
Abstract
Three monocot-infecting mastreviruses from Australia, all found primarily in pasture and naturalised grasses, have been characterised at the molecular level. Here, we present the full genome sequence of a fourth, Paspalum striate mosaic virus (PSMV), isolated from Paspalum dilatatum from south-east Queensland. The genome was 2816 nt long and had an organisation typical of other monocot-infecting mastreviruses. Its nearest relative is Bromus cartharticus striate mosaic virus (BCSMV), with which it shares an overall genome identity of 75%. Phylogenetic analysis of the complete genome and each of the putative viral proteins places PSMV in a group with the other three Australian striate mosaic viruses. PSMV, BCSMV and Digitaria didactyla striate mosaic virus all contain a similar, small recombinant sequence in the small intergenic region.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Geminiviruses are among the most serious emerging plant pathogens. All geminiviruses have a circular, single-stranded DNA genome (c. 2.6-3.0 kb) and geminate virions (about 18 × 30 nm) and replicate via a rolling-circle mechanism [5]. The family Geminiviridae is divided into four genera, Mastrevirus, Curtovirus, Topocuvirus and Begomovirus, based on genome organisation, insect vector, host range and sequence relatedness [4]. Additionally, beet curly top Iran virus (BCTIV, [20]), Eragrostis curvula streak virus (ECSV) [21] and turnip curly top virus (TCTV) [2] have recently been described and do not fit into the current genera.
Mastreviruses are transmitted by leafhoppers and predominantly infect monocot hosts [4, 5]. The genome is bi-directionally transcribed, with the movement protein and coat protein translated from the virion-sense RNA (ORFs V1 and V2) and two replication-associated proteins translated from the complementary-sense RNA (ORFs C1 and C2) [5]. ORF C2 nearly always lacks an AUG start codon and is only expressed following the splicing of an intron, resulting in a single in-frame coding region including C1 and C2 [1, 11]. Bean yellow dwarf virus (BeYDV), chickpea chlorotic dwarf Pakistan virus (CpCDPKV), chickpea chlorotic dwarf Sudan virus (CpCDSDV), chickpea chlorotic dwarf Syria virus (CpCDSYV), tobacco yellow dwarf virus (TYDV), chickpea redleaf virus (CpRLV), and chickpea chlorosis virus (CpCV) -A and -B are mastreviruses that infect dicot hosts. They have one or two additional ORFs on the complementary-sense RNA that potentially encode proteins greater than 10 kDa in size, although it is unclear yet whether these ORFs are functional [8, 11].
Of the various mastreviruses that have thus far been described, only TYDV, CpRLV, CpCV -A and -B, Chloris striate mosaic virus (CSMV), Bromus catharticus striate mosaic virus (BCSMV), Digitaria didactyla striate mosaic virus (DDSMV) and Paspalum striate mosaic virus (PSMV) are known to occur in Australia [3, 6, 12, 14, 15]. The complete genome sequences of all viruses have been determined except for that of PSMV, and there is still some uncertainty about the taxonomy of this virus. Pinner et al. [12] observed that PSMV and BCSMV were serologically very closely related and on this basis considered them to be strains of the same virus species. In this paper, we present the full genome sequence of an isolate of PSMV and argue that they are in fact members of distinct virus species.
A Paspalum dilatatum plant showing symptoms typical of PSMV infection (DPI Plant Virus Isolate No. 1611) was collected from Anstead, south-east Queensland, Australia (27º32.461′ S, 152º51.755′ E) in 2003. Leaf lamina (c. 0.3–0.5 g) was sampled using a sterile scalpel blade, crushed in a 1.5-ml microfuge tube containing 200 μl of TPS buffer (100 mM Tris-HCl pH 8.0, 10 mM EDTA, 1 M KCl) and heated to 65°C for 10 min, followed by rapid chilling on ice [17]. Prior to amplification by TempliPhi (GE Healthcare, USA), a 1-in-10 dilution of the leaf extract in water was denatured at 95°C for 3 min and then chilled on ice. A mastermix of 5 μl of TempliPhi reaction buffer and 0.2 μl of TempliPhi enzyme mix containing Φ29 DNA polymerase was prepared on ice for each sample. Five μl of the mastermix was added to each sample, and the reactions were incubated overnight at 30°C. The Φ29 DNA polymerase was inactivated at 65°C for 10 min, and the reactions were stored at 4°C. Two to 5 μl of each TempliPhi DNA reaction was cut with BamHI, EcoRI, and Sau3AI (NEB), and the fragments were cloned into the pGEM-T Easy plasmid vector (Promega). Cloned DNA was sequenced at the Australian Genome Research Facility by primer walking. The sequences were edited and assembled using SEQUENCHER version 3. Within the assembled genome (2816 bp), we identified the canonical virion-strand origin of replication (v-ori) TAATATTAC sequence, two virion-strand open reading frames (ORFs) encoding a movement protein (MP) and a coat protein (CP), and a replication-associated protein (Rep) on the complementary strand (Supplementary Figures 1 and 2). Within the Rep, we identified the conserved catalytic, rolling-circle replication (RCR) motifs and ATP-binding domains (Supplementary Figure 2).
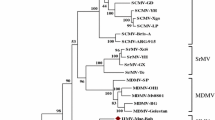
The entire PSMV genome sequence was aligned with all available mastreviruses genome sequences available in GenBank (11th April 2011) using Clustal W [16]; gap open penalty = 10; gap extension penalty = 5) with a representative sequence from each known mastrevirus species (Fig. 1) using MEGA version 4 [13]. A maximum-likelihood (ML) phylogenetic tree (Fig. 1) was constructed using PHYML [7], with GTR+I+G4 chosen as the best-fit model by RDP3 [9] and rooted with ECSV. Pairwise p-distance comparisons (pairwise deletion of alignment gaps) indicated that the PSMV genome shared 75.0, 55.4 and 57.6% nucleotide sequence identity with BCSMV, CSMV and DDMSV, respectively, and less than 48.5% nucleotide sequence identity with all other mastreviruses. The three proteins encoded by PSMV were most similar to the three characterised Australian mastreviruses (Fig. 2). The replication-associated protein shared 70% amino acid (aa) identity to BCSMV and 53–56% aa identity to DDSMV and CSMV (Fig. 2). The CP of PSMV shared 75.5% aa identity to BCSMV and 58.1–58.9% aa identity to DDSMV and CSMV (Fig. 2). The movement protein shared 65.5% aa identity to BCSMV and c. 40% aa identity to DDSMV and CSMV.

Maximum-likelihood phylogenetic tree (model GTR + I + G4) and a two-dimensional pairwise sequence similarity plot of 46 virus isolates representing known mastrevirus species and strains. The branches of the maximum-likelihood tree are coloured according to the geographical origins of the viruses. Branches marked with solid and open circles were supported in >90% and 70–89% of the bootstrap replicates, respectively, and the branches are coloured by country or region of sampling. The phylogenetic tree is rooted using the full genome sequence of Eragrostis curvula streak virus (ECSV; not shown)

Maximum-likelihood phylogenetic tree of CP (A) and Rep (B) proteins, drawn using the LG amino acid substitution model. Pairwise distance plot (C) of CP (above the diagonal) and Rep (below the diagonal). Branches marked with solid and open circles were supported in >90% and 70–89%, respectively, of the bootstrap replicates, and branches are coloured by country or region of sampling. The phylogenetic tree is rooted using the full genome sequence of beet curly top Iran virus (BCTIV; not shown). ECSV is included in both trees, since it has a very mastrevirus-like coat protein gene
A full-length nucleotide sequence identity of <75% is generally indicative of a distinct mastrevirus species, and when differences between two related viruses are not clear-cut, as is the case for PSMV and BCSMV, then biological properties must also be taken into consideration in taxonomic decisions [5]. The work of Greber, which is summarised below, supports recognition of PSMV as a member of a distinct species, as it is apparent that it occupies a unique ecological niche. CSMV can be experimentally transmitted to P. dilatatum and PSMV to Chloris gayana, but only with great difficulty. Part of this host specificity appears to be due to the feeding preferences of their vectors: Nesoclutha pallida biotype P (the vector of PSMV) survives poorly on C. gayana and does not breed on this host, and vice versa, C. gayana does not sustain N. pallida biotype C (the vector of CSMV) populations. However, there also appear to be intrinsic properties of PSMV that facilitate transmission by N. pallida biotype P, as when wheat is used as an acquisition source, N. pallida biotype C transmits PSMV very inefficiently when compared with biotype P, even though biotype C can readily acquire CSMV from wheat. Both biotypes of N. pallida transmit BCSMV and DDSMV with equal efficiency, although Bromus catharticus and Digitaria didactyla, respectively, are the only known natural hosts of these viruses, and P. dilatatum is resistant to infection by both. Furthermore, B. catharticus and D. didactyla are both resistant to infection by PSMV and CSMV.
Since recombination is common amongst mastreviruses [9, 18, 19], we analysed the four Australian monocot-infecting mastreviruses for evidence of recombination using the RDP, BOOTSCAN, GENECONV, MAXCHI, CHIMAERA, 3SEQ and SiScan methods implemented in RDP3 [9] with program default settings. The analysis revealed a small recombination-derived tract (in the short intergenic region) in DDSMV, BCSMV and PSMV derived from Miscanthus streak virus (MiSV) and Eragrostis minor streak virus (EMSV)–like sequences (p-value: RDP 7.165 × 10−12; GENECOV 8.985 × 10−12; BootScan 2.615 × 10−7, MaxChi 4.778 × 10−4; Chimaera 9.583 × 10−4). Phylogenetic support for this event is provided in Supplementary Figure 3.
In this report, we describe the characterisation of a fourth monocot-infecting mastrevirus from Australia, and it is becoming evident that the genetic diversity of monocot-infecting mastreviruses in Australia could rival that found in Africa. A recent report [10] has suggested that the centre of diversity of dicot-infecting mastreviruses is within the Pacific Rim, and that the recombination patterns observed in dicot-infecting mastreviruses mirror those found in moncot-infecting mastreviruses. To date, no Australian monocot-infecting mastreviruses have been linked to any crop loss despite BCSMV, CSMV, PSMV and DDSMV being transmissible to wheat, oats, barley and maize under controlled conditions, and in some cases, as natural infections [6]. The rapid increase in known Australian mastreviruses as a result of increased research efforts has already allowed the detection of distinct recombination events and has demonstrated the rapid evolution of these viruses. The jump to commercially important hosts in the near future is plausible and should warrant the continuation of research into these viruses to prevent a situation analogous to that which has occurred with the streak viruses in Africa.
GenBank accession - PSMV –[AU:QL:2003]; accession JF905486
References
Accotto GP, Donson J, Mullineaux PM (1989) Mapping of Digitaria streak virus transcripts reveals different RNA species from the same transcription unit. EMBO J 8:1033–1039
Briddon RW, Heydarnejad J, Khosrowfar F, Massumi H, Martin DP, Varsani A (2010) Turnip curly top virus, a highly divergent geminivirus infecting turnip in Iran. Virus Res 152:169–175
Briddon RW, Martin DP, Owor BE, Donaldson L, Markham PG, Greber RS, Varsani A (2010) A novel species of mastrevirus (family Geminiviridae) isolated from Digitaria didactyla grass from Australia. Arch Virol 155:1529–1534
Fauquet CM, Stanley J (2003) Geminivirus classification and nomenclature: progress and problems. Ann Appl Biol 142:165–189
Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (2005) Virus taxonomy: classification and nomenclature of viruses. Eighth report of the international committee on taxonomy of viruses. Elsevier Academic Press, San Diego
Greber RS (1989) Biological characteristics of grass geminiviruses from eastern Australia. Ann Appl Biol 114:471–480
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Liu L, van Tonder T, Pietersen G, Davies JW, Stanley J (1997) Molecular characterization of a subgroup I geminivirus from a legume in South Africa. J Gen Virol 78:2113–2117
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463
Martin DP, Briddon RW, Varsani A (2011) Recombination patterns in dicot-infecting mastreviruses mirror those found in monocot-infecting mastreviruses. Arch Virol 156:1463–1469
Morris BA, Richardson KA, Haley A, Zhan X, Thomas JE (1992) The nucleotide sequence of the infectious cloned DNA component of tobacco yellow dwarf virus reveals features of geminiviruses infecting monocotyledonous plants. Virology 187:633–642
Pinner MS, Markham PG, Rybicki EP, Greber RS (1992) Serological relationships of geminivirus isolates from Gramineae in Australia. Plant Pathol 41:618–625
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thomas JE, Bowyer JW (1980) Properties of tobacco yellow dwarf and bean summer death viruses. Phytopathology 70:214–217
Thomas JE, Parry JN, Schwinghamer MW, Dann EK (2010) Two novel mastreviruses from chickpea (Cicer arietinum) in Australia. Arch Virol 155:1777–1788
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Thomson D, Dietzgen RG (1995) Detection of DNA and RNA plant viruses by PCR and RT-PCR using a rapid virus release protocol without tissue homogenization. J Virol Methods 54:85–95
Varsani A, Shepherd DN, Monjane AL, Owor BE, Erdmann JB, Rybicki EP, Peterschmitt M, Briddon RW, Markham PG, Oluwafemi S, Windram OP, Lefeuvre P, Lett JM, Martin DP (2008) Recombination, decreased host specificity and increased mobility may have driven the emergence of maize streak virus as an agricultural pathogen. J Gen Virol 89:2063–2074
Varsani A, Monjane AL, Donaldson L, Oluwafemi S, Zinga I, Komba EK, Plakoutene D, Mandakombo N, Mboukoulida J, Semballa S, Briddon RW, Markham PG, Lett JM, Lefeuvre P, Rybicki EP, Martin DP (2009) Comparative analysis of Panicum streak virus and Maize streak virus diversity, recombination patterns and phylogeography. Virol J 6:194
Varsani A, Shepherd DN, Dent K, Monjane AL, Rybicki EP, Martin DP (2009) A highly divergent South African geminivirus species illuminates the ancient evolutionary history of this family. Virol J 6:36
Yazdi HR, Heydarnejad J, Massumi H (2008) Genome characterization and genetic diversity of beet curly top Iran virus: a geminivirus with a novel nonanucleotide. Virus Genes 36:539–545
Acknowledgments
AV is supported by the Marsden Fund of New Zealand (UOC0903). We thank Stuart Skabo for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2011_1129_MOESM1_ESM.pdf
Supplementary material 1 (PDF 22 kb). Supplementary Figure 1 Genome sequence annotations of PSMV-[AU:QL:2003] together with all other Australian monocot-infecting mastreviruses. Sequence regions or domains that are either known or believed to have a biological function are marked on the nucleotide sequence alignment. Gaps are represented with “-” characters
705_2011_1129_MOESM2_ESM.pdf
Supplementary material 2 (PDF 19 kb). Supplementary Figure 2 Annotated replication-associated protein, coat protein and movement protein amino acid sequences of PSMV-[AU:QL:2003] together with all other Australian monocot-infecting mastreviruses. The rolling-circle replication motifs (Rep), potential nuclear localisation signals (CP), DNA-binding domains (CP and Rep) and hydrophobic membrane-spanning domain (MP) are highlighted
705_2011_1129_MOESM3_ESM.ppt
Supplementary material 3 (PPT 186 kb). Supplementary Figure 3: A. Pairwise distance plot of EMSV, BCSMV and CSMV showing the recombinant region and recombination breakpoints. B. Maximum phylogenetic trees of non-recombinant and recombinant region supporting the recombination detection
Rights and permissions
About this article
Cite this article
Geering, A.D.W., Thomas, J.E., Holton, T. et al. Paspalum striate mosaic virus: an Australian mastrevirus from Paspalum dilatatum . Arch Virol 157, 193–197 (2012). https://doi.org/10.1007/s00705-011-1129-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-011-1129-2