Wood CE, Ruiz JC et al (2018) Niche adaptation and viral transmission of human papillomaviruses from archaic hominins to modern humans. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1007352
Silvestre RV, de Souza AJ, Júnior EC, Silva AK, de Mello WA, Nunes MR et al (2016) First new world primate papillomavirus identification in the Atlantic Forest, Brazil: Alouatta guariba papillomavirus 1. Genome Announc. https://doi.org/10.1128/genomeA.00725-16
Köhler A, Gottschling M, Manning K, Lehmann MD, Schulz E, Krüger-Corcoran D et al (2011) Genomic characterization of ten novel cutaneous human papillomaviruses from keratotic lesions of immunosuppressed patients. J Gen Virol. https://doi.org/10.1099/vir.0.030593-0
Zunino GE, Kowalewski MM (2008) Primate research and conservation in northern Argentina: the field station Corrientes (Estación Biológica de Usos Múltiples—EBCo). Trop Conserv Sci. https://doi.org/10.1177/194008290800100206
Kowalewski MM, Salzer JS, Deutsch JC, Raño M, Kuhlenschmidt MS, Gillespie TR (2011) Black and gold howler monkeys (Alouatta caraya) as sentinels of ecosystem health: patterns of zoonotic protozoa infection relative to degree of human-primate contact. Am J Primatol. https://doi.org/10.1002/ajp.20803
International Primatological Society, 2014. Code of best practices for field primatology. https://www.asp.org/resources/docs/Code%20of_Best_Practices%20Oct%202014.pdf. Accessed 10 Jan 2022
Morales MA, Fabbri CM, Zunino GE, Kowalewski MM, Luppo VC, Enría DA et al (2017) Detection of the mosquito-borne flaviviruses, West Nile, Dengue, Saint Louis Encephalitis, Ilheus, Bussuquara, and Yellow Fever in free-ranging black howlers (Alouatta caraya) of Northeastern Argentina. PLoS Negl Trop Dis. https://doi.org/10.1371/journal.pntd.0005351
Nieves M, Remis MI, Sesarini C, Hassel DL, Argüelles CF, Mudry MD (2021) Assessment of genetic variability in captive capuchin monkeys (Primates: Cebidae). Sci Rep. https://doi.org/10.1038/s41598-021-86734-w
Chouhy D, Gorosito M, Sánchez A, Serra EC, Bergero A, Fernandez Bussy R, Giri AA (2010) New generic primer system targeting mucosal/genital and cutaneous human papillomaviruses leads to the characterization of HPV 115, a novel Beta-papillomavirus species 3. Virology. https://doi.org/10.1016/j.virol.2009.11.020
Bolatti EM, Hošnjak L, Chouhy D, Re-Louhau MF, Casal PE, Bottai H et al (2018) High prevalence of Gammapapillomaviruses (Gamma-PVs) in pre-malignant cutaneous lesions of immunocompetent individuals using a new broad-spectrum primer system, and identification of HPV210, a novel Gamma-PV type. Virology. https://doi.org/10.1016/j.virol.2018.09.006
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. https://doi.org/10.1093/nar/gkh340
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. https://doi.org/10.1038/nmeth.4285
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. https://doi.org/10.1093/molbev/msu300
Minh BQ, Nguyen MA, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. https://doi.org/10.1093/molbev/mst024
Rambaut, A. (2010) FigTree v1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 10 Jan 2022.
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A (2018) Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. https://doi.org/10.1093/ve/vey016
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst Biol. https://doi.org/10.1093/sysbio/syy032
Charleston (2011) TreeMap 3, which is freely available at https://sites.google.com/site/cophylogeny/software. Accessed 10 Jan 2022.
Page RD, Charleston MA (1997) From gene to organismal phylogeny: reconciled trees and the gene tree/species tree problem. Mol Phylogenet Evol. https://doi.org/10.1006/mpev.1996.0390
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. https://doi.org/10.1093/sysbio/syq010
D’arc M, Moreira FRR, Dias CA, Souza AR, Seuánez HN, Soares MA, Tavares MCH, Santos AFA (2020) The characterization of two novel neotropical primate papillomaviruses supports the ancient within-species diversity model. Virus Evol. https://doi.org/10.1093/ve/veaa036
Forslund O (2007) Genetic diversity of cutaneous human papillomaviruses. J Gen Virol. https://doi.org/10.1099/vir.0.82911-0
Bolatti EM, Hošnjak L, Chouhy D, Casal PE, Re-Louhau MF, Bottai H et al (2020) Assessing Gammapapillomavirus infections of mucosal epithelia with two broad-spectrum PCR protocols. BMC Infect Dis. https://doi.org/10.1186/s12879-020-4893-3
Bottalico D, Chen Z, Dunne A, Ostoloza J, McKinney S, Sun C et al (2011) The oral cavity contains abundant known and novel human papillomaviruses from the Betapapillomavirus and Gammapapillomavirus genera. J Infect Dis. https://doi.org/10.1093/infdis/jir383
Schino G, Di Giuseppe F, Visalberghi E (2009) Grooming, rank, and agonistic support in tufted capuchin monkeys. Am J Primatol. https://doi.org/10.1002/ajp.20627
Murahwa AT, Nindo F, Onywera H, Meiring TL, Martin DP, Williamson AL (2019) Evolutionary dynamics of ten novel Gamma-PVs: insights from phylogenetic incongruence, recombination and phylodynamic analyses. BMC Genom. https://doi.org/10.1186/s12864-019-5735-9
Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MA et al (2011) A molecular phylogeny of living primates. PLoS Genet. https://doi.org/10.1371/journal.pgen.1001342
Lynch Alfaro JW, Boubli JP, Olson LE, Di Fiore A, Wilson B, Gutierrez-Espeleta GA et al (2011) Explosive pleistocene range expansion leads to widespread Amazonian sympatry between robust and gracile capuchin monkeys. J Biogeogr. https://doi.org/10.1111/j.1365-2699.2011.02609.x
Cortés-Ortiz L, Bermingham E, Rico C, Rodríguez-Luna E, Sampaio I, Ruiz-García M (2003) Molecular systematics and biogeography of the Neotropical monkey genus, Alouatta. Mol Phylogenet Evol. https://doi.org/10.1016/S1055-7903(02)00308-1
Williams JH, van Dyk E, Nel PJ, Lane E, Van Wilpe E, Bengis RG et al (2011) Pathology and immunohistochemistry of papillomavirus-associated cutaneous lesions in Cape mountain zebra, giraffe, sable antelope and African buffalo in South Africa. J S Afr Vet Assoc 82(3):185
Munday JS, Hanlon EM, Howe L, Squires RA, French AF (2007) Feline cutaneous viral papilloma associated with human papillomavirus type 9. Vet Pathol. https://doi.org/10.1354/vp.44-6-924
Arroyo LS, Smelov V, Bzhalava D, Eklund C, Hultin E, Dillner J (2013) Next generation sequencing for human papillomavirus genotyping. J Clin Virol. https://doi.org/10.1016/j.jcv.2013.07.013
Acknowledgements
We thank Maria Patricia Casco and Ester Bernaldo de Quirós for their help with sample collection during fieldwork. We are also thankful to María Elina Totaro for her technical assistance in the laboratory.
Funding
This work was supported through a doctoral fellowship from the Consejo Nacional de Investigaciones Científico y Tecnológicas (CONICET) (10320130101208CO) to CSF. IB, MMK, EB, DC, and AAG are members of CONICET. This study was partially supported by research Grant PIP IU 0355 CONICET (MMK). The equipment and software used in the study were partially supported by an Idea Wilds grant (CSF) (501c(3)) and a Codon Code Aligner Grants license program (IB). None of the funding agencies have been involved in the study design, data collection, analysis, or paper writing and submission.
Author information
Authors and Affiliations
Contributions
CS-F: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, writing—review and editing, visualization, funding acquisition. EMB: methodology, validation, formal analysis, investigation, data curation, writing—review and editing. ACAC: methodology, formal analysis, visualization, writing—review and editing. DC: methodology, formal analysis, writing—review and editing. MMK: conceptualization, resources, supervision, project administration, writing—review and editing. EJS: investigation, writing—review and editing. TGS: formal analysis, resources, writing—review and editing. MAR, DJL, and RHC: resources, writing—review and editing. AAG: resources; project administration, funding acquisition, writing—review and editing. IB: conceptualization, methodology, formal analysis, resources, data curation, writing—review and editing, supervision, project administration, funding acquisition
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Handling Editor: Graciela Andrei.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sanchez-Fernandez, C., Bolatti, E.M., Culasso, A.C.A. et al. Identification and evolutionary analysis of papillomavirus sequences in New World monkeys (genera Sapajus and Alouatta) from Argentina. Arch Virol 167, 1257–1268 (2022). https://doi.org/10.1007/s00705-022-05420-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05420-y