
Abstract
Papillomaviruses (PVs) are host-species-specific and tissue-specific viruses that infect a diverse array of vertebrate hosts, including humans and non-human primates, with varying pathogenic outcomes. Although primate PVs have been studied extensively, no complete genome sequences of PVs from lemurs have been determined to date. Saliva samples from three critically endangered, captive black-and-white ruffed lemurs (Varecia variegata variegata) at the Duke Lemur Center (USA) were analyzed, using high-throughput sequencing, for the presence of oral papillomaviruses. We identified three PVs from two individuals, one of which had a coinfection with two different PVs. Two of the three PVs share 99.6% nucleotide sequence identity, and we have named these isolates "Varecia variegata papillomavirus 1" (VavPV1). The third PV shares ~63% nucleotide sequence identity with VavPV1, and thus, we have named it "Varecia variegata papillomavirus 2" (VavPV2). Based on their E1 + E2 + L1 protein sequence phylogeny, the VavPVs form a distinct clade. This clade likely represents a novel genus, with VavPV1 and VavPV2 belonging to two distinct species. Our findings represent the first complete genome sequences of PVs found in lemuriform primates, with their presence suggesting the potential existence of diverse PVs across the over 100 species of lemurs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Papillomaviruses (PVs) are a family of viruses with a small, circular, double-stranded DNA genome of about 6-8 kb and a non-enveloped, icosahedral (T=7) capsid [1, 2]. The PV genome encodes at least six proteins: E1, E2, L1, and L2 [3]. L1 and L2 are structural proteins: the major capsid protein and the minor capsid protein [4,5,6]. E1 and E2 are regulatory proteins involved in replication and transcription [4, 7,8,9], whereas E6 and E7 specifically associate with cell cycle regulators [10, 11], serving to stimulate the progression of the cell cycle, with the potential to disrupt important regulatory pathways and be factors in the development of PV-associated cancers [12].
The family Papillomaviridae has two subfamilies: Firstpapillomavirinae and Secondpapillomavirinae [3]. Within these subfamilies, PVs are classified into 53 genera, 133 species and 343 types (see curation at PaVE [13]). PVs have been identified in samples from mucosal and epithelial cells, which are the primary sites of PV infection. They have also been identified in fecal samples from various animals as a result of epithelial cell shedding [12, 14]. PVs have co-evolved with their hosts, developing into a highly species- and tissue-specific viral family [2, 12]. Despite the diversity of PVs and their hosts, PVs show conservation in their genome organization and particularly high conservation in the L1 protein [14].
Non-human primate PVs have been identified in howler monkeys, squirrel monkeys, colobus monkeys, macaques, baboons, gorillas, bonobos, black-tufted marmosets, and chimpanzees [2, 14,15,16,17,18,19]. Clinical manifestations of non-human primate PVs include focal epithelial hyperplasia in bonobos, cervical dysplasia in baboons, and aggressive warts and cervical cancer in macaques [12, 15, 18]. Despite the characterization of PV genomes in the great apes and New and Old World monkeys, relatively little is known about those in lemuriform primates. Lemuriform primates are of particular interest because of their unique evolutionary history, phylogenetic relatedness to humans, and rapidly declining populations (~98% of lemurs are threatened with extinction) [20]. There are over 100 species of lemurs; thus, species-specific lemuriform PVs are likely to be extremely diverse. Antonsson and Hansson [17] detected a gammapapillomavirus-like PV in a black-and-white ruffed lemur (Varecia variegata); however, challenges encountered during sequencing efforts prevented characterization of this PV genome.
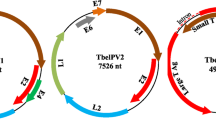
To address the significant gap in our knowledge of lemuriform PVs, we undertook a pilot project at the Duke Lemur Center in Durham, North Carolina, USA, to identify the oral PVs of black-and-white ruffed lemurs. We collected lemur saliva samples from three black-and-white ruffed lemurs (H1, H2, and H3) at the Duke Lemur Center under IACUC #A161-21-08 during July 2021 and March 2022 by allowing the lemurs to chew on a SalivaBio Children’s Swab (Salivametrics, USA). No oral abnormalities were noted. The saturated swabs were then placed into a SalivaBio Swab Storage Tube (Salivametrics, USA) and centrifuged to collect the saliva. Saliva samples were stored at -80°C until viral DNA extraction. We added SM buffer (0.1 M NaCl, 50 mM Tris-HCl [pH 7.4]) to each saliva sample up to a final volume of 400 µl, and 200 µl of the diluted sample was used to extract viral DNA using a High Pure Viral Nucleic Acid Kit (Roche Diagnostics, USA). The circular DNA in the viral DNA extract was amplified by rolling-circle amplification using an Illustra TempliPhi Kit (GE Healthcare, USA) and used to generate Illumina sequencing libraries using an Illumina DNA Prep Kit (with Tagmentation). Samples were sequenced on an Illumina NovaSeq 6000 system at the Duke Center for Genomic and Computational Biology facility. The paired-end reads (2 × 150 bp), with a Q30% of 90.58-92.58 and an average quality score of 35.33-35.64, were trimmed using Trimmomatic-0.39 [21] and assembled de novo using MEGAHITv.12.9 [22]. Circular contigs were identified based on terminal redundancy. Contigs >1000 nt in length were analyzed for viral-like sequences, using Diamond [23] to perform a BLASTx search of a local viral protein RefSeq database (release 210; downloaded March 2022). Contigs identified as potential PV-like sequences were confirmed using BLASTn [24]. We identified three contigs that represented complete genome sequences (based on terminal redundancy), ranging in size from 7953 to 7769 nt, to which ~0.0041-0.1475% of the total reads mapped. These three contigs had a read depth of 16.9×, 99.2×, and 103.2× and 881, 5191, and 5315 mapped reads, respectively, based on BBmap [25] read mapping analysis. The mapped reads have been deposited at SRA under BioProject: PRJNA874427; BioSample: SAMN30547858, SAMN30547860; SRA: SRR21284443, SRR21284445. The genomes were annotated (Fig. 1) using CenoteTaker2 [26] and refined using PaVE [13].

(A) Genome organization map of VavPV1 and VavPV2. (B) Identification of the conserved zinc-binding motifs in the E6 protein and the CR1, CR2, and zinc-binding motif in the E7 protein of the VavPVs. (C) Pairwise amino acid sequence identity values for the various proteins of VavPV1 and VavPV2. (D) Pairwise identity values for the proteins of VavPV1 and VavPV2 in comparison with their closest homologues from other PVs. The closest homologues were identified using BLASTp search of the complete PV protein sequence database, and pairwise identities were determined using SDT v1.2 [28]
Two of the PV genome sequences (accession nos. OP376965 and OP376966), obtained from black-and-white ruffed lemur individuals H1 and H2, share 99.4% identity. Thus, they are likely of the same PV type. We have named this virus "Varecia variegata papillomavirus 1" (VavPV1). The third PV genome sequence (accession no. OP376964), from black-and-white ruffed lemur H1, shares 63.0-63.2% pairwise identity with the VavPV1 isolates. Thus, it is clearly a different PV type, and we have named this virus "Varecia variegata papillomavirus 2" (VavPV2). Black-and-white ruffed lemur H1 appears to have been coinfected with VavPV1 and VavPV2. We did not detect any PV sequences in black-and-white ruffed lemur H3.
In VavPV1 and VavPV2, we identified all six core PV genes as well as putative E1^E4 and E8^E2 coding regions (Fig. 1). In the E6 and E7 proteins, we identified the conserved zinc-binding domains (CxxC), and in the E7 of VavPV2, we identified the pRB-binding motif (Lx[C/S]xE). We note that the lack of a pRB-binding motif in the E7 protein of VavPV1 is unusual. However, other previously described PVs also lack this motif (i.e., PVs in the genera Deltapapillomavirus, Dyochipapillomavirus, Dyoetapapillomavirus, Dyoiotapapillomavirus, Dyolambdapapillomavirus, Dyopsipapillomavirus, Dyorhopapillomavirus, Dyotaupapillomavirus, Dyoxipapillomavirus, Dyozetapapillomavirus, Epsilonpapillomavirus, Nupapillomavirus, Pipapillomavirus, Sigmapapillomavirus, and Zetapapillomavirus). In E7, two additional regions (CR1 and CR2) have been identified [10]. These regions share similarities to those in the E1A protein of human adenovirus 5 (family Adenoviridae) and the large tumor antigen of simian virus 40 (family Polyomaviridae). These two regions are also clearly recognizable in the E7 protein of the VavPV isolates (Fig. 1).
PV species classification is based on L1 nucleotide sequence similarity [27], with 70% identity being the species demarcation threshold. A query of the L1 genes of VavPV1 and VavPV2 revealed that they share <64% identity with each other and <66% identity with all other PV L1 sequences, as determined using SDT v1.2 [28]. Thus, VavPV1 and VavPV2 each represent a new PV species. To accurately determine the genus assignment for these two new PVs, we assembled a dataset of firstpapillomavirus E1, E2, and L1 protein sequences from PaVE [13] that included reference and non-reference sequences. The datasets were aligned with the sequences of the VavPVs using MAFFT v7.113 in AUTO mode [29]. The alignments were trimmed using TrimAL [30] with a gap threshold of 0.2. For each dataset, we determined the best-fit amino acid substitution model using ProtTest 3 [31]. The E1, E2, and L1 amino acid sequence alignments were concatenated, and the resulting alignment was used to construct a partitioned maximum-likelihood phylogenetic tree using IQtree 2 [32] with the model LG+I+G for E1, LG+I+G+F for E2, and LG+I+G+F for L1. The tree was rooted using avian and reptilian PV sequences and viewed and annotated in iTOL v6 [33]. The E1+E2+L1 sequences of the VavPVs clustered together and were part of a broader, well-supported clade containing sequences of viruses of the genera Dyoxipapillomavirus, Gammapapillomavirus, Pipapillomavirus, Taupapillomavirus, as well as Treisetapapillomavirus, and six unclassified viruses (Fig. 2). Although the results of L1 nucleotide sequence analysis using PaVE [13] would place the two VavPVs in the genus Gammapapillomavirus (60% pairwise identity threshold), the E1+E2+L1 protein phylogeny clearly shows that (1) the current gammapapillomaviruses, in particular gammapapillomavirus 6 and 7, are not monophyletic and that (2) phylogenetically, they are more closely related to taupapillomaviruses (Fig. 2). Therefore, VavPV1 and VavPV2 represent two new species in a putative new genus. Interestingly, this identifies another phylogenetic clade containing primate viruses, further demonstrating that the true diversity of the family Papillomaviridae is still not completely understood. The phylogenetic position of VavPV1 and VavPV2 further argues that other evolutionary mechanisms, in addition to cospeciation, must be in play [34].

Partitioned maximum-likelihood phylogenetic tree of concatenated amino acid sequences of E1, E2, and L1. Branches with >0.8 aLRT branch support are shown. PV genera are shown, and currently unclassified PVs that are unique at the species level were also included in the phylogeny
In this report, we describe the first complete genome sequences of PVs in lemuriform primates, and these novel viruses constitute a potential new genus. This contributes to our knowledge of non-human primate PVs, with particular significance for the virology, animal husbandry, and animal conservation communities. Continued research in this area will reveal the evolution and diversity of potential species-specific PVs present across the over 100 species of lemurs.
Data availability
The sequences described in this study have been deposited in the GenBank database under accession numbers OP376964-OP376966. Raw reads have been deposited at SRA under BioProject: PRJNA874427; BioSample: SAMN30547858; SAMN30547860; SRA: SRR21284443; SRR21284444.
References
Silvestre RV, de Souza AJ, Junior EC, Silva AK, de Mello WA, Nunes MR, Junior JL, Cardoso JF, de Vasconcelos JM, de Oliveira LF, da Silva SP, da Silva AM, Fries BG, Summa ME, de Sa LR (2016) First new world primate Papillomavirus identification in the Atlantic Forest, Brazil: Alouatta guariba papillomavirus 1. Genome Announc 4:e00725-e816
Joh J, Hopper K, Van Doorslaer K, Sundberg JP, Jenson AB, Ghim SJ (2009) Macaca fascicularis papillomavirus type 1: a non-human primate betapapillomavirus causing rapidly progressive hand and foot papillomatosis. J Gen Virol 90:987–994
Van Doorslaer K, Chen Z, Bernard HU, Chan PKS, DeSalle R, Dillner J, Forslund O, Haga T, McBride AA, Villa LL, Burk RD, Ictv Report C (2018) ICTV virus taxonomy profile: papillomaviridae. J Gen Virol 99:989–990
Chen Z, Long T, Wong PY, Ho WCS, Burk RD, Chan PKS (2019) Non-human primate papillomaviruses share similar evolutionary histories and niche adaptation as the human counterparts. Front Microbiol 10:2093
Buck CB, Day PM, Trus BL (2013) The papillomavirus major capsid protein L1. Virology 445:169–174
Wang JW, Roden RB (2013) L2, the minor capsid protein of papillomavirus. Virology 445:175–186
Middleton K, Peh W, Southern S, Griffin H, Sotlar K, Nakahara T, El-Sherif A, Morris L, Seth R, Hibma M, Jenkins D, Lambert P, Coleman N, Doorbar J (2003) Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J Virol 77:10186–10201
Bergvall M, Melendy T, Archambault J (2013) The E1 proteins. Virology 445:35–56
McBride AA (2013) The papillomavirus E2 proteins. Virology 445:57–79
Roman A, Munger K (2013) The papillomavirus E7 proteins. Virology 445:138–168
Vande Pol SB, Klingelhutz AJ (2013) Papillomavirus E6 oncoproteins. Virology 445:115–137
Long T, Burk RD, Chan PKS, Chen Z (2022) Non-human primate papillomavirus E6-mediated p53 degradation reveals ancient evolutionary adaptation of carcinogenic phenotype to host niche. PLoS Pathog 18:e1010444
Van Doorslaer K, Li Z, Xirasagar S, Maes P, Kaminsky D, Liou D, Sun Q, Kaur R, Huyen Y, McBride AA (2017) The Papillomavirus Episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res 45:D499–D506
Rector A, Van Ranst M (2013) Animal papillomaviruses. Virology 445:213–223
Bergin IL, Bell JD, Chen Z, Zochowski MK, Chai D, Schmidt K, Culmer DL, Aronoff DM, Patton DL, Mwenda JM, Wood CE, Burk RD (2013) Novel genital alphapapillomaviruses in baboons (Papio hamadryas Anubis) with cervical dysplasia. Vet Pathol 50:200–208
Chen Z, van Doorslaer K, DeSalle R, Wood CE, Kaplan JR, Wagner JD, Burk RD (2009) Genomic diversity and interspecies host infection of alpha12 Macaca fascicularis papillomaviruses (MfPVs). Virology 393:304–310
Antonsson A, Hansson BGR (2002) Healthy skin of many animal species harbors papillomaviruses which are closely related to their human counterparts. J Virol 76:12537–12542
Hoffmann M, Schütze E, Bernhard A, Schlaphoff L, Kaul A, Schöniger S, Pöhlmann S (2019) Disease manifestation and viral sequences in a bonobo more than 30 years after papillomavirus infection. Pathogens 8:13
D’Arc M, Moreira FRR, Dias CA, Souza AR, Seuanez HN, Soares MA, Tavares MCH, Santos AFA (2020) The characterization of two novel neotropical primate papillomaviruses supports the ancient within-species diversity model. Virus Evol 6:veaa036
Barrett MA, Brown JL, Junge RE, Yoder AD (2013) Climate change, predictive modeling and lemur health: assessing impacts of changing climate on health and conservation in Madagascar. Biol Cons 157:409–422
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W (2015) MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bushnell B (2014) BBMap: a fast, accurate, splice-aware aligner. Lawrence Berkeley National Lab (LBNL), Berkeley
Tisza MJ, Belford AK, Dominguez-Huerta G, Bolduc B, Buck CB (2021) Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evol 7:veaa100
Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM (2010) Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401:70–79
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9:e108277
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:W293–W296
Willemsen A, Bravo IG (2019) Origin and evolution of papillomavirus (onco)genes and genomes. Philos Trans R Soc Lond B Biol Sci 374:20180303
Acknowledgements
We particularly thank the Duke Lemur Center (DLC) staff members was scheduled and aided in sample collection. This is DLC publication #1530.
Funding
The work described here was supported by TriCEM (Triangle Center for Evolutionary Medicine), Duke Biology, and Sigma Xi grants awarded to ENP.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Permits
Animal handling and sample collection protocols were approved by the Office of Animal Welfare Assurance at Duke University, USA (IACUC #A161-21-08).
Additional information
Handling Editor: Ana Cristina Bratanich.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Paietta, E.N., Kraberger, S., Custer, J.M. et al. Identification of diverse papillomaviruses in captive black-and-white ruffed lemurs (Varecia variegata). Arch Virol 168, 13 (2023). https://doi.org/10.1007/s00705-022-05679-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-022-05679-1