
Abstract
Recent advances in the ease with which the genomes of small circular single-stranded DNA viruses can be amplified, cloned, and sequenced have greatly accelerated the rate at which full genome sequences of mastreviruses (family Geminiviridae, genus Mastrevirus) are being deposited in public sequence databases. Although guidelines currently exist for species-level classification of newly determined, complete mastrevirus genome sequences, these are difficult to apply to large sequence datasets and are permissive enough that, effectively, a high degree of leeway exists for the proposal of new species and strains. The lack of a standardised and rigorous method for testing whether a new genome sequence deserves such a classification is resulting in increasing numbers of questionable mastrevirus species proposals. Importantly, the recommended sequence alignment and pairwise identity calculation protocols of the current guidelines could easily be modified to make the classification of newly determined mastrevirus genome sequences significantly more objective. Here, we propose modified versions of these protocols that should substantially minimise the degree of classification inconsistency that is permissible under the current system. To facilitate the objective application of these guidelines for mastrevirus species demarcation, we additionally present a user-friendly computer program, SDT (species demarcation tool), for calculating and graphically displaying pairwise genome identity scores. We apply SDT to the 939 full genome sequences of mastreviruses that were publically available in May 2012, and based on the distribution of pairwise identity scores yielded by our protocol, we propose mastrevirus species and strain demarcation thresholds of >78 % and >94 % identity, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The family Geminiviridae includes insect-transmitted, plant-infecting viruses with circular, single-stranded DNA (ssDNA) genomes that are encapsidated within geminate particles. The genus Mastrevirus of this family consists of viruses that are transmitted by leafhoppers and have a single genome component with a conserved arrangement of three genes (encoding a movement protein, a coat protein and two versions of a replication-associated protein) and two non-coding regions (the large and small intergenic regions).
A variety of International Committee on Taxonomy of Viruses (ICTV)–endorsed guidelines currently exist for the classification and naming of new mastreviruses [7, 13–15]. Primary among these guidelines is the application of carefully selected genome-wide pairwise sequence identity thresholds, either to assign newly determined mastreviruses to existing species or strains, or as the basis for proposing that newly determined sequences correspond to new species or strains.
There are, however, quite a few different ways in which pairwise nucleotide sequence identities could be calculated and, importantly, the identity score that one gets for any given pair of sequences can vary quite substantially depending on exactly how it was calculated. One obvious example of how pairwise identity scores could be either inflated or deflated involves the treatment of gap characters that are inserted during the sequence alignment process. If gap characters are treated as a fifth possible nucleotide state, positions in the sequence alignment where one sequence has a nucleotide but the other does not will be (and quite reasonably so) counted as a mismatch. There exists a problem, however, in determining, firstly, how much such mismatches should count relative to standard nucleotide mismatches and, secondly, how much each gap character in runs of several gaps should count relative to isolated gaps. Side-stepping this problem altogether by simply ignoring all alignment sites at which one or the other sequence has a gap character is the approach of choice when, for example, calculating genetic and evolutionary distances in applications such as phylogenetic tree construction [16, 35]. Whereas ignoring positions where one sequence has a gap and the other does not will inflate pairwise identity scores, evenly scoring every one of these sites as a “normal” nucleotide mismatch will deflate identity scores over methods that include runs of gaps as a single mismatch.
An additional important factor that can cause fluctuations in pairwise identity scores of a given pair of sequences is the method used for sequence alignment. An optimal pairwise alignment of the sequences will generally yield a higher pairwise identity score than if the sequences were aligned within the context of a multiple sequence alignment that includes one or more additional sequences. Also, as the number and diversity of sequences in a multiple sequence alignment increases, so it is expected that the pairwise identity scores of any individual pair of sequences in the alignment will decrease [25]. What this means is that pairwise identity scores will tend downwards with increasing alignment size. Finally, different multiple sequence alignments generated either by different multiple sequence alignment programs (such as ClustalV, ClustalW, MAFFT or MUSCLE), or by a single program with different alignment settings (such as gap open and extension penalties) will not all be equally accurate [11, 12, 25, 44, 55].
Despite these various issues, pairwise-identity-based virus classification criteria are extremely popular amongst virus taxonomists and are likely to grow in importance due to how easy they are to use and the fact that, once properly validated, they accurately reflect the biology of these organisms [2, 32, 33]. We have therefore devised a pairwise-identity-based approach for mastrevirus classification that almost completely removes all the alignment and gap-handling problems of the current ICTV-endorsed mastrevirus classification protocol. We apply an approach that is almost identical to that described by Bao et al. [2] for their pairwise sequence comparison (PASC) method. Rather than relying on multiple sequence alignments and the counting of gap characters as a fifth nucleotide state (as is done in the currently recommended approach), our method and that of Bao et al. [2] rely on accurate and highly repeatable/reproducible pairwise sequence alignments and the complete exclusion of sites with gap characters from the pairwise identity calculations.
We apply this approach to determine the distribution of mastrevirus genome-wide pairwise identity scores and identify logical mastrevirus strain and species demarcation thresholds. We then apply these new mastrevirus species and strain demarcation criteria to all full mastrevirus genome sequences that were publically available in May 2012 and propose updates to all mastrevirus isolate names to make these consistent with the proposed criteria. Although the modified protocol yields a classification that is very similar to the current mastrevirus classification (sequences belonging to three proposed and one accepted species are “demoted” to the level of strains of pre-existing species), it is extremely objective (i.e., the pairwise identity scores it yields are almost completely un-manipulable) and is applied within a freely available and easy-to-use computer program that should tremendously simplify the classification of any new mastrevirus full genome sequence.
A new approach to calculating pairwise identity scores
Given a set of mastrevirus full genome sequences that have all been linearised at the same position, the approach that we have chosen for pairwise identity score calculations is very simple and essentially involves two steps. In the first step, every unique pair of sequences is individually aligned, essentially using the Needleman-Wunsch algorithm [43] as applied in multiple sequence alignment programs such as ClustalW [9, 31], MUSCLE [11] and MAFFT [25]. For a set of S sequences, this will yield [S × (S−1)]/2 pairwise alignments. For each of these alignments, the identity of each pair of sequences is calculated as 1−M/N, where M is the number of mismatched nucleotides and N is the total number of columns along the alignment where neither aligned sequence has a gap character. Our identity score is therefore simply one minus the ratio of the Hamming distance over the length of the pairwise-aligned sequences.
Since we knew of no computer programs that would perform pairwise alignments and output a table containing identity scores, we produced a computer program, called SDT (species demarcation tool), to largely automate this process (available from http://web.cbio.uct.ac.za/SDT). SDT will take as input a FASTA file with up to 1,000 sequences (either aligned or unaligned) and, in a single step, “calculate”, sort and display a colour-coded matrix of pairwise identity scores (Fig. 1a). It will additionally produce both plots of these pairwise identity scores and text files containing the plotted data to facilitate the identification of rational pairwise-identity-based taxonomic demarcation criteria (Fig. 1b).

The SDT interface. a Colour-coded matrix of pairwise identity scores. b Distribution plot of pairwise identity scores. (1) Command menus used to load FASTA files, save analysis results in various graphical and text formats, terminate the program and rerun analyses with different settings. (2) Command buttons used to switch between the matrix display and the pairwise identity distribution display, to zoom in and out of the two displays and to switch between the full-colour mode and the three-colour mode. (3) Spin controls used to adjust defined pairwise similarity demarcation cutoffs that can be used to, for example, colour pairwise similarity scores of viruses within a species differently from scores between viruses that are in different species. (4) The horizontal order of sequence names from left to right is the same as the vertical order from top to bottom and reflects the vertical ordering of sequences that would occur within a neighbour-joining phylogenetic tree constructed from the pairwise identity matrix. (5) Each coloured cell at the intersection of two sequence names represents the percent identity between those two sequences such that, for example, the three red triangles represent three clusters of closely related sequences (having >95 % identity). (6) A key indicating the correspondence between pairwise identities and the colours displayed in the matrix. (7) The horizontal axis indicates the percentage pairwise identity, and the vertical axis indicates the proportion of pairwise identities. (8) The valleys between peaks in the plot indicate percentage pairwise identities that would make relatively conflict-free pairwise-identity-score-based taxonomic demarcation thresholds
Rational mastrevirus species and strain demarcation criteria
Using SDT, we performed pairwise alignments of 939 full genome sequences of mastreviruses and calculated a total of 440,391 pairwise identity scores (Fig. 2). The distribution of these scores has notable peaks at ~48–71 %, 78–92 %, and 94–100 % pairwise identity and clear valleys at 72–77 % and 93 % pairwise identity. Whereas peaks indicate demarcation thresholds that would likely yield classifications with high degrees of conflict (i.e., where large numbers of sequences could justifiably be classified as belonging to two or more different species), valleys indicate thresholds that would likely yield classifications with minimal conflict.

The new mastrevirus strain and species demarcation criteria. A Distribution of pairwise identity scores of full genome sequences of mastreviruses as determined using three different multiple sequence alignment programs: MUSCLE in blue, ClustalW in red and MAFFT in green (all with default settings). The vertical grey lines indicate the position of the 78 % species demarcation cutoff and the 94 % strain demarcation cutoff. B The new strain and species demarcation criteria yield, with only one exception, a series of species and strains where the degree of identity shared by the two most genetically different isolates within these species and strains are, respectively, within the 78 % (gray bars) and the 94 % (white bars) demarcation cutoffs. The only exceptions are the degrees of identity between 22 pairs of MSV-B isolates (out of a total of 1327 pairs), which are between 93.14 % and 94.0 %
Irrespective of the alignment program used (blue, red and green plots in Fig. 2a denoting MUSCLE, ClustalW and MAFFT, respectively), the method applied in SDT yields reasonably consistent distributions of pairwise identity scores for sequences that share >71 % pairwise identity. Overall, the MUSCLE method yields the highest pairwise identity scores (notice the rightward shift of the blue plot relative to the red and green plots) implying that, of the three alignment methods applied in SDT, its use in the classification of novel mastreviruses will yield the most conservative test of whether these sequences should represent species or strains. For our standardised mastrevirus classification protocol, we have therefore opted to recommend MUSCLE as the preferred alignment method.
Given that a species demarcation threshold of 78 % identity yields a species list that has a very low degree of conflict and requires only minor reclassifications of currently accepted mastrevirus species (i.e., it is mostly consistent with the currently prescribed classification system), we propose that mastrevirus genomes that are calculated to be >78 % similar with our new approach should be considered members of the same species.
Similarly, our analysis indicates that 94 % would be a relatively robust mastrevirus-wide strain demarcation threshold that would additionally be consistent with the informal strain demarcation systems currently in place for approved and tentative mastrevirus species such as “Chickpea chlorosis Australia virus” (CpCAV), “Chickpea chlorosis virus” (CpCV), “Chickpea chlorotic dwarf virus” (CpCDV), Chloris striate mosaic virus (CSMV), Digitaria didactyla striate mosaic virus (DDSMV), Maize streak virus (MSV), Panicum streak virus (PanSV), “Paspalum dilatatum striate mosaic virus” (PDSMV), “Paspalum striate mosaic virus” (PSMV), Sugarcane streak Reunion virus (SSRV), Sugarcane streak virus (SSV), Tobacco yellow dwarf virus (TYDV), and Wheat dwarf virus (WDV). We therefore propose that mastrevirus genomes that are calculated to be >94 % similar with our new approach should be considered members (or variants) of the same strain.
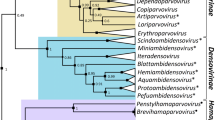
Importantly, there is strong phylogenetic support for almost all of the species and strains identified with the proposed classification system (Fig. 3 for all mastrevirus species other than MSV and Fig. 4 for MSV). The only cases where there is not strong phylogenetic support for the clustering of sequences within the identified strain and species groupings are those where only a single isolate has been identified as representatives of a given species or strain Table 1.

Phylogenetic support for the proposed mastrevirus species and strain demarcation criteria. Maximum-likelihood phylogenetic tree (constructed using full genome sequences and with the nucleotide substitution model GTR + I+G4 [19, 48]) depicting the likely evolutionary relationships of mastrevirus species and proposed strain groupings. Note that due to the large number of available maize streak virus (MSV) genome sequences, these sequences are represented on a separate tree presented in Fig. 4. The African, European, Asian and Australasian origins of the various isolates are indicated. BCSMV, bromus catharticus striate mosaic virus; CpCAV, chickpea chlorosis Australia virus; CpCV, chickpea chlorosis virus; CpCDV, chickpea chlorotic dwarf virus; CpRLV, chickpea redleaf virus; CpYV, chickpea yellows virus; CSMV, chloris striate mosaic virus; DCSMV, digitaria ciliaris striate mosaic virus; DDSMV, digitaria didactyla striate mosaic virus; DSV, digitaria streak virus; ESV, eragrostis streak virus; MiSV, miscanthus streak virus; MSRV, maize streak Reunion virus; ODV, oat dwarf virus; PanSV, panicum streak virus; PDSMV, paspalum dilatatum striate mosaic virus; PSMV, paspalum striate mosaic virus; SacSV, saccharum streak virus; SSMV-1, sporobolus striate mosaic virus-1; SSMV-2, sporolobus striate mosaic virus-2; SSEV, sugarcane streak Egypt virus; SSRV, sugarcane streak Reunion virus; SSV, sugarcane streak virus; TYDV, tobacco yellow dwarf virus; USV, urochloa streak virus; WDIV, wheat dwarf India virus; WDV, wheat dwarf virus

Phylogenetic support for the proposed maize streak virus (MSV) species and strain demarcation criteria. Maximum-likelihood phylogenetic tree (constructed using full genome sequences and with the nucleotide substitution model GTR + I+G4 [19, 48]) depicting the likely evolutionary relationships of the proposed MSV strain groupings
It is also worth pointing out that in the case of MSV and WDV, there are notable biological differences between viral isolates that, according to this proposal, would be classified into different strains. For example, whereas the “A” strain of MSV is clearly the only group of MSV variants that cause severe disease in maize [37, 58], the “A” strain of WDV preferentially infects barley, whereas the “C” strain preferentially infects wheat [50].
Updating the names of known mastrevirus isolates to reflect the new classification criteria
We applied these classification criteria to the 939 full genome sequences of mastreviruses available in public databases in May 2012 (Supplementary Figure 1) and have updated the various sequence names accordingly in Supplementary Table 1. Briefly, the names of these viruses now have the following form:
<virus name>-<strain name>[<country/territory code>-<lab codes/old names/host species of origin/sample number/location of origin>-<year of sampling>]
Virus name
For a newly obtained isolate the <virus name> is simply the ICTV-accepted name (or the acronym thereof) of the group of viruses to which the genome sequence has >78 % identity. If the sequence has >78 % identity to sequences classified as belonging to more than one established species, it is our recommendation that it simply be given the virus name of whichever sequences it is most similar to. Obviously, if the sequence has <78 % identity to any known mastrevirus genome sequences, it belongs to a new species, and it should be given a unique name (i.e., a name not shared by any other currently named virus) containing the name of the host from which the sequence was isolated and a succinct symptom descriptor. For example, “maize fine streak virus” and “maize stippled streak virus” could all be suitable names for viruses isolated from maize that produce symptoms resembling those of maize streak virus.
A number of mastrevirus species names that are currently accepted contain the name of the country/territory from which the first representative of that species was isolated. For example, isolates of sugarcane streak Reunion virus are only distantly related to those of sugarcane streak virus but produce similar symptoms in sugarcane. Since such names can be very misleading when such viruses are subsequently isolated in different countries/territories (for example, maize streak Reunion virus is also found in Southern Africa), we would suggest that this practice be discontinued and additional descriptors relating to symptoms be used, as outlined in the previous paragraph.
Strain name
Although we have chosen here to simply name strains alphabetically, this should not preclude anyone from naming strains based on consistently observable biological differences between the members of different strains. For example, suitable alternative names for MSV-A and MSV-B that reflect the different host preferences of viruses belonging to these strains would be MSV-Maize and MSV-Digitaria, respectively [58]. Although a descriptive strain name could potentially be useful, it should be borne in mind that unless it genuinely reflects the characteristics of all members of a strain, it also could be quite misleading. If symptom descriptors such as “mild” or “severe” are used as strain names, they should be based on symptom phenotypes observed in multiple independently isolated members of a strain.
It should also be noted that in many cases ad hoc “subtype” classification systems have been used to further categorise the members of certain strains. For, example MSV-A strains have been categorised into subtypes MSV-A1, -A2, A3, A4, A5 and A6. Although such sub-strain classifications are beyond the scope of this paper, it is appreciated that they can serve a practical purpose and, should such classifications be used, it is recommended that, as has been done with MSV, the sub-strain classifications be denoted with a subscript after the strain name.
Isolate descriptor field
Bounded by square brackets (i.e., “[ ]”) the isolate descriptor field may contain any number of sub-fields, each separated by a hyphen (i.e., “-”) but should, wherever possible, have as the first sub-field the two-letter international code of the country/territory of origin (Supplementary table 2) and as the last sub-field the year of isolation. Between these first and last sub-fields can be placed any additional useful descriptors, such as the district or city from which an isolate was obtained or the host species in which it was found. These “in-between” sub-fields can also contain additional information such as sample code numbers or former names. Crucially, the recommended format is “machine readable” in that various sequence analysis programs will be able to extract country/territory and sampling date information from such sequence names. Also note that we have broken with the Ninth ICTV Report’s recommendations for geminivirus nomenclature [7] and have avoided use of the “:” symbol to separate the isolate descriptor fields. We have done this because this symbol is specifically used to indicate branch-length information in the Newick phylogenetic tree file format (see http://en.wikipedia.org/wiki/Newick_format), and its use within sequence names can therefore cause problems for many computer programs that infer and/or render phylogenetic trees.
Resolving conflicts within the new mastrevirus classification system
Although the species and strain demarcation thresholds that we have chosen minimise the number of ambiguous classifications that might be made with the currently available full genome sequences, it is important to point out that situations are likely to arise where there is some uncertainty over the proper species or strain assignments of some isolates. The four possible reasons why a newly sequenced genome might be difficult to classify will be:
-
1.
Although >78 % identical to some isolates from a particular species, the new genome is <78 % identical to other isolates of that same species.
-
2.
The new genome is >78 % identical to isolates from two or more different species.
-
3.
Although >94 % identical to some isolates from a particular strain, the new genome is <94 % identical to other isolates of that same strain.
-
4.
The new genome is >94 % identical to some isolates from two or more different strains.
Among the mastrevirus genomes analysed here, we encountered only one instance of conflict type (3) – i.e., in some cases, MSV-B isolates were between 93.2 % and 94 % identical to other MSV-B isolates (Fig. 2b). We therefore recommend that each of the four above-mentioned conflict situations be resolved as follows:
-
1.
The new isolate should be classified as belonging to any species with which it shares >78 % identity to any one isolate formerly classified as belonging to that species, even if it is <78 % identical to other isolates classified as belonging to that species.
-
2.
The new isolate should be considered as belonging to the species containing the isolate with which it shares the highest degree of identity.
-
3.
The new isolate should be classified as belonging to any strain with which it shares >94 % identity to any one isolate formerly classified as belonging to that strain, even if it is <94 % identical to other isolates classified as belonging to that strain.
-
4.
The new isolate should be considered as belonging to the strain containing the isolate with which it shares the highest degree of identity.
A step-by-step guide to classifying new full genome sequences of mastreviruses
Following the determination of the full genome sequence a new mastrevirus it is recommended that:
-
1.
The new sequence should be used to perform a “nucleotide BLAST” search (accessible via http://blast.ncbi.nlm.nih.gov/Blast.cgi) of the NCBI “Nucleotide collection” database to, firstly, obtain the set of currently deposited sequences that most closely resemble the new sequence and, secondly, to identify the species to which the new sequence is most closely related.
-
2.
The set of sequences returned by the NCBI BLAST search should be saved in FASTA format and added to any other set of mastrevirus reference sequences (which, if also in FASTA file format, can simply be cut and pasted into the same FASTA file using a standard text editor). Such a mastrevirus reference sequence dataset is included with the SDT installation package in the file “mastrevirus references.sdt”, and updated versions of this file will be kept on the SDT web page (http://web.cbio.uct.ac.za/SDT). Alternatively, searching the nucleotide database at the NCBI website (http://www.ncbi.nlm.nih.gov/nuccore/) using the search term “<virus species/genus/family name> AND 2500:4000[SLEN]” will return all genomes indicated in the “<virus species/genus/family name> field that are between 2500 and 4000 nucleotides long. These can then also be saved to a FASTA file. Regardless of how datasets are compiled, sub-genome-length sequences should ultimately be removed from FASTA files that are intended for use in pairwise sequence identity analysis. Also, care should be taken to ensure that all the sequences being analysed all start at the same genomic coordinate. In the case of mastreviruses, there remain a small number of sequences in the database that do not begin at the virion strand origin of replication, and these should either be removed or edited so that they begin at this site prior to analysis.
-
3.
The FASTA file should be opened with the computer program SDT and, following selection of the MUSCLE method and calculation of the pairwise identity score matrix (Fig. 1a), it should be decided whether the sequence falls within a previously ICTV-accepted or proposed species (i.e., shares >78 % identity with isolates of that species) and, if so, whether it falls within a previously identified strain (i.e., shares >94 % identity with isolates previously classified as belonging to a named strain). If it falls within a previously named species and strain, the name and strain of the new sequence should be reflected in the species and strain name fields of its name. Similarly, if use is to be made of the reference mastrevirus dataset, the “mastrevirus references.sdt” file should be loaded first, and the user-generated FASTA file containing the researcher’s own newly determined sequence(s) (and perhaps also the nearest relatives of these sequences that were revealed by a BLAST search) should be appended to the mastrevirus references set using the “Append” command button (the SDT program will prompt this whole process once the “.sdt” file is selected rather than a FASTA file). From this point on, the analysis would be carried out as above for the analysis of a FASTA file.
-
4.
If the new sequence belongs to a new species (i.e., it is <78 % identical to any other known mastrevirus sequence), an appropriate species name should be proposed (see above for details) and the sequence should be given the strain name “A”.
-
5.
If the new sequence belongs to an existing species but is a new strain (i.e., it is <94 % identical to all isolates described for that species), the strain name proposed should follow our alphabetical naming convention, with pertinent details of the new sequence being added to the isolate descriptor field of the name, and not the strain field. If, in the future, multiple variants of the new strain have some unique, well-defined biological property, the strain could then be given a more descriptive name.
Conclusions
A pairwise-identity-based mastrevirus species demarcation criterion has been proposed that, while almost entirely consistent with the current mastrevirus classification, includes a very strict pairwise identity calculation protocol that should, if widely applied, significantly reduce the numbers of inappropriate new species proposals that are submitted for consideration by the ICTV. Also proposed is a new mastrevirus-wide pairwise-identity-based strain demarcation threshold. The standardised strain-level classification scheme should provide a consistent framework within which mastrevirus strains belonging to different species can be meaningfully compared with respect to, for example, their relative host and geographical ranges. The main strength of the proposal is that the prescribed pairwise identity calculation protocol is very difficult to manipulate (either intentionally or unintentionally) and will yield identical pairwise identity scores for a given pair of sequences, irrespective of how many other sequences are being compared within a dataset. This means that as the number of deposited full genome sequences of mastreviruses increases, there will be no need in the future to continuously revise the classification of already established species and strains. Perhaps most important from the perspective of the broader virology community with a general interest in virus diversity, however, is the fact that the computer program implementing our pairwise identity calculation protocol, SDT, can also very easily be adopted in standardised protocols for the classification of other virus groups.
The genome-wide pairwise-identity-based proposal for the classification of mastreviruses has been approved by the executive committee of the ICTV, and the document is available at http://talk.ictvonline.org/files/proposals/taxonomy_proposals_plant1/m/plant04/4399.aspx (2012.019abP.A.v3.Mastrevirus-17sp,rem-2sp.pdf).
References
Andersen MT, Richardson KA, Harbison SA, Morris BA (1988) Nucleotide sequence of the geminivirus chloris striate mosaic virus. Virology 164:443–449
Bao Y, Chetvernin V, Tatusova T (2012) PAirwise sequence comparison (PASC) and its application in the classification of Filoviruses. Viruses 4:1318–1327
Behjatnia A, Afsharifar A, Tahan V, Motlagh VA, Gandomani OE, Niazi A, Izadpanah K (2008) Widespread occurrence and molecular characterization of barley dwarf geminivirus in Iran. Phytopathology 98:S20–S20
Bendahmane M, Schalk HJ, Gronenborn B (1995) Identification and characterization of wheat dwarf virus from France using a rapid method for geminivirus DNA preparation. Phytopathology 85:1449–1455
Bigarre L, Salah M, Granier M, Frutos R, Thouvenel J, Peterschmitt M (1999) Nucleotide sequence evidence for three distinct sugarcane streak mastreviruses. Arch Virol 144:2331–2344
Briddon RW, Martin DP, Owor BE, Donaldson L, Markham PG, Greber RS, Varsani A (2010) A novel species of mastrevirus (family Geminiviridae) isolated from Digitaria didactyla grass from Australia. Arch Virol 155:1529–1534
Brown JK, Fauquet CM, Briddon RW, Zerbini M, Moriones E, Navas-Castillo J (2012) Virus taxonomy: ninth report of the international committee on taxonomy of viruses. Academic Press, Waltham
Chatani M, Matsumoto Y, Mizuta H, Ikegami M, Boulton MI, Davies JW (1991) The nucleotide sequence and genome structure of the geminivirus miscanthus streak virus. J Gen Virol 72:2325–2331
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500
Donson J, Accotto GP, Boulton MI, Mullineaux PM, Davies JW (1987) The nucleotide sequence of a geminivirus from Digitaria sanguinalis. Virology 161:160–169
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:1–19
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Fauquet CM, Maxwell DP, Gronenborn B, Stanley J (2000) Revised proposal for naming geminiviruses. Arch Virol 145:1743–1761
Fauquet CM, Stanley J (2003) Geminivirus classification and nomenclature: progress and problems. Ann Appl Biol 142:165–189
Fauquet CM, Briddon RW, Brown JK, Moriones E, Stanley J, Zerbini M, Zhou X (2008) Geminivirus strain demarcation and nomenclature. Arch Virol 153:783–821
Felsenstein J (2004) Inferring phylogenies. Sinauer Associates, Sunderland
Geering AD, Thomas JE, Holton T, Hadfield J, Varsani A (2012) Paspalum striate mosaic virus: an Australian mastrevirus from Paspalum dilatatum. Arch Virol 157:193–197
Greber R (1989) Biological characteristics of grass geminiviruses from eastern Australia. Ann App Biol 114:471–480
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Hadfield J, Martin DP, Stainton D, Kraberger S, Owor BE, Shepherd DN, Lakay F, Markham PG, Greber RS, Briddon RW, Varsani A (2011) Bromus catharticus striate mosaic virus: a new mastrevirus infecting Bromus catharticus from Australia. Arch Virol 156:335–341
Hadfield J, Thomas JE, Schwinghamer MW, Kraberger S, Stainton D, Dayaram A, Parry JN, Pande D, Martin DP, Varsani A (2012) Molecular characterisation of dicot-infecting mastreviruses from Australia. Virus Res 166:13–22
Halley-Stott RP, Tanzer F, Martin DP, Rybicki EP (2007) The complete nucleotide sequence of a mild strain of Bean yellow dwarf virus. Arch Virol 152:1237–1240
Harkins GW, Martin DP, Duffy S, Monjane AL, Shepherd DN, Windram OP, Owor BE, Donaldson L, van Antwerpen T, Sayed RA, Flett B, Ramusi M, Rybicki EP, Peterschmitt M, Varsani A (2009) Dating the origins of the maize-adapted strain of maize streak virus, MSV-A. J Gen Virol 90:3066–3074
Hughes FL, Rybicki EP, Kirby R (1993) Complete nucleotide sequence of sugarcane streak monogeminivirus. Arch Virol 132:171–182
Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518
Koklu G, Ramsell JN, Kvarnheden A (2007) The complete genome sequence for a Turkish isolate of Wheat dwarf virus (WDV) from barley confirms the presence of two distinct WDV strains. Virus Genes 34:359–366
Kraberger S, Thomas JE, Geering AD, Dayaram A, Stainton D, Hadfield J, Walters M, Parmenter KS, van Brunschot S, Collings DA, Martin DP, Varsani A (2012) Australian monocot-infecting mastrevirus diversity rivals that in Africa. Virus Res 169:127–136
Kumar J, Singh SP, Tuli R (2012) A novel mastrevirus infecting wheat in India. Archi Virol 257:2031–2034
Kundu JK, Gadiou S, Cervena G (2009) Discrimination and genetic diversity of wheat dwarf virus in the Czech Republic. Virus genes 38:468–474
Kvarnheden A, Lindblad M, Lindsten K, Valkonen JP (2002) Genetic diversity of wheat dwarf virus. Arch Virol 147:205–216
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lauber C, Gorbalenya AE (2012) Partitioning the genetic diversity of a virus family: approach and evaluation through a case study of picornaviruses. J Virol 86:3890–3904
Lauber C, Gorbalenya AE (2012) Toward genetics-based virus taxonomy: comparative analysis of a genetics-based classification and the taxonomy of picornaviruses. J Virol 86:3905–3915
Lawry R, Martin DP, Shepherd DN, van Antwerpen T, Varsani A (2009) A novel sugarcane-infecting mastrevirus from South Africa. Arch Virol 154:1699–1703
Lemey P, Salemi M, Vandamme A-M (2009) The phylogenetic handbook: a practical approach to phylogenetic analysis and hypothesis testing, 2nd edn. Cambridge University Press, New York
MacDowell SW, Macdonald H, Hamilton WD, Coutts RH, Buck KW (1985) The nucleotide sequence of cloned wheat dwarf virus DNA. EMBO journal 4:2173–2180
Martin DP, Willment JA, Billharz R, Velders R, Odhiambo B, Njuguna J, James D, Rybicki EP (2001) Sequence diversity and virulence in Zea mays of maize streak virus isolates. Virology 288:247–255
Martin DP, Linderme D, Lefeuvre P, Shepherd DN, Varsani A (2011) Eragrostis minor streak virus: an Asian streak virus in Africa. Arch Virol 156:1299–1303
Monjane AL, Harkins GW, Martin DP, Lemey P, Lefeuvre P, Shepherd DN, Oluwafemi S, Simuyandi M, Zinga I, Komba EK, Lakoutene DP, Mandakombo N, Mboukoulida J, Semballa S, Tagne A, Tiendrebeogo F, Erdmann JB, van Antwerpen T, Owor BE, Flett B, Ramusi M, Windram OP, Syed R, Lett JM, Briddon RW, Markham PG, Rybicki EP, Varsani A (2011) Reconstructing the history of maize streak virus strain a dispersal to reveal diversification hot spots and its origin in southern Africa. J Virol 85:9623–9636
Morris BA, Richardson KA, Haley A, Zhan X, Thomas JE (1992) The nucleotide sequence of the infectious cloned DNA component of tobacco yellow dwarf virus reveals features of geminiviruses infecting monocotyledonous plants. Virology 187:633–642
Mumtaz H, Kumari SG, Mansoor S, Martin DP, Briddon RW (2011) Analysis of the sequence of a dicot-infecting mastrevirus (family Geminiviridae) originating from Syria. Virus genes 42:422–428
Nahid N, Amin I, Mansoor S, Rybicki EP, van der Walt E, Briddon RW (2008) Two dicot-infecting mastreviruses (family Geminiviridae) occur in Pakistan. Arch Virol 153:1441–1451
Needleman SB, Wunsch CD (1970) A general method applicable to search for similarities in amino acid sequence of 2 proteins. J Mol Biol 48:443
Ogdenw TH, Rosenberg MS (2006) Multiple sequence alignment accuracy and phylogenetic inference. Syst Biol 55:314–328
Oluwafemi S, Varsani A, Monjane AL, Shepherd DN, Owor BE, Rybicki EP, Martin DP (2008) A new African streak virus species from Nigeria. ArchVirol 153:1407–1410
Owor BE, Martin DP, Shepherd DN, Edema R, Monjane AL, Rybicki EP, Thomson JA, Varsani A (2007) Genetic analysis of maize streak virus isolates from Uganda reveals widespread distribution of a recombinant variant. J Gen Virol 88:3154–3165
Pande D, Kraberger S, Lefeuvre P, Lett JM, Shepherd DN, Varsani A, Martin DP (2012) A novel maize-infecting mastrevirus from La Reunion Island. Arch Virol 157:1617–1621
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Ramsell JNE, Lemmetty A, Jonasson J, Andersson A, Sigvald R, Kvarnheden A (2008) Sequence analyses of wheat dwarf virus isolates from different hosts reveal low genetic diversity within the wheat strain. Plant Pathol 57:834–841
Ramsell JNE, Boulton MI, Martin DP, Valkonen JPT, Kvarnheden A (2009) Studies on the host range of the barley strain of Wheat dwarf virus using an agroinfectious viral clone. Plant Pathol 58:1161–1169
Rybicki EP (1994) A phylogenetic and evolutionary justification for three genera of Geminiviridae. Arch Virol 139:49–77
Schubert J, Habekuss A, Kazmaier K, Jeske H (2007) Surveying cereal-infecting geminiviruses in Germany—diagnostics and direct sequencing using rolling circle amplification. Virus Res 127:61–70
Shepherd DN, Varsani A, Windram OP, Lefeuvre P, Monjane AL, Owor BE, Martin DP (2008) Novel sugarcane streak and sugarcane streak reunion mastreviruses from southern Africa and La Reunion. Arch Virol 153:605–609
Thomas JE, Parry JN, Schwinghamer MW, Dann EK (2010) Two novel mastreviruses from chickpea (Cicer arietinum) in Australia. Arch Virol 155:1777–1788
Thompson JD, Plewniak F, Poch O (1999) A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res 27:2682–2690
Tobias I, Shevchenko O, Kiss B, Bysov A, Snihur H, Polischuk V, Salanki K, Palkovics L (2011) Comparison of the nucleotide sequences of wheat dwarf virus (WDV) isolates from Hungary and Ukraine. Polish J Microbiol/Polskie Towarzystwo Mikrobiologow = Polish Soc Microbiologists 60:125–131
Varsani A, Oluwafemi S, Windram OP, Shepherd DN, Monjane AL, Owor BE, Rybicki EP, Lefeuvre P, Martin DP (2008) Panicum streak virus diversity is similar to that observed for maize streak virus. Arch Virol 153:601–604
Varsani A, Shepherd DN, Monjane AL, Owor BE, Erdmann JB, Rybicki EP, Peterschmitt M, Briddon RW, Markham PG, Oluwafemi S, Windram OP, Lefeuvre P, Lett JM, Martin DP (2008) Recombination, decreased host specificity and increased mobility may have driven the emergence of maize streak virus as an agricultural pathogen. J Gen Virol 89:2063–2074
Varsani A, Monjane AL, Donaldson L, Oluwafemi S, Zinga I, Komba EK, Plakoutene D, Mandakombo N, Mboukoulida J, Semballa S, Briddon RW, Markham PG, Lett JM, Lefeuvre P, Rybicki EP, Martin DP (2009) Comparative analysis of Panicum streak virus and maize streak virus diversity, recombination patterns and phylogeography. Virol J 6:194
Wu BL, Melcher U, Guo XY, Wang XF, Fan LJ, Zhou GH (2008) Assessment of codivergence of Mastreviruses with their plant hosts. BMC Evol Biol 8:335
Acknowledgments
BM is funded by the University of Cape Town, South Africa. JNC and EM are members of the Research Group AGR-214, partially funded by Consejería de Economía, Innovación y Ciencia, Junta de Andalucía, Spain, cofinanced by FEDER-FSE. The authors would like to thank the Center for High Performance Computing in Cape Town and the Information Communication Technology Services Department at the University of Cape Town for use of their high-performance computing clusters. The authors would additionally like to thank Claude Fauquet for reading through and commenting on the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2012_1601_MOESM1_ESM.docx
Supplementary material 1 Updated names of full genome sequences of mastreviruses. Sequences that have been assigned to new species are highlighted in yellow. (DOCX 121 kb)
Rights and permissions
About this article
Cite this article
Muhire, B., Martin, D.P., Brown, J.K. et al. A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch Virol 158, 1411–1424 (2013). https://doi.org/10.1007/s00705-012-1601-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-012-1601-7