
Abstract
We use all the currently known 405 Papillomavirus (PV) sequences, 343 curated PV sequences from both humans and animals from the PAVE data base, to analyse the recombination dynamics of these viruses at the whole genome levels. After showing some evidence of human and non-human primate PV recombination, we report a comprehensive recombination analysis of all currently known 82 Alphapapillomaviruses (Alpha-PVs). We carried out an exploratory study and found novel recombination events between High-Risk HPV Types and Macaca fascicularis PV1 (MfPV1), Macaca Fuscata PV2 (MfuPV2) and Pan Paniscus PV1 (PpPV1) Papillomaviruses. This is the first evidence of interactions between PVs from different hosts and hence postulates the likelihood of ancient host switching among Alpha-PVs. Notwithstanding these results should be interpreted with caution because the major and minor parents indicated by RDP4 program are simply the sequences in the alignment that most closely resemble the actual parents. We found statistically significant differences between the phylogenies of the PV sequences with recombination regions and PV sequences without recombination regions using the Shimodaira–Hasegawa phylogenetic incongruence testing. We show that not more than 76MYA Alpha-PVs were in the same biological niche, a pre-requisite for recombination, and as the hosts evolved and diversified, the viruses adapted to specific host niches which eventually led to coevolution with specific hosts before speciation of primate species. Thus providing evidence that in ancient times no earlier than the Cretaceous period of the Mesozoic age, Alpha-PVs recombined and switched hosts, but whether this host switching is occurring currently is unknown. However, a clearer picture of the PVs evolutionary landscape can only be achieved with the incremental discovery of PV sequences, especially from the animal kingdom.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Papillomaviridae (PV) family was once part of the larger family of Papovaviridae which was split into Polyomaviridae and Papillomaviridae by the International Committee on Taxonomy of Viruses (Van Regenmortel 2000). According to the most recent ICTV classification, the PV family includes two subfamilies Firstpapillomavirinae with 52 genera and Secondpapillomavirinae with one genus and one species (Van Doorslaer et al. 2018). Genera are named according to the Greek alphabet from alpha to omega, and following exhaustion of the alphabet the term dyo and treis (Greek for second time and third, respectively) coined to accommodate the extra genera e.g. dyo-deltapapillomaviruses (Bernard et al. 2010). There are about 405 known reference PVs uptodate as listed in the curated PAVE database (https://pave.niaid.nih.gov/), 226 of these are human papillomaviruses (HPVs) and 179 from different animal species (Van Doorslaer and Dillner 2019). HPVs are the most studied and are distributed over 5 genera (Alpha, Beta, Gamma, Mu and Nu). The other PV genera are from other mammals, birds and reptiles (Bernard et al. 2010). Below the genus level are species and below the species level are PV types (de Villiers et al. 2004). Different genera have less than 60% similarity within the L1 gene, while species share between 60 and 70% similarity and types share between 71 and 89% similarity. The ICTV is responsible for nomenclature of viruses down to species level, and below species level, the International Human Papillomavirus (HPV) Reference Centre (Karonliska Institute, Sweden) and the Animal Papillomavirus Reference Centre (University of Arizona, USA) assigns unique PV-type numbers after the complete genome has been sequenced, cloned and confirmed by the Centre (Mühr et al. 2018).
Alphapapillomaviruses (Alpha-PVs) genus is the most widely studied of all the PV genera. According to the PAVE (https://pave.niaid.nih.gov/) database, there are currently 82 Alpha-PV types classified into 14 species (Alpha-1 to Alpha-14). Of the 82 known types, 17 are animal Alpha-PVs but only from non-human primates and 65 from humans.
Traditionally, PVs have been thought to evolve slowly because they replicate by co-opting high-fidelity host cellular DNA polymerases that have an error rate of about 4.3 × 10− 5 substitutions per nucleotide site per year (Korona et al. 2011). The general assumption is that PVs have co-evolved with their hosts (Chen et al. 2009a; Bravo and Felez-Sanchez 2015; Dube Mandishora et al. 2018). Selection pressures due to host immune responses differ among PV genes and result in these genes displaying different substitution rates. Further, the cellular polymerases of different host species may differ in their degree of fidelity such that virus lineages infecting different hosts might display different substitution rates (de Oliveira et al. 2015). The popular views about PV evolution have been (1) they are static and slow evolving viruses with very low mutation rates (2) host linked co-divergence (3) strict tissue tropism (4) host specific and (5) lack of recombination (Bravo et al. 2010). However, the understanding of PV evolution has advanced over the years, with various alternative mechanisms such as inter-species transmission, adaptive radiation and recombination (Angulo and Carvajal-Rodriguez 2007; Carvajal-Rodriguez 2008; Gottschling et al. 2011; Robles-Sikisaka et al. 2012).
The strict definition of recombination incorporates reciprocation, meaning the recipient of a genome portion acts as a donor of the replaced portion in the source, which is not the case with PV recombination (Pérez-Losada et al. 2015). PV recombination could be more appropriately named, gene conversion, but it has been so widely used that changing it would introduce confusion to the whole subject. Recombination has a potential major impact on PV evolution, pharmacogenomics and vaccine development. In other viruses, recombination has been associated with emergence of novel viruses, increase in virulence and pathogenesis, changes in tissue tropisms and expansion of viral host ranges (Martin et al. 2011; Simon-Loriere and Holmes 2011).
The biological plausibility of PV recombination is occasioned by the genetic multiplicity of PVs and the high frequencies of observed HPV co-infections (Angulo and Carvajal-Rodriguez 2007). However, the study of PV recombination has been hampered by technical difficulties associated with the accurate alignment of highly diverse PV gene sequences (Posada and Crandall 2001). One of the most commonly used approaches to recombination detection is the use of the various recombination analysis tools implemented within the RDP4 software package (Martin and Rybicki 2000). During recombination detection, RDP4 rigorously tests the quality of sequence alignments to guard against the detection of false-positive recombination signals that arise due to sequence misalignment (Varsani et al. 2006).
We report here the use of all 405 known PV sequences, 343 curated PV sequences from both humans and animals, to analyse the recombination dynamics of these viruses at the whole genome levels. Specifically, we use these sequences to identify recombination and to determine whether there is intra-genus, inter-species and inter-host species recombination. After stumbling upon evidence of human and non-human primate PV recombination, we report also a comprehensive recombination analysis of all currently known 82 Alpha-PVs. Consequently, we carried out the final test for recombination, phylogenetic incongruence, to prove a statistically significant difference between the phylogenies of the PV sequences used.
Methods
Design
This was an exploratory study to investigate the likelihood of ancient host-switching events among PVs as previously postulated in literature (Chen et al. 2018, 2019).
Source of Sequence Data
All the 405 currently known PV whole-genome reference sequences from the PAVE database were downloaded in Fasta format (https://pave.niaid.nih.gov/#search/search_database). See also Supplementary Material 1a for the fasta format of the sequences. Additionally, Fasta format of the concatenated E1-E2-L1-L2 alignment of 343 sequences was downloaded from the ICTV website (https://talk.ictvonline.org/ictv-reports/ictv_online_report/dsdna-viruses/w/papillomaviridae/1214/papillomaviridae---v201911), (see also Supplementary Material 1b for the fasta alignment of the sequences) and all the 82 currently known Alpha-PVs whole-genome sequences were obtained from PAVE database see also Supplementary Material 1c for the fasta format of the sequences.
Recombination Analysis
This was a hierarchical approach in which we started with the whole genomes of all the currently known PVs (405)—R1, then we used a set of all curated genomes 343 from the ICTV database where the alignment was actually downloaded as mentioned in Table 1, R2, with concatenated gene regions in order to improve the alignment. Then, lastly, we use all the 82 known Alpha-PVs, R3, based on the fact that R1 and R2 had shown recombination among Alpha-PVs.
R1: Recombination Analysis 1
All the 405 currently known whole-genome PV reference sequences from the PAVE database were included in the initial recombination analysis (R1). We then constructed an alignment of the 405 PV sequences using MUSCLE and the CLUSTALW output was used for the R1 recombination analysis. This alignment was analysed using RDP v4.95 (Martin and Rybicki 2000) (with default settings) which implements analysis of recombination using a suite of 7 recombination detection methods or algorithms: RDP (Martin and Rybicki 2000), BOOTSCAN (Martin et al. 2005a, b), CHIMAERA (Martin et al. 2005a, b), GENECONV (Padidam et al. 1999), MAXIMUM X2 (Smith 1992) and SISCAN (Gibbs et al. 2000). Only recombination events that were identified by at least four methods and no showed warning flags (messages generated by the software to warn the reader of the possible reasons for each outcome, and to interpret them with caution) in the RDP software were considered.
R2: Recombination Analysis 2
FASTA format of the concatenated E1-E2-L1-L2 alignment of the 343 sequences (downloaded from ICTV, https://talk.ictvonline.org/ictv-reports/ictv_online_report/dsdna-viruses/w/papillomaviridae/1214/papillomaviridae---v201911) were used in the recombination analysis R2. This alignment was also analysed using RDP v4.95 (Martin and Rybicki 2000) (with default settings) as was done for R1 above, and only recombination events that were identified by at least four methods and no showed warning flags in the RDP software were considered.
R3: Recombination Analysis 3
Eighty-two (82) Alpha-PVs complete genomes (65 Alpha-HPVs and 17 non-human primate Alpha-PVs), which are of all the currently known Alpha-PVs, were obtained from the PAVE database. We then constructed an alignment containing the 82 Alpha-PVs using MUSCLE, the CLUSTALW output was used for the R3 recombination analysis as a follow-up investigation to findings from R1 and R2. This alignment was also analysed using RDP v4.95 (Martin and Rybicki 2000) (with default settings) as was done for R1 and R2 above, and only recombination events that were identified by at least four methods and no showed warning flags in the RDP software were considered.
Phylogenetic Incongruence Testing
The Shimodaira–Hasegawa (SH) test (Shimodaira and Hasegawa 1999) using W-IQ-TREE (Trifinopoulos et al. 2016). We used CLUSTALW alignments (Sievers et al. 2011; McWilliam et al. 2013; Li et al. 2015) of all the 405 currently known PV whole-genome reference sequences, 343 curated E1-E2-L1-L2 concatenated sequences and 82 currently known Alpha-PVs complete genomes to compute the log-likelihoods of phylogenetic trees in W-IQ-TREE (https://iqtree.cibiv.univie.ac.at) (Trifinopoulos et al. 2016). The tool tests tree topology, estimates model parameters such as substitution rates and optimizes tree branch lengths to lessen computational usage. We used default settings of the W-IQ-Tree, including best fit model (Kalyaanamoorthy et al. 2017) and ultra-fast bootstrap analysis (1000 alignments) (Minh et al. 2013) to run tree topology analysis including the Kishino–Hasegawa (KH) test (Kishino and Hasegawa 1989), Shimodaira–Hasegawa (SH) test (Shimodaira and Hasegawa 1999) and approximately unbiased (AU) test (Shimodaira 2002) to test if there is a difference in evolutionary patterns among trees generated after removing the recombinant regions from the original sequences.
All the trees performed were denoted A1 and A2, A1 representing the trees generated from the original sequence alignments and A2 denoting the trees generated after removing the recombinant regions from the original sequences, after the recombination analyses. Alignments without recombinant regions were generated automatically from RDP v4.95 (Martin and Rybicki 2000) after each of the recombination analyses.
Phylogenetic Analysis of 82 Known Alpha-PV Genomes
The sequences of 82 human and animal Alpha-PVs whole genomes (Alphapapillomavirus genus) were aligned using MUSCLE v7.221 (Edgar 2004). A maximum-likelihood tree of the nucleotide sequences was generated in PhyML using the optimal model of evolution (GTR+G) as determined within MEGA 7 (Kumar et al. 2016). The Newick format of the tree was uploaded and modified in iTOL https://itol.embl.de/tree/.
Results
Recombination Analysis 1 (R1)
There were a total of 393 recombination events but only 4 were sufficiently supported by at least 4 algorithms as indicated in the priori criterion set in the methodology and no software flags, see Table 2 below for a summary of the 4 events, showing parent and recombinant PV types, the number of methods/algorithms to support the event and positions of the breaking points were the gene conversions occurred the same is true for Tables 3 and 4 for R2 and R3, respectively. (See also Supplementary Fig. 2).
Event 1 is between cetacean PVs and involve TtPV1 of the Upsilonpapillomavirus genus as a major parent to recombinants TtPV5 and TtPV6 of the Ominkronpapillomvirus genus. This becomes the first evidence of inter-genus recombination, in this case between Upsilon- and Omikronpapillomaviruses. Event 3 is between low-risk human Alpha-PVs. Event 4 is between an Alpha-HPV 68 of the alpha-7 species as a recombinant and a non-human primate PV PpPV1 (Pan Paniscus PV1 from pygmy chimpanzees commonly known as Bonobos, alpha-10 species) as the major parent. This becomes the first evidence of inter-host PV recombination, yet still inter-genus. All the events were sufficiently supported by at least 5 methods. Event 4 prompted further investigation into other recombination events that can occur across PVs that infect different host species. A well-curated set of 343 sequences was thus used to explore this possibility.
Recombination Analysis 2 (R2)
Recombination signals were detected across both the early regions (E genes) and the late regions (L genes) of the concatenated alignment of the PV genomes. There were a total of 456 events detected by the software but only 7 events were sufficiently supported by at least 4 recombination detection algorithms. See Table 3 below for a summary of the 7 events and Supplementary Fig. 3.
Events 1, 2 and 4 are between cetacean PVs. DdPV1 (Delphinus delphis PV1) from the common short-nose dolphin belongs to the Upsilonpapillomavirus-1 species together with TtPV1 (Tursiops truncatus PV1) from the common bottlenose dolphin. TtPV5, TtPV6, PphPV1 (Phocoena phocoena PV1; harbour porpoise) and PsPV1 (Phocoena spinipinnis PV1; Burmeister porpoise) all belong to the Omikronpapillomavirus-1 species. It is apparent from events 1, 2 and 4 that TtPV1 is a minor parent in all the recombination events. Events 1 and 2 are well supported by 7 recombination algorithms while event 4 is supported by 4 algorithms. Event 4 is also an exception as it spans from the late and a small portion of the early region.
Events 3, 5 and 6 are between HPV types. HPV54 a low-risk HPV type is a major parent in all the events, and only in event 6 it is a parent to HPV82 which is classified as a high-risk HPV type. All the HPV recombination events are intra-genus.
Event 7 is between HPV70 as the recombinant and a non-human primate, MfPV7 (Macaca Fascicularis PV7, commonly known as cynomolgus macaque) as the major parent. MfPV7 is from the alpha-12 species while HPV70 is of the alpha-7 species. Event 7 also spans about 692 bp in the late region, about half the size of either the L1 or L2. Event 4 of R1 and event 7 of R2 all pointed to the fact that recombination occurred only among Alpha-PVs, this prompted a third analysis R3, of all currently known Alpha-PVs.
Recombination Analysis 3 (R3)
There were a total of 117 events detected by the software but only 10 events were sufficiently supported by at least 4 recombination detection algorithms. See Table 4 below for a summary of the 10 events and Supplementary Fig. 4.
Events 1, 3, 4, 5, 6, 7 and 10 are solely between HPV types, with events 3, 4, 6, 7 and 10 involving high-risk HPV (HR-HPV) types; HPV82, HPV51, HPV45, HPV53 and HPV59 as recombinants, respectively. Events 2, 8 and 9 involve recombination between HPVs and non-human primate Alpha-PVs. In event 2 and 8, h-HPV types HPV39 and HPV68 of the alpha-7 species are recombinants of one major parent PpPV1 (Pan Paniscus PV1, alpha-10 species). In event 9, h-HPV 66 of the alpha-6 species is a recombinant of LR-HPV54 of the alpha-13 species as a major parent and minor parent MfuPV2 (Macaca Fuscata PV2, from Japanese macaques) of the alpha-12 species. All the recombination events are intra-genus but mostly cross-species. All the events but event 8 were supported by at least 5 algorithms in the RDP v4.95 software (Martin and Rybicki 2000).
Generally, based on the analysis from R1, R2 and R3 there are 21 recombination events and of these 16 are among high-risk Alpha-PVs, 5 events are between HPVs and non-primate PVs and 5 among cetacean PVs. Event 3 in R1 is the same as event 5 in R2 (save that the minor parent is HPV71), also the same as event 1 in R3. This common recombination event shows HPV30 as the recombinant and HPV54 as the major parent. HR-HPV54 is also a major parent in 3 other recombination events, event 3 and 6 in R2 and event 9 in R3. Thus, HPV54 is a major parent in a total of six recombination events in all the three analyses. HPV82 is a recombinant in 2 events (event 6 in R2 and event 3 in R3) and HPV44 is a major parent in 2 events (event 7 and event 10 in R3).
Phylogenetic Incongruence Testing
To determine if the phylogenetic trees for different Gamma-PV genes were congruent, we used a more conclusive test, the SH test (Shimodaira and Hasegawa 1999). The null hypothesis of the SH test states that the difference between trees (branch length, topology or likelihoods) is zero. The observed differences for the 405A1 and A2, 343A1 and A2 trees (deltaL values) are significantly greater than zero and the null hypothesis was rejected, thus declaring that these trees are significantly different i.e. incongruent (p < 0.05). The deltaL values of Alpha A1 and A2 trees were < 30 and hence not significantly greater than zero and we thus failed to reject the null hypothesis, thus declaring the Alpha A1 and A2 trees to be similar. The null hypothesis of the SH test states that the difference between trees (branch length, topology or likelihoods) is zero. The observed differences (deltaL values) were significantly greater than zero and rejected the null hypothesis and declare that the trees are significantly different i.e. incongruent (p < 0.05). Table 5 shows the results of the SH test using W-IQ-Tree, indicating that there is substantial phylogenetic incongruence between the 405A1 and A2, 343A1 and A2 trees but not for the Alpha A1 and A2 trees as shown by the p-values (p-SH).
Phylogenetic Analysis of 82 Known Alpha-PV Genomes
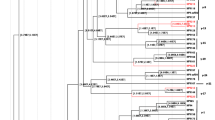
After demonstrating five recombination events between human and non-human primate Alpha-PVs, we sought to show how the 82 Alpha-PV sequences from both humans and animals cluster together by constructing a phylogenetic tree as described below. Members of the alpha-12 species (non-human primate PVs) are closely related to the members of the alpha-9 species (High-risk HPVs) and alpha-11 species as also shown in Fig. 1. MmPV2 and MmPV6 (Macaca mulatta PVs from the Rhesus macaque monkeys) cluster with HPV54 of the alpha-13 species, while MfuPV2 and MmPV3 cluster with the alpha-2 HPVs. CgPV1 from the old world monkey, Colobus guereza, cluster with alpha-14 HPVs.

Alphapapillomavirus genus. Phylogenetic analysis of all the 82 human and animal Alpha-PVs whole genomes. The sequences were aligned using MUSCLE v7.221 (Edgar 2004). A maximum-likelihood tree of the nucleotide sequences was generated in PhyML using the optimal model of evolution (GTR+G) as determined within MEGA 7 (Kumar et al. 2016). The Newick format of the tree was uploaded and modified in iTOL https://itol.embl.de/tree/
Discussion
Ancient Recombination Events to Explain Ancient Host Switching
We report five novel recombination events between HPVs and non-human primate PVs using 3 different sequence sets. These recombination events were between High-Risk HPV Types and Macaca fascicularis PV1 (MfPV1), Macaca Fuscata PV2 (MfuPV2) and Pan Paniscus PV1 (PpPV1) PVs. This observation provides the first evidence of interactions between PVs from different hosts and the likelihood of ancient host switching among Alpha-PVs, thus refuting, host specificity, a central dogma in PV evolution. Chen et al 2009b also postulated that an overlapping set of MfPVs in rhesus and cynomolgus macaques indicates non-human primate Alpha-PVs may not strictly be host specific (Chen et al. 2009b). The same authors also report that members of the alpha-12 species (non-human primate PVs) are closely related to the members of the alpha-9 species (High-risk HPVs) and alpha-11 species as also shown in Fig. 1.
The most important biological prerequisite for recombination to occur is that two virus sequences be in the same space at the same time. So ancestral non-human primate PVs and HPVs should have been in the same space (biological niche) and time if recombination events had to occur as shown above. This unlikely event of cross-host infection by PVs challenges one of PVs central evolutionary tenets. Our findings provide the first likely evidence of such, but we are not the first authors to hypothesize this. Chen et al 2019 showed that non-human primate PVs share similar evolutionary histories and niche adaptation as the human counterparts (Chen et al. 2019), using the example of HPV16 and MfPV3 that evolved from a most recent common ancestor containing the determinants of carcinogenicity. In another study, Chen et al 2018 also reported on the possibility of niche adaptation and viral transmission of HPVs from archaic hominins to modern humans, the archaic hominin-host-switch model (Chen et al. 2018).
It is important in the mathematical modelling of recombination to estimate the time when this recombination occurred. We predict, based on the time tree (https://www.timetree.org/about) that the recombination between High-Risk HPV Types, Cynomolgus & Japanese Macaques and Bonobos PVs may have occurred not more than 76 million years ago (Smelov et al. 2018), at the time of the last common ancestor of all primates. Chen et al. demonstrated specific host niche adaptation of primate PVs ensued thereafter by host co-divergence to have occurred at least 40MYA (Chen et al. 2018, 2019), which concurs with our prediction. Our observation suggests that more than 76MYA these viruses were in the same biological niche (same host). As the hosts evolved and diversified, the viruses adapted to specific host niches, which eventually led to coevolution with specific hosts, and this occurred before the speciation events of all primate host species including humans. Recombination events among Cetacean PVs detected in this study have also been previously reported elsewhere (Robles-Sikisaka et al. 2012), but were not the mainstay of this study.
Mechanisms of Recombination
The biological plausibility of Alpha-PVs recombination events is occasioned by the fact that recombination occurs only during viral replication (Pérez-Losada et al. 2015). Additionally, the high replication rate of Alpha-PVs as seen from their oncogenicity and prevalence in mucosal sites of both humans and non-human primates promote recombination events (Chen et al. 2009b). Multiple infections with HR-HPV types have been reported extensively in several studies (Vinodhini et al. 2012; Mbulawa et al. 2013; Murahwa et al. 2015; Teixeira et al. 2018). These infections create a conducive environment for recombination to occur within the same biological niche or anatomical site through availing or enhancing DNA viral load, which in turn increases the probability of genomes to recombine. Multiple infections have also been reported in Betapapillomaviruses (Murahwa et al. 2015) and Gammapapillomaviruses (Meiring et al. 2017) cutaneous infections, but there are limited data on recombination in these genera.
Phylogenetic Congruence/Incongruence
A1 trees generated from the original sequence alignments and A2 trees generated after removing the recombinant regions from the original sequences were incongruent for the 405 PV sequences and the 343 concatenated sequences (i.e. the trees showed different phylogenies), hence supporting recombination as an essential driving force in PV evolution. However, the Alpha-PVs A1 and A2 trees showed congruence, seemingly rendering recombination an unimportant driving force in their evolution. The congruence can be explained by the low number (82) of Alpha-PV sequences currently available for the analysis to make inferential conclusions. Thus, the low number of sequences imply a lack of power for detection of phylogenetic incongruence. Moreover, the fact that essentially 7 sequences (PpPV1, MfPV7, HPV54, HPV70, HPV68, HPV39 and HPV66) of the 82 Alpha-PVs showed recombination events explains how these could not have changed the phylogeny of the whole Alpha-PV genus, with or without recombination regions from these 7 sequences. Thus, recombination plays a role in their evolution as shown in the dispersion of non-human-primate PVs across the Alpha-PV phylogenetic tree in Fig. 1.
Caveats and Limitations to Understanding PV Evolution
Our current knowledge of PVs is limited and focused on a few medically important and closely related human PVs associated with anogenital cancers and warts (Bravo and Felez-Sanchez 2015), while the rest of the other plethora of PVs biology is largely understudied and unknown. Hence, assumptions made from studying these cannot be necessarily generalized and applied to all PVs.
Many PV sequences are still being discovered, and until a threshold number of representative sequences are attained, the PV community of researchers will remain underpowered to make assumptions closest to the reality of what happened in the evolution of this group of viruses. The addition of more PV sequences has a bearing on the understanding of the origin, evolution and clinical outcome prediction of given PV genomes. A recent discovery of the first fish PV, Sparus aurata PV1 from the gilthead bream fish showed the smallest PV genome consisting only of E1-E2-L1-L2 backbone (Lopez-Bueno et al. 2016). It is prudent to hypothesize at this juncture that the PV ancestor is of marine origin unless more sequences become available for further analysis (Puustusmaa et al. 2017). More fish PV sequences are needed to elucidate our understanding of PV evolution.
Conclusions
Recombination, without doubt, constitutes an important driving force in Alpha-PVs evolution. We show that not more than 76MYA Alpha-PVs were in the same biological niche, a pre-requisite for recombination, and as the hosts evolved and diversified, the viruses adapted to specific host niches which eventually led to coevolution with specific hosts. Thus providing evidence that in ancient times no earlier than the Cretaceous period of the Mesozoic age, Alpha-PVs recombined and switched hosts, but whether this host switching is occurring currently is unknown. It is important to fully understand the evolutionary history of different PVs to better inform carcinogenicity and novel vaccine development.
References
Angulo M, Carvajal-Rodriguez A (2007) Evidence of recombination within human alpha-papillomavirus. Virol J 4:33
Bernard HU, Burk RD, Chen Z, van Doorslaer K, Zur Hausen H, de Villiers EM (2010) Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401(1):70–79
Bravo IG, de Sanjose S, Gottschling M (2010) The clinical importance of understanding the evolution of papillomaviruses. Trends Microbiol 18(10):432–438
Bravo IG, Felez-Sanchez M (2015) Papillomaviruses: viral evolution, cancer and evolutionary medicine. Evol Med Public Health 2015(1):32–51
Carvajal-Rodriguez A (2008) Detecting recombination and diversifying selection in human alpha-papillomavirus. Infect Genet Evol 8(5):689–692
Chen Z, DeSalle R, Schiffman M, Herrero R, Burk RD (2009a) Evolutionary dynamics of variant genomes of human papillomavirus types 18, 45, and 97. J Virol 83(3):1443–1455
Chen Z, van Doorslaer K, DeSalle R, Wood CE, Kaplan JR, Wagner JD, Burk RD (2009b) Genomic diversity and interspecies host infection of alpha12 Macaca fascicularis papillomaviruses (MfPVs). Virology 393(2):304–310
Chen Z, DeSalle R, Schiffman M, Herrero R, Wood CE, Ruiz JC, Clifford GM, Chan PKS, Burk RD (2018) Niche adaptation and viral transmission of human papillomaviruses from archaic hominins to modern humans. PLoS Pathog 14(11):e1007352
Chen Z, Long T, Wong PY, Ho WCS, Burk RD, Chan PKS (2019) Non-human primate papillomaviruses share similar evolutionary histories and niche adaptation as the human counterparts. Front Microbiol 10:2093
de Oliveira CM, Bravo IG, Santiago e Souza NC, Genta ML, Fregnani JH, Tacla M, Carvalho JP, Longatto-Filho A, Levi JE (2015) High-level of viral genomic diversity in cervical cancers: a Brazilian study on human papillomavirus type 16. Infect Genet Evol 34:44–51
de Villiers EM, Fauquet C, Broker TR, Bernard HU, Zur Hausen H (2004) Classification of papillomaviruses. Virology 324(1):17–27
Dube Mandishora RS, Gjotterud KS, Lagstrom S, Stray-Pedersen B, Duri K, Chin'ombe N, Nygard M, Christiansen IK, Ambur OH, Chirenje MZ, Rounge TB (2018) Intra-host sequence variability in human papillomavirus. Papillomavirus Res 5:180–191
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Gibbs MJ, Armstrong JS, Gibbs AJ (2000) Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16(7):573–582
Gottschling M, Goker M, Stamatakis A, Bininda-Emonds OR, Nindl I, Bravo IG (2011) Quantifying the phylodynamic forces driving papillomavirus evolution. Mol Biol Evol 28(7):2101–2113
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14(6):587–589
Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol 29(2):170–179
Kishino H, Miyata T, Hasegawa M (1990) Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J Mol Evol 31(2):151–160
Korona DA, Lecompte KG, Pursell ZF (2011) The high fidelity and unique error signature of human DNA polymerase epsilon. Nucleic Acids Res 39(5):1763–1773
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43(W1):W580–584
Lopez-Bueno A, Mavian C, Labella AM, Castro D, Borrego JJ, Alcami A, Alejo A (2016) Concurrence of iridovirus, polyomavirus, and a unique member of a new group of fish papillomaviruses in lymphocystis disease-affected Gilthead Sea Bream. J Virol 90(19):8768–8779
Martin D, Rybicki E (2000) RDP: detection of recombination amongst aligned sequences. Bioinformatics 16(6):562–563
Martin DP, Biagini P, Lefeuvre P, Golden M, Roumagnac P, Varsani A (2011) Recombination in eukaryotic single stranded DNA viruses. Viruses 3(9):1699–1738
Martin DP, Posada D, Crandall KA, Williamson C (2005a) A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses 21(1):98–102
Martin DP, Williamson C, Posada D (2005b) RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 21(2):260–262
Mbulawa ZZ, Johnson LF, Marais DJ, Coetzee D, Williamson AL (2013) The impact of human immunodeficiency virus on human papillomavirus transmission in heterosexually active couples. J Infect 67(1):51–58. https://doi.org/10.1016/j.jinf.2013.1003.1009
McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R (2013) Analysis tool web services from the EMBL-EBI. Nucleic Acids Res 41(Web Server issue):W597–600
Meiring TL, Mbulawa ZZA, Lesosky M, Coetzee D, Williamson AL (2017) High diversity of alpha, beta and gamma human papillomaviruses in genital samples from HIV-negative and HIV-positive heterosexual South African men. Papillomavirus Res 3:160–167
Minh BQ, Nguyen MAT, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30(5):1188–1195
Mühr LSA, Eklund C, Dillner J (2018) Towards quality and order in human papillomavirus research. Virology 519:74–76
Murahwa AT, Muchemwa FC, Duri K, Kanyera RB, Tshabalala M, Manhanzva MT, Mapingure MP, Stray-Pedersen B (2015) Frequency of betapapillomavirus infections among HIV infected and uninfected Black Zimbabweans with cutaneous lesions. J Med Virol 87(3):478–484
Padidam M, Sawyer S, Fauquet CM (1999) Possible emergence of new geminiviruses by frequent recombination. Virology 265(2):218–225
Pérez-Losada M, Arenas M, Galán JC, Palero F, González-Candelas F (2015) Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infect Genet Evol 30:296–307
Posada D, Crandall KA (2001) Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci 98(24):13757–13762
Puustusmaa M, Kirsip H, Gaston K, Abroi A (2017) The enigmatic origin of papillomavirus protein domains. Viruses 9(9):240
Robles-Sikisaka R, Rivera R, Nollens HH, St Leger J, Durden WN, Stolen M, Burchell J, Wellehan JF Jr (2012) Evidence of recombination and positive selection in cetacean papillomaviruses. Virology 427(2):189–197
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51(3):492–508
Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16(8):1114–1114
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Simon-Loriere E, Holmes EC (2011) Why do RNA viruses recombine? Nat Rev Microbiol 9(8):617–626
Smelov V, Muwonge R, Sokolova O, McKay-Chopin S, Eklund C, Komyakov B, Gheit T (2018) Beta and gamma human papillomaviruses in anal and genital sites among men: prevalence and determinants. Sci Rep 8(1):8241
Smith JM (1992) Analyzing the mosaic structure of genes. J Mol Evol 34(2):126–129
Strimmer K, Rambaut A (2002) Inferring confidence sets of possibly misspecified gene trees. Proc Biol Sci 269(1487):137–142
Teixeira MF, Sabido M, Leturiondo AL, de Oliveira Ferreira C, Torres KL, Benzaken AS (2018) High risk human papillomavirus prevalence and genotype distribution among women infected with HIV in Manaus, Amazonas. Virol J 15(1):36
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44(W1):W232–235
Van Doorslaer K, Chen Z, Bernard HU, Chan PKS, DeSalle R, Dillner J, Forslund O, Haga T, McBride AA, Villa LL, Burk RD, Ictv Report C (2018) ICTV virus taxonomy profile: papillomaviridae. J Gen Virol 99(8):989–990
Van Doorslaer K, Dillner J (2019) The launch of an international animal papillomavirus reference center. Viruses 11(1):55
Van Regenmortel MHV, Fauquet CM, Bishop DHL, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGeoch DJ, Pringle CR, Wickner RB (2000) Virus Taxonomy. Seventh Report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego, NY, USA
Varsani A, van der Walt E, Heath L, Rybicki EP, Williamson AL, Martin DP (2006) Evidence of ancient papillomavirus recombination. J Gen Virol 87(Pt 9):2527–2531
Vinodhini K, Shanmughapriya S, Das BC, Natarajaseenivasan K (2012) Prevalence and risk factors of HPV infection among women from various provinces of the world. Arch Gynecol Obstet 285(3):771–777
Acknowledgements
We thank Prof Darren Martin for his help rendered with RDP4 software. Dr Alltalents T. Murahwa is the recipient of a Post Doc fellowship from the South African National Research Foundation. This work is based upon research supported by the South African Research Chairs Initiative of the Department of Science and Technology of South Africa and NRF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Handling Editor: Keith Crandall.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Murahwa, A.T., Tshabalala, M. & Williamson, AL. Recombination Between High-Risk Human Papillomaviruses and Non-Human Primate Papillomaviruses: Evidence of Ancient Host Switching Among Alphapapillomaviruses. J Mol Evol 88, 453–462 (2020). https://doi.org/10.1007/s00239-020-09946-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-020-09946-0