
Abstract
The abnormal accumulation of α-synuclein in the brain is a common feature of Parkinson’s disease (PD), PD dementia (PDD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), and synucleinopathies that present with overlapping but distinct clinical symptoms that include motor and cognitive deficits. Synapse degeneration is the crucial neuropathological event in these synucleinopathies and the neuropathological correlate of connectome dysfunction. The cognitive and motor deficits resulting from the connectome dysfunction are currently measured by scalar systems that are limited in their sensitivity and largely subjective. Ideally, a marker of synapse degeneration would correlate with measures of cognitive or motor impairment, and could therefore be used as a more objective, surrogate biomarker of the core clinical features of these diseases. Furthermore, an objective surrogate biomarker that can detect and monitor the progression of synapse degeneration would improve patient management and clinical trial design, and could provide a measure of therapeutic response. Here, we review the published findings relating to candidate biomarkers of synapse degeneration in PD, PDD, DLB, and MSA patient-derived biofluids and discuss the findings in the context of the mechanisms associated with α-synuclein-mediated synapse degeneration. Understanding these mechanisms is essential not only for discovery of biomarkers, but also to improve our understanding of the earliest changes in disease pathogenesis of synucleinopathies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term synucleinopathy refers to a group of adult-onset, progressive neurodegenerative diseases characterized by abnormal accumulation of α-synuclein in the brain (Spillantini et al. 1997; Baba et al. 1998; Jellinger 2003). The most prevalent diseases with underlying synucleinopathy include Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA). Neuropathological evaluation of these patients post-mortem reveals α-synuclein inclusions that can be found in neuronal cytoplasm (Lewy bodies), neurites (Lewy neurites), or in glial cytoplasm (glial cytoplasmic inclusions). Although synucleinopathy is common to these diseases, inclusion type and deposition pattern and degree of neuronal loss, as well as the temporal order and prevalence of clinical manifestations can differ substantially across synucleinopathies (see Box 2).
In the healthy brain, α-synuclein is expressed predominantly at presynaptic terminals (Iwai et al. 1995; Maroteaux et al. 1988) where it modulates neurotransmitter release (Burre et al. 2010). In synucleinopathies, α-synuclein exists in a thermodynamic equilibrium of soluble oligomers and insoluble fibrils (Baba et al. 1998; Jellinger 2003; Fusco et al. 2018) that can be propagated from neuron to neuron and from neuron to astrocytes (Hoffmann et al. 2016; Sorrentino et al. 2019; George et al. 2019; Olanow et al. 2019). It is increasingly recognized that the diffusible oligomeric assemblies, rather than the fibrillar form, are the driving factor underlying neurodegeneration, ultimately leading to connectome dysfunction and neuronal death (Mahul-Mellier et al. 2020; Bellucci et al. 2016). Studies using neuroimaging tracers of synaptic proteins reinforce the idea that synucleinopathies are a result of functional connectome deficits (Box 2).
Aggregation and propagation of α-synuclein at synapses are a major event in the pathogenesis of synucleinopathies (Calo et al. 2016), and may be exacerbated by activation of microglial cells (Hoffmann et al. 2016), which in turn induce the spread of α-synuclein pathology (George et al. 2019; Olanow et al. 2019). In addition, soluble α-synuclein can be transported from neurons into neighbouring astrocytes in response to stress and/or neurodegeneration (Sorrentino et al. 2019). The correlation between the spread of Lewy bodies and disease progression (Braak et al. 2003) suggests that synaptic propagation of α-synuclein is integral to disease pathogenesis.
Dopaminergic neurons of the substantia nigra are particularly vulnerable to synuclein pathology. Their extensive axonal projections (which can exceed 4 m in length) and abundance of synaptic contacts (estimated 1 million synapses per neuron) may make this neuronal population particularly susceptible to disruptions in energy (Pissadaki and Bolam 2013). It is this disruption of dopamine homeostasis in basal ganglia circuits (Bellucci et al. 2016; Lowther et al. 2014) and sensory–motor networks (Peraza et al. 2014) that underlies the motor symptoms that are a common feature of the synucleinopathies (Box 2). Non-motor symptoms on the other hand likely reflect progressive connective dysfunction in mesolimbic–striatal loops (Luo et al. 2014) or in visual and attentional, default mode, salience, and executive networks (Sourty et al. 2016; Lowther et al. 2014). The recurrent complex visual hallucinations that are prevalent in PD may be due to functional changes associated with GABA-ergic neurons in the primary visual cortex (Khundakar et al. 2016). Treatment of patients with the dopamine precursor, levodopa, or with dopamine agonists improves clinical symptoms, but does not modify or stop disease progression.
In summary, PD, PDD, DLB, and MSA are a result of functional connectome deficits induced by oligomeric α-synuclein at the synapse. Despite the clear evidence implicating synapse degeneration as an early event in synucleinopathy, there are currently no biomarkers of synapse degeneration that are established in the clinical routine. In this paper, we will first outline the need for synaptic biomarkers in synucleinopathies and discuss the use of α-synuclein species as synaptic biomarkers. We then review the current literature on biofluid markers of synapse degeneration in clinical cohorts of PD, DLB, and MSA patients, with a particular emphasis on placing the findings in the context the pathophysiological mechanisms of synapse degeneration that have been associated with α-synuclein. We will also discuss their potential added value in aiding the diagnosis of synucleinopathy-related diseases.
Need for synaptic biomarkers in synucleinopathies
Cognitive and motor deficits are currently measured by scalar systems that are limited in their sensitivity and can be subjective. As the neuropathological correlate of underlying connectome deficits, synapse density is a good predictor of the rate of cognitive decline in synucleinopathies (Bereczki et al. 2016). Therefore, a marker of synapse degeneration could represent a much needed objective, surrogate biomarker of the core clinical features of these diseases.
There is also a need for a synaptic biomarker in the clinical trial setting. There are currently no therapeutic interventions that can slow down or stop diseases associated with synucleinopathy. Although several clinical trials are on-going, such treatments could be futile if delivered after irreversible neuronal damage has already taken hold. Successful future clinical trials require robust and specific biomarkers that can detect and monitor early disease pathogenesis, such as synapse degeneration.
To address this need, both neuroimaging and biofluid markers of synapse degeneration have been investigated. It is important to note, however, that while the neuroimaging SV2A PET tracer informs on the synaptic density at the point of analysis, it does not necessarily indicate an active degeneration of synapses. Quantification of synaptic proteins in the cerebrospinal fluid (CSF), on the other hand, may reflect the active clearance (or lack of, due to synaptic internalization or aggregation in the parenchyma) of synaptic proteins from the brain interstitia. Consequently, both neuroimaging and biofluid markers can provide complementary information as to the cumulative and active effects of a toxic (or therapeutic) agent on the synapse, respectively. As the costs for wide implementation of neuroimaging tracers are high and the use of radioactive tracers precludes PET use in certain patients/centres, biofluid markers have a wider clinical potential.
As synapse degeneration is common to all neurodegenerative diseases, a synaptic biomarker may not be specific to any one disease. This should not be seen as a limitation of synaptic biomarkers. After all, this is also the case for biomarkers of neurodegeneration. Alternatively, a synaptic biomarker may reflect dysfunction of a specific mechanism or degeneration of a specific synaptic subpopulation and therefore may be better suited to a specific disease or neuropathological subtype. For this reason, here, we first focus on understanding the candidate synaptic biomarkers in terms of the biological mechanisms associated with these proteins and their source synaptic populations and later will discuss their utility in aiding clinical and differential diagnoses.
α-Synuclein species as biofluid markers of synapse degeneration in synucleinopathies
Both normal and pathological α-synuclein species play an important role at the synapse. As such, α-synuclein species could be considered a candidate biomarker of synapse degeneration. Different α-synuclein species (total and aggregated) have been studied in the CSF of PD and DLB patients. The majority of studies report a decrease in total α-synuclein concentration in the CSF of PD and DLB patients compared to healthy controls (Eusebi et al. 2017; Sako et al. 2014; Gao et al. 2015; Zhou et al. 2015; van Steenoven et al. 2018) with considerable overlap in CSF concentrations between PD and other synucleinopathies (Wennstrom et al. 2013; Backstrom et al. 2015; Magdalinou et al. 2015). Longitudinal studies found no change in CSF α-synuclein over 12 months in PD patients (Mollenhauer et al. 2016, 2017). The aggregated form of α-synuclein has been detected in the CSF of PD and DLB patients by real-time quaking-induced conversion (RT–QuIC) and protein misfolding cyclic amplification (PMCA) assays (Fairfoul et al. 2016; Shahnawaz et al. 2017; Quadalti et al. 2021). In contrast to total α-synuclein, several studies have shown that aggregated α-synuclein and phosphorylated α-synuclein are elevated in PD and DLB patients compared to controls (Eusebi et al. 2017; Zhou et al. 2015; van Steenoven et al. 2018). Initial studies reported that combination with other biomarkers was needed to show good diagnostic accuracy for PD, DLB, and MSA (Eusebi et al. 2017; Parnetti et al. 2014; Singer et al. 2020). More recently, studies are reporting a high sensitivity and specificity of CSF aggregated α-synuclein for PD and DLB, and to a lesser extent in MSA patients (Poggiolini et al. 2021; Rossi et al. 2021; Bongianni et al. 2019). In addition to its clinical utility, aggregated α-synuclein has been proposed as a promising biomarker to use in clinical trials that target α-synuclein pathology to stratify patients according to their α-synuclein aggregation profile (Brockmann et al. 2021).
These findings support alterations in CSF concentrations across synucleinopathies with good diagnostic accuracy, particularly in PD and DLB. While synuclein species may also prove to be useful surrogate markers of synapse degeneration in other neurodegenerative diseases (Oeckl et al. 2020), they may not be the most appropriate synaptic marker in patients with underling synucleinopathy where oligomerization, fibrillization, and sequestration of α-synuclein in the brain would impact on CSF concentrations. This may also be pertinent in clinical trials targeting synuclein pathology, where there is a need for an alternative surrogate measure of cognitive performance, independent of the drug target.
Biofluid markers of synapse degeneration in synucleinopathies
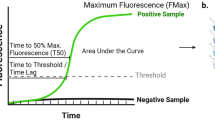
In this section, we review the literature on synaptic proteins, other than α-synuclein, in biofluids of patients diagnosed with any of the major synucleinopathies, namely PD, PDD, DLB, and MSA. We searched for studies in CSF and blood (plasma and serum). For the purposes of this review, we defined a protein as synaptic if it is included in the expert curated evidence-based database of synapse function and gene enrichment studies (Koopmans et al. 2019). We have only included biomarker studies that used quantitative methods (e.g., quantitative mass spectrometry or immunoassay) and, to improve diagnostic certainty, we only included that studies that used consensus diagnostic criteria or where neuropathological evaluation confirmed the diagnosis. We found 1 study reporting free concentrations of synaptic proteins in serum, 1 study of extracellular vesicles (EVs) in serum, and 17 studies in CSF. Table 1 summarizes the sample sizes and diagnostic criteria for each study as well as the principal findings for each synaptic protein including correlation to cognitive performance and motor scales. The functions of these synaptic proteins can all be related to mechanisms that are known to be associated with synuclein pathology, which provided a framework within which to discuss the findings. We first introduce the mechanism of synapse degeneration and its relationship to α-synuclein pathology and subsequently discuss the published findings of the candidate synaptic biomarker associated with this mechanism. A schematic representation of the mechanisms and associated candidate biomarker profiles in CSF is shown in the Fig. 1A–E. The numbers in parentheses in the text refer to corresponding process represented in Fig. 1.

Schematic representation of mechanisms related to synuclein-mediated synapse degeneration (A–E) and published findings of related biofluid candidate biomarkers. LB lewy body, DA dopamine, βSYN β-synuclein, NT neurotransmitter, DYN dynorphins, CaM calmodulin. Each mechanism of dysfunction is shown in a different colour, marked A–E. Explanation of each process can be found in the text. Created with BioRender.com
Disturbances in vesicular-mediated neurotransmitter transport and secretion (Fig. 1A)
One of the normal physiological functions of α-synuclein is in vesicle-mediated neurotransmitter transport and secretion for which soluble n-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) are fundamental to exocytosis and neurotransmitter release via synaptic vesicle recycling (Sauvola and Littleton 2021). In vitro studies have demonstrated that α-synuclein can bind directly to the SNARE protein, VAMP-2, forming a bridge between the membrane and synaptic vesicle, which facilitates SNARE complex formation and stabilizes the docked vesicle, thus allowing the exocytosis of neurotransmitters (Lou et al. 2017). An interaction between α-synuclein and the actin cytoskeleton further regulates vesicle-mediated trafficking and vesicle pool organisation (Jeannotte and Sidhu 2008; Rust and Maritzen 2015).
Human and animal studies show that disruptions to vesicle-mediated trafficking and secretory pathways are a key feature of synucleinopathy, with downstream effects on neurotransmitter signalling. For example, neuropathological evaluation of PDD and DLB patient brains post-mortem has revealed reduced neuropeptide levels as well as reduced expression of SNARE proteins, the neurosecretory peptide VGF, and proteins such as dynamin-1 (DNM1), that are critical for vesicle fission and control SNARE formation (1) (Alpadi et al. 2013; Anantharam et al. 2012; Fernandez et al. 1996; Bereczki et al. 2016) (Cocco et al. 2010; Vallortigara et al. 2014). The implication of this latter finding to disease pathogenesis is highlighted by the correlation between DNM1 expression post-mortem with the rate of global cognitive decline in DLB and PDD patients (Vallortigara et al. 2014). As SNARE proteins are abundant in Lewy bodies and glial cytoplasmic inclusions isolated from DLB and MSA patients (McCormack et al. 2019), it has even been proposed that vesicle-mediated transport may serve to selectively target aggregated α-synuclein to the Lewy body inclusions (2). These studies are supported by PD mouse models, which show defects in synaptic vesicle recycling and redistribution of SNARE proteins in the striatum (Garcia-Reitbock et al. 2010; Nemani et al. 2010). Evidence that these defects are related to alterations in dopamine neurotransmission comes from the finding that levodopa treatment in these models reversed the decrease in concentration of peptide neurotransmitters, such as dynorphin (Broderick et al. 2017).
These disruptions to neurotransmitter secretion maybe related to, or further exacerbated by, the disruption of actin cytoskeleton dynamics mediated by pathological α-synuclein (3) that has been observed in cells lines expressing α-synuclein harbouring mutations associated with familial PD (Sousa et al. 2009; Prots et al. 2018).
On the other hand, proteins such as β-synuclein and growth-associated protein-43 (GAP-43) may protect neurons against neurotoxicity by regulating and potentiating dopamine release at synaptic vesicles. For example, mouse models have shown that β-synuclein (4) can ameliorate motor deficits by inhibiting α-synuclein aggregation (Hashimoto et al. 2001) and potentiating vesicular monoamine transporter-2–dependent uptake of dopamine by synaptic vesicles (Ninkina et al. 2021). Whether alterations in β-synuclein expression and function are directly implicated in synucleinopathies has yet to be fully determined, but reduced β-synuclein expression has been reported across the cortex in DLB patients post-mortem with neuropathological confirmation of Lewy body pathology (Beyer et al. 2010) and a genetic study has reported two amino acid alterations in the β-synuclein gene of DLB patients. GAP-43 (5), a protein that regulates actin dynamics and induces axonal sprouting (He et al. 1997), can be sorted onto vesicles and transported down the axon by fast axonal transport to the growth cone or presynaptic membrane (Gonzalo and Linder 1998) where it has been implicated in dopamine release (Denny 2006; Dekker et al. 1989). Phosphorylation of GAP-43 by protein kinase C (PKC) facilitates synaptic vesicle recycling and neurotransmitter release via a direct interaction with vesicle proteins, SNAP-25, and synaptophysin (Holahan 2017). However, GAP-43 was nominally reduced in the brain of PDD patients post-mortem compared to controls (Bereczki et al. 2018), suggesting that altered GAP-43 expression may be a feature of synucleinopathy.
SNARE proteins
The principal components of the SNARE complex are synaptosomal-associated protein 25 (SNAP-25), the syntaxins 1A and 1B, syntaxin-binding protein-1, and the vesicle-associated membrane proteins (VAMP-1, VAMP-2) (Sevlever et al. 2015). Despite the growing evidence implicating vesicle-mediated transport in synuclein-mediated degeneration, a few studies have investigated the biofluid profile of the SNAREs in synucleinopathy-related diseases. Low concentrations of syntaxin-1A and VAMP-2, but not SNAP-25, have been reported in EVs isolated from the serum of PD patients compared to healthy controls (Fig. 1A, Table 1 Ai), thus supporting the idea that vesicle-mediated trafficking is impaired in PD (Agliardi et al. 2021). However, as shown in Table 1 Ai, the authors did not find any correlation between concentration of syntaxin-1A, VAMP-2, or SNAP-25 with age-at-onset, disease duration, tremor-dominant manifestation, postural instability/gait difficulty, levodopa dose, or cognitive score. Thus, a relationship between these potential biomarkers and cognitive and motor symptoms has yet to be established.
To our knowledge, CSF concentrations of VAMP and syntaxin proteins have yet to be investigated in synucleinopathies. On the other hand, one study has reported higher CSF SNAP-25 concentration in PD patients compared to controls (Fig. 1A, Table 1 Aiii), particularly in patients treated with levodopa (Bereczki et al. 2017). It is unclear whether this is due to the medication itself or whether it reflects the later disease stage of the treated compared to the untreated group. CSF SNAP-25 did not correlate with cognitive score or disease progression (Table 1 Aiii). The authors proposed that the increase in SNAP-25 in PD CSF reflects the continuous leakage of proteins into the brain interstitial fluid and subsequent clearance into the CSF due to on-going synapse degeneration.
β-Synuclein
β-Synuclein has been quantified in both serum (Table 1 Aii) and CSF (Fig. 1A, Table 1 Aiii) by targeted mass spectrometry (Oeckl et al. 2016, 2020). In both fluids, β-synuclein concentrations were comparable to controls in PD, and a pooled set of PDD/DLB patients. The same authors have since developed an ELISA assay for β-synuclein, which was used to successfully replicate the findings in CSF (Halbgebauer et al. 2020). In contrast to the synucleinopathies, β-synuclein may have utility as a synaptic biomarker in Alzheimer’s disease and Creutzfeldt–Jakob disease (Oeckl et al. 2016, 2020). These findings suggest that biofluid concentrations of β-synuclein may reflect amyloid rather than synuclein-mediated synaptotoxicity.
GAP-43
A few studies have investigated GAP-43 as a candidate biomarker in synucleinopathies and results have been conflicting (Fig. 1A, Table 1 Aiii). One study reported specifically decreased CSF concentrations of GAP-43 in PD patients compared to controls, a reduction that was not apparent in other neurodegenerative diseases (Sjogren et al. 2000) and was not replicated in two subsequent studies (Remnestal et al. 2016; Sandelius et al. 2019). However, one of the latter studies did report a weak negative correlation between CSF GAP-43 concentration and disease severity in the PD group (Table 1 Aiii), suggesting that low CSF GAP-43 concentrations may be a feature of advanced PD (Remnestal et al. 2016). Those same studies also reported no change in CSF GAP-43 concentration between DLB patients and controls. Studies showing elevated CSF GAP-43 concentrations in Alzheimer’s disease patients compared to controls and that CSF GAP-43 concentrations correlated better with Alzheimer pathological burden than with TDP-43 or α-synuclein burden post-mortem suggest the possibility that CSF GAP43 concentrations better reflect Alzheimer-related rather than synuclein-related changes (Remnestal et al. 2016; Sandelius et al. 2019).
Secretory vesicle proteins
A study of CSF DNM1 concentrations in DLB and PDD patients (Fig. 1A, Table 1 Aiii) reported elevated DNM1 compared to subjects with subjective cognitive decline, but concentrations did not correlate with global cognition (Enache et al. 2020). While these results are promising, validation of these findings in independent cohorts is needed.
Secretory granule proteins such as neurosecretory peptide VGF and secretogranin-2 (SCG2) are exclusively expressed in neuronal and neuroendocrine and endocrine cells. A proteomic study of the CSF (Fig. 1A, Table 1 Aiii) revealed that PD and DLB patients as well as patients with other neurodegenerative diseases had significantly lower CSF concentration of VGF and SCG2 compared to controls (van Steenoven et al. 2019, 2020). The finding in DLB was validated in an independent cohort in the same study. Lower CSF concentrations of both proteins were associated with worse global cognitive performance and were lower in DLB compared to all other diseases, including PD (Table 1 Aiii). In summary, preliminary studies suggest that low CSF concentrations of these secretory proteins may be a common feature of neurodegeneration. The potential of these proteins as objective markers of cognitive performance in DLB is promising, but requires follow-up in further independent studies. The potential of secretory proteins as surrogate markers of cognitive decline in synucleinopathies other than DLB and PD also warrants further investigation.
Prodynorphin
One study has reported reduced CSF concentrations of the dense core vesicle protein PDYN in two populations of DLB patients and one population of PD patients compared to healthy controls (Fig. 1A, Table 1 Aiii), albeit that PDYN concentrations were lower in DLB patients compared to PD and other neurodegenerative diseases, suggesting a degree of specificity for DLB, the reason for which is unclear (van Steenoven et al. 2020). They also reported that CSF PDYN concentrations were associated with global cognitive performance in DLB and controls combined (Table 1 Aiii). PDYN is processed at the presynapse to dynorphin opioid peptides in response to potassium-induced depolarization of the neuron (Yakovleva et al. 2006) and is expressed in hypocretin/orexin hypothalamic neurons, which are affected in PD and may be implicated in the sleep disturbances that are a common feature of synucleinopathies (Crocker et al. 2005). Whether this alteration in CSF concentration of PDYN reflects a loss of hypocretin/orexin hypothalamic neurons is an interesting avenue worth pursuing.
Deficits in synaptic plasticity (Fig. 1B)
Another reported mechanism by which α-synuclein oligomers may induce synaptotoxicity is by impairing long-term potentiation (LTP). Specifically, α-synuclein oligomers can reduce N-methyl-d-aspartate (NMDA) receptor-mediated synaptic currents producing deficits in visuospatial learning mice (Durante et al. 2019). The kinase activity of protein kinase C (PKC) is necessary for LTP (Abeliovich et al. 1993). Two important substrates of PKC include the presynaptic growth-associated protein-43 (GAP-43) (5) and the postsynaptic protein, neurogranin (Ng). In addition to GAP-43 (previously discussed), there is accumulating evidence directly implicating Ng dysfunction in synucleinopathy (6). For example, a direct interaction between Ng and α-synuclein has been reported in human brain extracts from the temporal cortex of healthy controls, an interaction that was reduced in PD but not DLB patients, while both PD and DLB patients showed decreased levels of phosphorylated Ng in the temporal cortex compared to controls (Koob et al. 2014).
Neurogranin
Several studies have evaluated CSF Ng as a potential surrogate marker of synapse dysfunction in synucleinopathy-related diseases but with somewhat conflicting results (Fig. 1B, Table 1 B). On one hand, increased CSF Ng concentration has been reported in drug naïve PD patients compared to healthy controls, correlating with both cognitive impairment and motor disease stage (Bereczki et al. 2017). Even more promising, the same study showed that PD patients on dopaminergic therapies showed statistically comparable CSF Ng concentration to controls. On the other hand, another study has reported differences in CSF Ng in the opposite direction; namely, reduced CSF Ng concentration in PD patients compared to controls, correlating with CSF α-synuclein levels, cortical glucose metabolism, and motor stage (Selnes et al. 2017). This was partially supported by another study, which reported lower CSF Ng concentration in patients diagnosed with PD, PDD, and MSA, but not DLB, compared to controls, correlating with markers of tau-mediated and axonal neurodegeneration but not with cognitive dysfunction, motor impairment, or subsequent development of dementia (Hall et al. 2020). Further studies reported no change in CSF Ng concentration between MSA, PD, PDD, or LBD patients and controls (Portelius et al. 2018; Wellington et al. 2016; Janelidze et al. 2016; Remnestal et al. 2016). Thus, the relationship of CSF Ng to clinical diagnosis of synucleinopathy-related diseases is unclear. Studies determining the relationship between CSF Ng and synuclein pathology could shed further light on this matter, but unfortunately are scarce. One study has reported a lack of association between antemortem CSF Ng concentration and post-mortem α-synuclein burden in the amygdala or hippocampus of autopsy-confirmed patients with DLB (Portelius et al. 2018). No data relating to post-mortem α-synuclein burden were available for PD, PDD, or MSA patients.
When interpreting data on CSF Ng, it is important to consider the abundance of studies showing a strong correlation (r values ranging from 0.5 to 0.9) of CSF Ng with markers of tau-mediated neurodegeneration that is not restricted to Alzheimer’s disease and the tauopathies but is also apparent in synucleinopathies such as PD (with and without dementia) and DLB (Portelius et al. 2018; Hall et al. 2020). This raises the possibility that tau comorbidity (likely, Alzheimer-related), which is common in PD, PDD, DLB, and MSA, may be a confounding factor that could explain the conflicting findings for CSF Ng concentrations reported in synucleinopathies. If so, CSF Ng may reflect different processes of synapse degeneration depending on the underlying tau morbidity and could explain the conflicting findings in the literature. Further studies that take into account tau comorbidity in synucleinopathies are needed to fully determine the clinical potential of CSF Ng in synucleinopathy-related diseases. To our knowledge, Ng has yet to be evaluated in plasma or serum from patients with underlying synucleinopathy. However, as plasma Ng concentrations were detectable but unchanged in Alzheimer’s disease patients, Ng shows little promise as a blood-based biomarker (De Vos et al. 2015).
Deficits in synapse adhesion (Fig. 1C)
The presence of synapse adhesion molecules, such as the contactins (7), in nigral and cortical Lewy bodies in PD patients post-mortem (Chatterjee et al. 2020), suggests that they may play a role in synucleinopathy-mediated degeneration. Synapse adhesion molecules form trans-complexes that span the synaptic cleft (Rudenko 2017) and their expression is regulated by synaptic activity to dynamically scale up or scale down synaptic communication and regulate synaptic plasticity (Fu and Huang 2010; Friedman et al. 2015; Pierre et al. 2001; Tai et al. 2007; Yamada et al. 2007). Contactin-1 (CNTN-1) has a crucial function in regulating synaptic plasticity by promoting neurogenesis in the hippocampus (Puzzo et al. 2013) and cerebellar interneurons (Berglund et al. 1999). Contactin-2 (CNTN-2) regulates axon guidance and synaptogenesis (Stoeckli 2010). Sequestration of synapse adhesion molecules in Lewy bodies (7) could therefore affect synapse development and synaptic plasticity, although this has yet to be demonstrated experimentally.
Contactins
Lower CSF CNTN-1 concentrations (Fig. 1A, Table 1 Aii) have been reported in PD patients compared to controls, correlating with CSF α-synuclein and markers of tau-mediated neurodegeneration but not disease stage or duration or global cognitive performance (Chatterjee et al. 2020). The same study showed no statistical difference in CSF CNTN-1 concentration between DLB patients and controls. To explain the apparent specificity of CSF CNTN-1 for PD over DLB, the authors proposed that as contactins are absent or reduced in and around amyloid plaques in Alzheimer’s disease brain tissue (Chatterjee et al. 2018), that CNTN-1 sequestration in Lewy bodies may be less prevalent in DLB patients due to more abundant Alzheimer pathology in DLB compared to PD. The idea that CSF CNTN-1 concentrations may be influenced by Alzheimer pathology is supported by the positive correlation between CSF CNTN-1 and CSF markers of tau-mediated neurodegeneration in the DLB patients (r = 0.58) and controls (r = 0.67). CSF concentrations of CNTN-2 were nominally elevated but statistically comparable to controls in PD and DLB patients (Chatterjee et al. 2020). In summary, the finding of low CSF CNTN-1 concentrations in PD patients is promising, but the lack of association with cognitive performance or motor scales may limit the utility as a marker of synuclein-mediated synapse degeneration.
Excitatory/inhibitory homeostatic imbalance (Fig. 1D)
α-Synuclein expression in the adult mouse brain is found predominantly at excitatory synapses (Zhang et al. 2019) but is also expressed at inhibitory synapses, particularly in regions most vulnerable to synuclein pathology, such as the olfactory bulb, medulla oblongata, pons, substantia nigra, amygdala, and piriform cortex (Taguchi et al. 2016). In PD patients, both elevated expression of glutamate receptors (NMDA, AMPA, kainite, and metabotropic GluR5) and a loss of GABA-ergic interneurons have been reported (8) (Zhang et al. 2019; Khundakar et al. 2016). These findings suggest that over excitation and/or a loss of inhibition are also a feature in synucleinopathy. The importance of regulating excitatory transmission to PD pathogenesis is evident in that anti-PD therapies based on negative allosteric modulation of glutamate receptors improve PD motor symptoms and reduce levodopa-induced dyskinesia (Zhang et al. 2019). Meanwhile, the loss of GABA-ergic interneurons in the visual cortex could explain the recurrent visual hallucinations that are a clinical feature of synucleinopathies (Khundakar et al. 2016). Neuronal pentraxin-2 (NPTX2) is critical to regulating excitatory-inhibitory circuit function (8). Specifically, NPTX2 is recruited to the presynapse by the neuronal pentraxin receptor (NPTXR) where increased neural activity stimulates NPTX2 secretion (Lee et al. 2017; Kirkpatrick et al. 2000). NPTX2 then recruits and clusters AMPA receptors at the post-synapse via direct binding to the GluA4 subunit, thus strengthening the excitatory synapse onto GABA-ergic parvalbumin interneurons (Tsui et al. 1996; O'Brien et al. 1999; Xu et al. 2003). Whole-genome expression profiling of the substantia nigra of PD patients and controls revealed NPTX2 as the most highly upregulated gene in PD patients (Moran et al. 2008). Moreover, the same study showed that NPTX2 expression was present in a subset of Lewy bodies in the substantia nigra and cerebral cortex of PD patients and appeared to form a bridge between Lewy bodies, thus implicating NPTX2 in inclusion growth. Whether NPTX2 expression is also upregulated in DLB and MSA patient brains has yet to be reported. Similar to PDYN, NPTX2 is expressed in hypocretin/orexin hypothalamic nerve cells that are known to be affected in PD and are implicated in the sleep disturbances that are a common feature of synucleinopathies (Crocker et al. 2005). Taken together, disruption of the excitatory/inhibitory homeostatic balance represents another potential mechanism relating pathological α-synuclein to synaptic dysfunction.
Neuronal pentraxins
Studies in CSF (Fig. 1D, Table 1 D) have reported lower CSF NPTX2 and NPTXR concentrations in several cohorts of DLB patients compared to healthy controls, with CSF NPTX2 concentration correlating with global cognitive decline and decline in the visual spatial domain (Boiten et al. 2020; van Steenoven et al. 2020). Although low CSF NPTX2 and NPTXR concentrations were also reported in PD patients, they were significantly lower in DLB compared to PD patients (van Steenoven et al. 2020). In the cognitively normal aged population, low CSF NPTX2 concentrations were associated with less functional connectivity in the salience/ventral attention networks, an association that was stronger among individuals with lower levels of cognitive reserve (Soldan et al. 2019). This may be relevant to DLB where dysfunction in salience and attentional networks have been reported (Sourty et al. 2016; Lowther et al. 2014). The observation that low CSF NPTX2 concentration in DLB patients correlated with low CSF α-synuclein and VGF concentrations (Boiten et al. 2020) is consistent with evidence that NPTX2 expression may be regulated by the VGF/BDNF pathway (Beckmann et al. 2020). Potential mechanisms to explain the low CSF NPTX2 concentrations include a loss of glutamatergic synapses onto GABA-ergic interneurons or, as suggested in post-mortem studies of PD patients, that NPTX2 may accumulate in the brain in Lewy bodies. Taken together, these studies support CSF NPTX2 as a promising biomarker of excitatory/inhibitory homeostatic balance in DLB and PD that requires validation in independent cohorts and is worthy of further investigation in other synucleinopathies. Of particular interest could be further study of the relationship to functional changes in the visuospatial domain in neuroimaging cohorts.
Glutamate receptors
To our knowledge, GluA3 is the only glutamate receptor to be investigated in patient-derived biofluids of the synucleinopathies (Fig. 1D, Table 1 D). CSF concentrations of GluA3 were comparable between PDD and DLB patients and subjects with subjective cognitive impairment (Enache et al. 2020). Studies in other synucleinopathies are lacking.
Altered metal ion homeostasis (Fig. 1E)
Disturbance to metal ion homeostasis represents another potential mechanism involved in α-synuclein-mediated synapse degeneration. α-Synuclein has an exceptionally high binding capacity for Ca2+ and an imbalance in calcium or α-synuclein can cause synaptic vesicle clustering in vitro (Lautenschlager et al. 2018). The same study showed that lowering either the levels of α-synuclein or calcium prevented dopamine toxicity. Other metal ions, particularly, Cu2+ and Zn2+, induce self-oligomerization of α-synuclein in vitro (Paik et al. 1999) (9). Long-term exposure to copper in the workplace is a risk factor for developing PD due to its detrimental effect on dopamine metabolism, oxidative stress, and α-synuclein oligomerization (Bisaglia and Bubacco 2020). Similarly, zinc exposure is an environmental risk factor for PD and zinc deposition has been reported in the substantia nigra and striatum of PD patients post-mortem (Sikora and Ouagazzal 2021). In rodents, elevated zinc causes degeneration of the nigrostriatal dopaminergic pathway and locomotor deficits (Sikora and Ouagazzal 2021). Zinc homeostasis is also critical to synaptic plasticity, which, as discussed above, is impaired by α-synuclein oligomers. The zinc transporter (ZnT3) (10), which is specifically expressed within Zn2+-containing synaptic glutamatergic, GABA-ergic and dopaminergic neurons of the hippocampus, cortex, olfactory bulb, and spinal cord (Ruiz et al. 2004; Wang et al. 2002), is essential for the correct sequestration of Zn2+ ions and subsequent release into the synaptic cleft (Lee et al. 2011; Palmiter et al. 1996). The binding of Zn2+ ions to the NMDA receptor can potentiate allosteric inhibition of NMDA receptor currents in low extracellular Zn2+concentrations and act as a pore blocker of NMDA receptors at high concentrations (Sensi et al. 2009; Paoletti et al. 1997) (10). There is also evidence that zinc homeostasis may modulate GABA-ergic synaptic transmission (10). ZnT3 knockout mice show diminished GABAA-mediated inhibitory postsynaptic potentials and are resistant to dopamine-induced locomotor deficits and memory impairment (Lopantsev et al. 2003; Sikora and Ouagazzal 2021). Taken together, synaptically released Zn2+ from corticostriatal terminals could underlie the motor and cognitive deficits associated with PD and could be related to α-synuclein-mediated effects on synaptic plasticity (Sikora and Ouagazzal 2021).
Ion transporters
A study of CSF concentrations of ZnT3 in DLB and PDD patients (Fig. 1E, Table 1 E) reported significantly elevated ZnT3 concentrations in DLB (and a trend towards elevated ZnT3 in PDD) compared to subjects with subjective cognitive decline (Enache et al. 2020). CSF ZnT3 concentrations did not correlate with global cognition in either DLB or PDD. The utility of CSF ZnT3 as a biomarker of zinc dyshomeostasis in DLB and PDD requires further investigation.
Synaptic proteins as diagnostic fluid biomarkers in syuncleinopathies
Although not a necessary requirement of a surrogate marker of synapse degeneration CSF and blood concentrations of synaptic proteins could potentially aid in the clinical and differential diagnoses of synucleinopathy-related diseases. Table 2 shows the diagnostic accuracies for the synaptic proteins included in this review (either alone or in combination), where reported. In terms of clinical diagnosis (Table 2 a), the highest reported accuracy for a model including synaptic proteins in PD was the oligomeric α-synuclein/VAMP-2 ratio in serum EVs (Agliardi et al. 2021), which gave an area-under-the-curve (AUC) of 0.88 and performed nominally better than serum EV oligomeric α-synuclein alone (AUC = 0.82). CSF biomarkers performed worse than serum EV markers (all AUC < 0.73). A few synaptic proteins have been evaluated in PDD and all showed poor diagnostic accuracy (all AUC < 0.68). In DLB, a combination of CSF Ng, NPTX2, total α-synuclein, and age gave an AUC of 0.94 (Boiten et al. 2020), surpassing any biomarker alone. If validated in other cohorts, this level of accuracy suggests that this model could be used as an additional diagnostic maker for DLB. However, it may be difficult to incorporate into the clinical setting due to the number of analytes needed. We found no studies reporting on diagnostic accuracy of synaptic markers in MSA.
A biomarker that can aid in the differential diagnosis of distinct synucleinopathies and parkinsonian disorders (Table 2b) would also be a welcome addition to the biomarker arsenal. Several studies have tested the performance of various combinations of synaptic biomarkers to distinguish PD from DLB. The best reported model was a combination CNTN-1, total α-synuclein, total tau, phosphorylated tau, and Aβ1-42, which gave an AUC of 0.88 (Chatterjee et al. 2020). Again, the complexity of this combined model may be difficult to implement, but is worthy of further investigation in independent cohorts. The performance of CSF Ng to distinguish PD/PDD and atypical parkinsonian disorders was also reported but performed poorly (AUC = 0.56).
The clinical manifestations of synucleinopathy-related diseases, particularly DLB, can overlap with other neurodegenerative disorders, thus further complicating diagnosis. Thus, a biomarker that can aid the differential diagnosis from other neurodegenerative diseases (Table 2c) would also be useful. Most of the studies included in this review, focused on the differential diagnosis between DLB and AD or DLB/PDD and AD, for which the best reported model was for a combination of VGF, SCG2, and PDYN, with an AUC of 0.89.
In summary, these studies suggest that synaptic proteins, particularly when used in combination with synuclein species, can provide added value as diagnostic biomarkers.
What have we learned from studies of synaptic biomarkers in synucleinopathies?
We have performed a comprehensive review of candidate biomarkers of synapse degeneration in PD, DLB, and MSA patient-derived CSF (14 proteins), serum (1 protein), and serum extracellular vesicles (3 proteins), and placed these findings in the context of the mechanisms associated with α-synuclein-mediated synapse degeneration. We also have discussed their diagnostic accuracy. Based on the findings discussed in this review, we conclude the following:
-
1.
The data provided by neuroimaging tracers provide further evidence that synapse degeneration is a major event in synucleinopathy. Neuroimaging and biofluid markers may provide complementary information as to the cumulative and active effects of a toxic (or therapeutic) agent on the synapse, respectively.
-
2.
Synuclein species may have greater utility as diagnostic or prognostic markers rather than as surrogate markers of synapse degeneration in patients with underlying synucleinopathy.
-
3.
Despite the evidence for synapse degeneration as a driving factor in disease pathogenesis, the synucleinopathies have received relatively less attention than other neurodegenerative diseases. This is particularly true for MSA. For comparison, over 31 candidate CSF synaptic markers have been investigated in over 50 studies of Alzheimer’s disease patients to-date. Of all the markers included in this review, only Ng has been evaluated in MSA patient CSF and the results were not replicated across the two studies, so few conclusions can be taken as to the utility of these synaptic markers in MSA patients. This relative lack of attention towards MSA compared to the more common synucleinopathies likely reflects the relative scarcity of MSA cohorts, something that will hopefully be addressed as the diagnostic accuracy of MSA improves (Miki et al. 2019). The predominant deposition of α-synuclein in GCI as opposed to Lewy bodies could mean that the mechanisms underlying synapse degeneration in MSA are different to those in PD and DLB thus further highlighting the need for studies in this relatively neglected synucleinopathy.
-
4.
In CSF, the most promising candidate synaptic biomarkers are related to neurotransmitter transport and secretion and excitatory/inhibitory dyshomeostasis. Within the vesicle-mediated transport/secretory pathway, the most promising findings were for VGF, SCG2, and PDYN (which were all reduced in all reported DLB and PD cohorts). CSF concentrations of all three proteins correlated with measures of global cognition. One caveat of this finding is that the analyses were performed in the total dataset, rather than stratified by clinical diagnoses, and as such may merely reflect the stratification of cognitive performance between the healthy controls and the disease groups. As neurotransmitter secretion is common to all synapse populations, these CSF changes likely reflect global synapse loss in affected regions. Data relating to the pentraxins are also promising. Both pentraxins were consistently reduced in multiple DLB cohorts and in one PD cohort and both proteins were associated with cognitive performance. Particularly relevant is the association for NPTX2, which was observed in the DLB patients rather in the pooled dataset and which was specific to the visual domain. NPTX2 forms synapses onto parvalbumin GABA interneurons, a loss of which has been related to visual hallucinations in PD. Whether low CSF NPTX2 in DLB CSF reflects a loss of GABA synapses in the visual cortex is an interesting avenue worth pursuing in neuroimaging cohorts. Although these findings need replication in further independent cohorts, they provide further support to the data implicating dysfunction in the transport and secretion of neurotransmitters and for excitatory/inhibitory dyshomeostasis in synucleinopathies (PD and DLB).
-
5.
Alzheimer comorbidity in synucleinopathies may confound interpretation of some synaptic markers. The reported findings for proteins related to synaptic plasticity (Ng and GAP-43) were inconsistent across studies. Neither protein was altered in DLB CSF compared to controls in any of the 7 cohorts, while in PD and MSA, conflicting results were reported across studies of similar sample size. Nevertheless, elevated CSF concentrations of Ng and SNAP-25 recapitulate findings reported for these biomarkers in Alzheimer’s disease CSF and may be explained by the strong correlation of these proteins with CSF markers of tau-mediated neurodegeneration, a hallmark of Alzheimer’s disease, and a common feature in synucleinopathies. We found similar inconsistencies in reported findings relating to proteins involved in synapse adhesion (contactins), which also correlated with CSF markers of tau-mediated neurodegeneration. Future studies of Ng, SNAP-25, and CNTN-1 that take underlying Alzheimer’s disease comorbidity into account (e.g., using the CSF phosphorylated tau / Aβ42 ratio) could be particularly valuable for minimizing neuropathologic heterogeneity across studies.
-
6.
All biofluid studies discussed here report findings in patients with full-blown disease, when neurodegeneration is likely to have already taken hold. As discussed above, the elevated clearance of neuronal proteins into the CSF due to neuronal degeneration may mask more subtle changes in synaptic proteins attributed to synapse degeneration. The recently published research criteria for prodromal PD (Heinzel et al. 2019) and prodromal DLB (McKeith et al. 2020) may pave the way for future studies in subjects at the prodromal disease stage, which may shed further light on synapse degeneration in earlier disease stages.
-
7.
The only study published to-date reporting free concentrations of a synaptic protein in blood was a study of β-synuclein in serum, which reported negative findings. One study has reported low concentrations of SNARE proteins in serum EVs in PD patients, but the technical limitations of preparing EVs from patient biofluid limit their utility in the clinical setting. Thus, blood-based biomarkers of synapse degeneration in synucleinopathies remain elusive.
-
8.
Combination of synaptic proteins with existing diagnostic biomarkers can give provide good diagnostic accuracy for PD and DLB. More studies comparing CSF synaptic biomarkers across synucleinopathies are needed to fully explore their potential.
In summary, there are several promising candidate biomarkers of synapse dysfunction or degeneration in PD and DLB. The most promising candidates include proteins related to neurotransmitter transport and secretion (ubiquitous across synapses) or to a specific subpopulation of synapses that form excitatory inputs onto GABA-ergic interneurons.
Availability of data and materials
Not applicable.
References
Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S (1993) Modified hippocampal long-term potentiation in PKC gamma-mutant mice. Cell 75(7):1253–1262. https://doi.org/10.1016/0092-8674(93)90613-u
Agliardi C, Meloni M, Guerini FR, Zanzottera M, Bolognesi E, Baglio F, Clerici M (2021) Oligomeric alpha-Syn and SNARE complex proteins in peripheral extracellular vesicles of neural origin are biomarkers for Parkinson’s disease. Neurobiol Dis 148:105185. https://doi.org/10.1016/j.nbd.2020.105185
Alpadi K, Kulkarni A, Namjoshi S, Srinivasan S, Sippel KH, Ayscough K, Zieger M, Schmidt A, Mayer A, Evangelista M, Quiocho FA, Peters C (2013) Dynamin-SNARE interactions control trans-SNARE formation in intracellular membrane fusion. Nat Commun 4:1704. https://doi.org/10.1038/ncomms2724
Anantharam A, Axelrod D, Holz RW (2012) Real-time imaging of plasma membrane deformations reveals pre-fusion membrane curvature changes and a role for dynamin in the regulation of fusion pore expansion. J Neurochem 122(4):661–671. https://doi.org/10.1111/j.1471-4159.2012.07816.x
Andersen KB, Hansen AK, Damholdt MF, Horsager J, Skjaerbaek C, Gottrup H, Klit H, Schacht AC, Danielsen EH, Brooks DJ, Borghammer P (2021) Reduced synaptic density in patients with lewy body dementia: an [(11) C]UCB-J PET imaging study. Mov Disord 36(9):2057–2065. https://doi.org/10.1002/mds.28617
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 152(4):879–884
Backstrom DC, Eriksson Domellof M, Linder J, Olsson B, Ohrfelt A, Trupp M, Zetterberg H, Blennow K, Forsgren L (2015) Cerebrospinal fluid patterns and the risk of future dementia in early incident parkinson disease. JAMA Neurol 72(10):1175–1182. https://doi.org/10.1001/jamaneurol.2015.1449
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117(6):613–634. https://doi.org/10.1007/s00401-009-0538-8
Beckmann ND, Lin WJ, Wang M, Cohain AT, Charney AW, Wang P, Ma W, Wang YC, Jiang C, Audrain M, Comella PH, Fakira AK, Hariharan SP, Belbin GM, Girdhar K, Levey AI, Seyfried NT, Dammer EB, Duong D, Lah JJ, Haure-Mirande JV, Shackleton B, Fanutza T, Blitzer R, Kenny E, Zhu J, Haroutunian V, Katsel P, Gandy S, Tu Z, Ehrlich ME, Zhang B, Salton SR, Schadt EE (2020) Multiscale causal networks identify VGF as a key regulator of Alzheimer’s disease. Nat Commun 11(1):3942. https://doi.org/10.1038/s41467-020-17405-z
Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F, Pizzi M, Missale C, Spano P (2016) Review: Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol 42(1):77–94. https://doi.org/10.1111/nan.12297
Berardelli A, Wenning GK, Antonini A, Berg D, Bloem BR, Bonifati V, Brooks D, Burn DJ, Colosimo C, Fanciulli A, Ferreira J, Gasser T, Grandas F, Kanovsky P, Kostic V, Kulisevsky J, Oertel W, Poewe W, Reese JP, Relja M, Ruzicka E, Schrag A, Seppi K, Taba P, Vidailhet M (2013) EFNS/MDS-ES/ENS [corrected] recommendations for the diagnosis of Parkinson’s disease. Eur J Neurol 20(1):16–34. https://doi.org/10.1111/ene.12022
Bereczki E, Francis PT, Howlett D, Pereira JB, Hoglund K, Bogstedt A, Cedazo-Minguez A, Baek JH, Hortobagyi T, Attems J, Ballard C, Aarsland D (2016) Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimers Dement 12(11):1149–1158. https://doi.org/10.1016/j.jalz.2016.04.005
Bereczki E, Bogstedt A, Hoglund K, Tsitsi P, Brodin L, Ballard C, Svenningsson P, Aarsland D (2017) Synaptic proteins in CSF relate to Parkinson’s disease stage markers. NPJ Parkinsons Dis 3:7. https://doi.org/10.1038/s41531-017-0008-2
Bereczki E, Branca RM, Francis PT, Pereira JB, Baek JH, Hortobagyi T, Winblad B, Ballard C, Lehtio J, Aarsland D (2018) Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain 141(2):582–595. https://doi.org/10.1093/brain/awx352
Berglund EO, Murai KK, Fredette B, Sekerkova G, Marturano B, Weber L, Mugnaini E, Ranscht B (1999) Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron 24(3):739–750. https://doi.org/10.1016/s0896-6273(00)81126-5
Beyer K, Domingo-Sabat M, Santos C, Tolosa E, Ferrer I, Ariza A (2010) The decrease of beta-synuclein in cortical brain areas defines a molecular subgroup of dementia with Lewy bodies. Brain 133(Pt 12):3724–3733. https://doi.org/10.1093/brain/awq275
Bisaglia M, Bubacco L (2020) Copper ions and parkinson’s disease: why is homeostasis so relevant? Biomolecules. https://doi.org/10.3390/biom10020195
Boiten WA, van Steenoven I, Xiao M, Worley PF, Lemstra AW, Teunissen CE (2020) Pathologically decreased csf levels of synaptic marker NPTX2 in DLB are correlated with levels of alpha-synuclein and VGF. Cells. https://doi.org/10.3390/cells10010038
Bongianni M, Ladogana A, Capaldi S, Klotz S, Baiardi S, Cagnin A, Perra D, Fiorini M, Poleggi A, Legname G, Cattaruzza T, Janes F, Tabaton M, Ghetti B, Monaco S, Kovacs GG, Parchi P, Pocchiari M, Zanusso G (2019) alpha-Synuclein RT-QuIC assay in cerebrospinal fluid of patients with dementia with Lewy bodies. Ann Clin Transl Neurol 6(10):2120–2126. https://doi.org/10.1002/acn3.50897
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Brockmann K, Quadalti C, Lerche S, Rossi M, Wurster I, Baiardi S, Roeben B, Mammana A, Zimmermann M, Hauser AK, Deuschle C, Schulte C, Waniek K, Lachmann I, Sjodin S, Brinkmalm A, Blennow K, Zetterberg H, Gasser T, Parchi P (2021) Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun 9(1):175. https://doi.org/10.1186/s40478-021-01276-6
Broderick PA, Wenning L, Li YS (2017) Neuromolecular imaging, a nanobiotechnology for Parkinson’s disease: advancing pharmacotherapy for personalized medicine. J Neural Transm (vienna) 124(1):57–78. https://doi.org/10.1007/s00702-016-1633-3
Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329(5999):1663–1667. https://doi.org/10.1126/science.1195227
Calo L, Wegrzynowicz M, Santivanez-Perez J, Grazia Spillantini M (2016) Synaptic failure and alpha-synuclein. Mov Disord 31(2):169–177. https://doi.org/10.1002/mds.26479
Chatterjee M, Del Campo M, Morrema THJ, de Waal M, van der Flier WM, Hoozemans JJM, Teunissen CE (2018) Contactin-2, a synaptic and axonal protein, is reduced in cerebrospinal fluid and brain tissue in Alzheimer’s disease. Alzheimers Res Ther 10(1):52. https://doi.org/10.1186/s13195-018-0383-x
Chatterjee M, van Steenoven I, Huisman E, Oosterveld L, Berendse H, van der Flier WM, Del Campo M, Lemstra AW, van de Berg WDJ, Teunissen CE (2020) Contactin-1 is reduced in cerebrospinal fluid of Parkinson’s disease patients and is present within lewy bodies. Biomolecules. https://doi.org/10.3390/biom10081177
Cocco C, D’Amato F, Noli B, Ledda A, Brancia C, Bongioanni P, Ferri GL (2010) Distribution of VGF peptides in the human cortex and their selective changes in Parkinson’s and Alzheimer’s diseases. J Anat 217(6):683–693. https://doi.org/10.1111/j.1469-7580.2010.01309.x
Crocker A, Espana RA, Papadopoulou M, Saper CB, Faraco J, Sakurai T, Honda M, Mignot E, Scammell TE (2005) Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology 65(8):1184–1188. https://doi.org/10.1212/01.wnl.0000168173.71940.ab
De Vos A, Jacobs D, Struyfs H, Fransen E, Andersson K, Portelius E, Andreasson U, De Surgeloose D, Hernalsteen D, Sleegers K, Robberecht C, Van Broeckhoven C, Zetterberg H, Blennow K, Engelborghs S, Vanmechelen E (2015) C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement 11(12):1461–1469. https://doi.org/10.1016/j.jalz.2015.05.012
Dekker LV, De Graan PN, Versteeg DH, Oestreicher AB, Gispen WH (1989) Phosphorylation of B-50 (GAP43) is correlated with neurotransmitter release in rat hippocampal slices. J Neurochem 52(1):24–30. https://doi.org/10.1111/j.1471-4159.1989.tb10893.x
Delva A, Van Weehaeghe D, Koole M, Van Laere K, Vandenberghe W (2020) Loss of presynaptic terminal integrity in the substantia nigra in early Parkinson’s disease. Mov Disord 35(11):1977–1986. https://doi.org/10.1002/mds.28216
Denny JB (2006) Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr Neuropharmacol 4(4):293–304. https://doi.org/10.2174/157015906778520782
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8(12):1150–1157. https://doi.org/10.1016/S1474-4422(09)70238-8
Durante V, de Iure A, Loffredo V, Vaikath N, De Risi M, Paciotti S, Quiroga-Varela A, Chiasserini D, Mellone M, Mazzocchetti P, Calabrese V, Campanelli F, Mechelli A, Di Filippo M, Ghiglieri V, Picconi B, El-Agnaf OM, De Leonibus E, Gardoni F, Tozzi A, Calabresi P (2019) Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain 142(5):1365–1385. https://doi.org/10.1093/brain/awz065
Emre M (2003) Dementia associated with Parkinson’s disease. Lancet Neurol 2(4):229–237. https://doi.org/10.1016/s1474-4422(03)00351-x
Enache D, Pereira JB, Jelic V, Winblad B, Nilsson P, Aarsland D, Bereczki E (2020) Increased cerebrospinal fluid concentration of ZnT3 is associated with cognitive impairment in Alzheimer’s disease. J Alzheimers Dis 77(3):1143–1155. https://doi.org/10.3233/JAD-200498
Eusebi P, Giannandrea D, Biscetti L, Abraha I, Chiasserini D, Orso M, Calabresi P, Parnetti L (2017) Diagnostic utility of cerebrospinal fluid alpha-synuclein in Parkinson’s disease: a systematic review and meta-analysis. Mov Disord 32(10):1389–1400. https://doi.org/10.1002/mds.27110
Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, Joachim C, Esiri M, Evetts SG, Rolinski M, Baig F, Ruffmann C, Wade-Martins R, Hu MT, Parkkinen L, Green AJ (2016) Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 3(10):812–818. https://doi.org/10.1002/acn3.338ACN3338
Fernandez A, de Ceballos ML, Rose S, Jenner P, Marsden CD (1996) Alterations in peptide levels in Parkinson’s disease and incidental Lewy body disease. Brain 119(Pt 3):823–830. https://doi.org/10.1093/brain/119.3.823
Friedman LG, Benson DL, Huntley GW (2015) Cadherin-based transsynaptic networks in establishing and modifying neural connectivity. Curr Top Dev Biol 112:415–465. https://doi.org/10.1016/bs.ctdb.2014.11.025
Fu Y, Huang ZJ (2010) Differential dynamics and activity-dependent regulation of alpha- and beta-neurexins at developing GABAergic synapses. Proc Natl Acad Sci USA 107(52):22699–22704. https://doi.org/10.1073/pnas.1011233108
Fusco G, Sanz-Hernandez M, De Simone A (2018) Order and disorder in the physiological membrane binding of alpha-synuclein. Curr Opin Struct Biol 48:49–57. https://doi.org/10.1016/j.sbi.2017.09.004
Gao L, Tang H, Nie K, Wang L, Zhao J, Gan R, Huang J, Zhu R, Feng S, Duan Z, Zhang Y (2015) Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: a systematic review and meta-analysis. Int J Neurosci 125(9):645–654. https://doi.org/10.3109/00207454.2014.961454
Garcia-Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della Corte L, Spano P, Tofaris GK, Goedert M, Spillantini MG (2010) SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 133(Pt 7):2032–2044. https://doi.org/10.1093/brain/awq132
Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56(1):33–39. https://doi.org/10.1001/archneur.56.1.33
George S, Rey NL, Tyson T, Esquibel C, Meyerdirk L, Schulz E, Pierce S, Burmeister AR, Madaj Z, Steiner JA, Escobar Galvis ML, Brundin L, Brundin P (2019) Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol Neurodegener 14(1):34. https://doi.org/10.1186/s13024-019-0335-3
Geser F, Wenning GK, Poewe W, McKeith I (2005) How to diagnose dementia with Lewy bodies: state of the art. Mov Disord 20(Suppl 12):S11-20. https://doi.org/10.1002/mds.20535
Gilman S, Low P, Quinn N, Albanese A, Ben-Shlomo Y, Fowler C, Kaufmann H, Klockgether T, Lang A, Lantos P, Litvan I, Mathias C, Oliver E, Robertson D, Schatz I, Wenning G (1998) Consensus statement on the diagnosis of multiple system atrophy. American Autonomic Society and American Academy of Neurology. Clin Auton Res 8(6):359–362. https://doi.org/10.1007/BF02309628
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71(9):670–676. https://doi.org/10.1212/01.wnl.0000324625.00404.15
Gonzalo S, Linder ME (1998) SNAP-25 palmitoylation and plasma membrane targeting require a functional secretory pathway. Mol Biol Cell 9(3):585–597. https://doi.org/10.1091/mbc.9.3.585
Halbgebauer S, Oeckl P, Steinacker P, Yilmazer-Hanke D, Anderl-Straub S, von Arnim C, Froelich L, Gomes LA, Hausner L, Huss A, Jahn H, Weishaupt J, Ludolph AC, Thal DR, Otto M (2020) Beta-synuclein in cerebrospinal fluid as an early diagnostic marker of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2020-324306
Hall S, Janelidze S, Zetterberg H, Brix B, Mattsson N, Surova Y, Blennow K, Hansson O (2020) Cerebrospinal fluid levels of neurogranin in Parkinsonian disorders. Mov Disord 35(3):513–518. https://doi.org/10.1002/mds.27950
Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E (2001) beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 32(2):213–223. https://doi.org/10.1016/s0896-6273(01)00462-7
He Q, Dent EW, Meiri KF (1997) Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J Neurosci 17(10):3515–3524
Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB (2019) Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord 34(10):1464–1470. https://doi.org/10.1002/mds.27802
Hoffmann A, Ettle B, Bruno A, Kulinich A, Hoffmann AC, von Wittgenstein J, Winkler J, Xiang W, Schlachetzki JCM (2016) Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem Biophys Res Commun 479(4):881–886. https://doi.org/10.1016/j.bbrc.2016.09.109
Holahan MR (2017) A shift from a pivotal to supporting role for the growth-associated protein (GAP-43) in the coordination of axonal structural and functional plasticity. Front Cell Neurosci 11:266. https://doi.org/10.3389/fncel.2017.00266
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55(3):181–184. https://doi.org/10.1136/jnnp.55.3.181
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8(1):1–13. https://doi.org/10.1016/j.jalz.2011.10.007
Iwai N, Shimoike H, Ohmichi N, Kinoshita M (1995) Angiotensinogen gene and blood pressure in the Japanese population. Hypertension 25(4 Pt 2):688–693
Janelidze S, Hertze J, Zetterberg H, Landqvist Waldo M, Santillo A, Blennow K, Hansson O (2016) Cerebrospinal fluid neurogranin and YKL-40 as biomarkers of Alzheimer’s disease. Ann Clin Transl Neurol 3(1):12–20. https://doi.org/10.1002/acn3.266ACN3266
Jeannotte AM, Sidhu A (2008) Regulated interactions of the norepineprhine transporter by the actin and microtubule cytoskeletons. J Neurochem 105(5):1668–1682. https://doi.org/10.1111/j.1471-4159.2008.05258.x
Jellinger KA (2003) Neuropathological spectrum of synucleinopathies. Mov Disord 18(Suppl 6):S2-12. https://doi.org/10.1002/mds.10557
Khundakar AA, Hanson PS, Erskine D, Lax NZ, Roscamp J, Karyka E, Tsefou E, Singh P, Cockell SJ, Gribben A, Ramsay L, Blain PG, Mosimann UP, Lett DJ, Elstner M, Turnbull DM, Xiang CC, Brownstein MJ, O’Brien JT, Taylor JP, Attems J, Thomas AJ, McKeith IG, Morris CM (2016) Analysis of primary visual cortex in dementia with Lewy bodies indicates GABAergic involvement associated with recurrent complex visual hallucinations. Acta Neuropathol Commun 4(1):66. https://doi.org/10.1186/s40478-016-0334-3
Kirkpatrick LL, Matzuk MM, Dodds DC, Perin MS (2000) Biochemical interactions of the neuronal pentraxins. Neuronal pentraxin (NP) receptor binds to taipoxin and taipoxin-associated calcium-binding protein 49 via NP1 and NP2. J Biol Chem 275(23):17786–17792. https://doi.org/10.1074/jbc.M002254200
Koob AO, Shaked GM, Bender A, Bisquertt A, Rockenstein E, Masliah E (2014) Neurogranin binds alpha-synuclein in the human superior temporal cortex and interaction is decreased in Parkinson’s disease. Brain Res 1591:102–110. https://doi.org/10.1016/j.brainres.2014.10.013
Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, Cornelisse LN, Farrell RJ, Goldschmidt HL, Howrigan DP, Hussain NK, Imig C, de Jong APH, Jung H, Kohansalnodehi M, Kramarz B, Lipstein N, Lovering RC, MacGillavry H, Mariano V, Mi H, Ninov M, Osumi-Sutherland D, Pielot R, Smalla KH, Tang H, Tashman K, Toonen RFG, Verpelli C, Reig-Viader R, Watanabe K, van Weering J, Achsel T, Ashrafi G, Asi N, Brown TC, De Camilli P, Feuermann M, Foulger RE, Gaudet P, Joglekar A, Kanellopoulos A, Malenka R, Nicoll RA, Pulido C, de Juan-Sanz J, Sheng M, Sudhof TC, Tilgner HU, Bagni C, Bayes A, Biederer T, Brose N, Chua JJE, Dieterich DC, Gundelfinger ED, Hoogenraad C, Huganir RL, Jahn R, Kaeser PS, Kim E, Kreutz MR, McPherson PS, Neale BM, O’Connor V, Posthuma D, Ryan TA, Sala C, Feng G, Hyman SE, Thomas PD, Smit AB, Verhage M (2019) SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron 103(2):217–234. https://doi.org/10.1016/j.neuron.2019.05.002
Lautenschlager J, Stephens AD, Fusco G, Strohl F, Curry N, Zacharopoulou M, Michel CH, Laine R, Nespovitaya N, Fantham M, Pinotsi D, Zago W, Fraser P, Tandon A, St George-Hyslop P, Rees E, Phillips JJ, De Simone A, Kaminski CF, Schierle GSK (2018) C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat Commun 9(1):712. https://doi.org/10.1038/s41467-018-03111-4
Lee JY, Kim JS, Byun HR, Palmiter RD, Koh JY (2011) Dependence of the histofluorescently reactive zinc pool on zinc transporter-3 in the normal brain. Brain Res 1418:12–22. https://doi.org/10.1016/j.brainres.2011.08.055
Lee SJ, Wei M, Zhang C, Maxeiner S, Pak C, Calado Botelho S, Trotter J, Sterky FH, Sudhof TC (2017) Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J Neurosci 37(5):1062–1080. https://doi.org/10.1523/JNEUROSCI.2768-16.2016
Lopantsev V, Wenzel HJ, Cole TB, Palmiter RD, Schwartzkroin PA (2003) Lack of vesicular zinc in mossy fibers does not affect synaptic excitability of CA3 pyramidal cells in zinc transporter 3 knockout mice. Neuroscience 116(1):237–248. https://doi.org/10.1016/s0306-4522(02)00570-5
Lou X, Kim J, Hawk BJ, Shin YK (2017) alpha-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem J 474(12):2039–2049. https://doi.org/10.1042/BCJ20170200
Lowther ER, O’Brien JT, Firbank MJ, Blamire AM (2014) Lewy body compared with Alzheimer dementia is associated with decreased functional connectivity in resting state networks. Psychiatry Res 223(3):192–201. https://doi.org/10.1016/j.pscychresns.2014.06.004
Luo C, Chen Q, Song W, Chen K, Guo X, Yang J, Huang X, Gong Q, Shang HF (2014) Resting-state fMRI study on drug-naive patients with Parkinson’s disease and with depression. J Neurol Neurosurg Psychiatry 85(6):675–683. https://doi.org/10.1136/jnnp-2013-306237
Magdalinou NK, Paterson RW, Schott JM, Fox NC, Mummery C, Blennow K, Bhatia K, Morris HR, Giunti P, Warner TT, de Silva R, Lees AJ, Zetterberg H (2015) A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry 86(11):1240–1247. https://doi.org/10.1136/jnnp-2014-309562
Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW, Lashuel HA (2020) The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci 117(9):4971–4982. https://doi.org/10.1073/pnas.1913904117
Maroteaux L, Campanelli JT, Scheller RH (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8(8):2804–2815
Matuskey D, Tinaz S, Wilcox KC, Naganawa M, Toyonaga T, Dias M, Henry S, Pittman B, Ropchan J, Nabulsi N, Suridjan I, Comley RA, Huang Y, Finnema SJ, Carson RE (2020) Synaptic changes in parkinson disease assessed with in vivo imaging. Ann Neurol 87(3):329–338. https://doi.org/10.1002/ana.25682
McCormack A, Keating DJ, Chegeni N, Colella A, Wang JJ, Chataway T (2019) Abundance of synaptic vesicle-related proteins in alpha-synuclein-containing protein inclusions suggests a targeted formation mechanism. Neurotox Res 35(4):883–897. https://doi.org/10.1007/s12640-019-00014-0
McKeith IG, Galasko D, Kosaka K (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 47:1113–1124
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M, Consortium on DLB (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65(12):1863–1872. https://doi.org/10.1212/01.wnl.0000187889.17253.b1
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, El-Agnaf O, Feldman H, Ferman TJ, Ffytche D, Fujishiro H, Galasko D, Goldman JG, Gomperts SN, Graff-Radford NR, Honig LS, Iranzo A, Kantarci K, Kaufer D, Kukull W, Lee VMY, Leverenz JB, Lewis S, Lippa C, Lunde A, Masellis M, Masliah E, McLean P, Mollenhauer B, Montine TJ, Moreno E, Mori E, Murray M, O’Brien JT, Orimo S, Postuma RB, Ramaswamy S, Ross OA, Salmon DP, Singleton A, Taylor A, Thomas A, Tiraboschi P, Toledo JB, Trojanowski JQ, Tsuang D, Walker Z, Yamada M, Kosaka K (2017) Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 89(1):88–100. https://doi.org/10.1212/WNL.0000000000004058
McKeith IG, Ferman TJ, Thomas AJ, Blanc F, Boeve BF, Fujishiro H, Kantarci K, Muscio C, O’Brien JT, Postuma RB, Aarsland D, Ballard C, Bonanni L, Donaghy P, Emre M, Galvin JE, Galasko D, Goldman JG, Gomperts SN, Honig LS, Ikeda M, Leverenz JB, Lewis SJG, Marder KS, Masellis M, Salmon DP, Taylor JP, Tsuang DW, Walker Z, Tiraboschi P (2020) Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 94(17):743–755. https://doi.org/10.1212/WNL.0000000000009323
Miki Y, Foti SC, Asi YT, Tsushima E, Quinn N, Ling H, Holton JL (2019) Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain 142(9):2813–2827. https://doi.org/10.1093/brain/awz189
Mollenhauer B, Zimmermann J, Sixel-Doring F, Focke NK, Wicke T, Ebentheuer J, Schaumburg M, Lang E, Trautmann E, Zetterberg H, Taylor P, Friede T, Trenkwalder C (2016) Monitoring of 30 marker candidates in early Parkinson disease as progression markers. Neurology 87(2):168–177. https://doi.org/10.1212/WNL.0000000000002651
Mollenhauer B, Caspell-Garcia CJ, Coffey CS, Taylor P, Shaw LM, Trojanowski JQ, Singleton A, Frasier M, Marek K, Galasko D (2017) Longitudinal CSF biomarkers in patients with early Parkinson disease and healthy controls. Neurology 89(19):1959–1969. https://doi.org/10.1212/WNL.0000000000004609
Moran LB, Hickey L, Michael GJ, Derkacs M, Christian LM, Kalaitzakis ME, Pearce RK, Graeber MB (2008) Neuronal pentraxin II is highly upregulated in Parkinson’s disease and a novel component of Lewy bodies. Acta Neuropathol 115(4):471–478. https://doi.org/10.1007/s00401-007-0309-3
Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65(1):66–79. https://doi.org/10.1016/j.neuron.2009.12.023
Nicastro N, Holland N, Savulich G, Carter SF, Mak E, Hong YT, Milicevic Sephton S, Fryer TD, Aigbirhio FI, Rowe JB, O’Brien JT (2020) (11)C-UCB-J synaptic PET and multimodal imaging in dementia with Lewy bodies. Eur J Hybrid Imaging 4(1):25. https://doi.org/10.1186/s41824-020-00093-9
Ninkina N, Millership SJ, Peters OM, Connor-Robson N, Chaprov K, Kopylov AT, Montoya A, Kramer H, Withers DJ, Buchman VL (2021) beta-synuclein potentiates synaptic vesicle dopamine uptake and rescues dopaminergic neurons from MPTP-induced death in the absence of other synucleins. J Biol Chem 297(6):101375. https://doi.org/10.1016/j.jbc.2021.101375
O’Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P (1999) Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron 23(2):309–323. https://doi.org/10.1016/s0896-6273(00)80782-5
Oeckl P, Metzger F, Nagl M, von Arnim CA, Halbgebauer S, Steinacker P, Ludolph AC, Otto M (2016) Alpha-, beta-, and gamma-synuclein quantification in cerebrospinal fluid by multiple reaction monitoring reveals increased concentrations in alzheimer’s and creutzfeldt-jakob disease but no alteration in synucleinopathies. Mol Cell Proteom 15(10):3126–3138. https://doi.org/10.1074/mcp.M116.059915
Oeckl P, Halbgebauer S, Anderl-Straub S, von Arnim CAF, Diehl-Schmid J, Froelich L, Grimmer T, Hausner L, Denk J, Jahn H, Steinacker P, Weishaupt JH, Ludolph AC, Otto M (2020) Targeted mass spectrometry suggests beta-synuclein as synaptic blood marker in alzheimer’s disease. J Proteome Res 19(3):1310–1318. https://doi.org/10.1021/acs.jproteome.9b00824
Olanow CW, Savolainen M, Chu Y, Halliday GM, Kordower JH (2019) Temporal evolution of microglia and alpha-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain 142(6):1690–1700. https://doi.org/10.1093/brain/awz104
Paik SR, Shin HJ, Lee JH, Chang CS, Kim J (1999) Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem J 340(Pt 3):821–828
Palmiter RD, Cole TB, Quaife CJ, Findley SD (1996) ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci USA 93(25):14934–14939. https://doi.org/10.1073/pnas.93.25.14934
Paoletti P, Ascher P, Neyton J (1997) High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci 17(15):5711–5725
Parnetti L, Farotti L, Eusebi P, Chiasserini D, De Carlo C, Giannandrea D, Salvadori N, Lisetti V, Tambasco N, Rossi A, Majbour NK, El-Agnaf O, Calabresi P (2014) Differential role of CSF alpha-synuclein species, tau, and Abeta42 in Parkinson’s Disease. Front Aging Neurosci 6:53. https://doi.org/10.3389/fnagi.2014.00053
Peraza LR, Kaiser M, Firbank M, Graziadio S, Bonanni L, Onofrj M, Colloby SJ, Blamire A, O’Brien J, Taylor JP (2014) fMRI resting state networks and their association with cognitive fluctuations in dementia with Lewy bodies. Neuroimage Clin 4:558–565. https://doi.org/10.1016/j.nicl.2014.03.013
Pierre K, Dupouy B, Allard M, Poulain DA, Theodosis DT (2001) Mobilization of the cell adhesion glycoprotein F3/contactin to axonal surfaces is activity dependent. Eur J Neurosci 14(4):645–656. https://doi.org/10.1046/j.0953-816x.2001.01682.x
Pissadaki EK, Bolam JP (2013) The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front Comput Neurosci 7:13. https://doi.org/10.3389/fncom.2013.00013
Poggiolini I, Gupta V, Lawton M, Lee S, El-Turabi A, Querejeta-Coma A, Trenkwalder C, Sixel-Doring F, Foubert-Samier A, Le Traon AP, Plazzi G, Biscarini F, Montplaisir J, Gagnon JF, Postuma RB, Antelmi E, Meissner WG, Mollenhauer B, Ben-Shlomo Y, Hu MT, Parkkinen L (2021) Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain. https://doi.org/10.1093/brain/awab431
Portelius E, Olsson B, Hoglund K, Cullen NC, Kvartsberg H, Andreasson U, Zetterberg H, Sandelius A, Shaw LM, Lee VMY, Irwin DJ, Grossman M, Weintraub D, Chen-Plotkin A, Wolk DA, McCluskey L, Elman L, McBride J, Toledo JB, Trojanowski JQ, Blennow K (2018) Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol 136(3):363–376. https://doi.org/10.1007/s00401-018-1851-x
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30(12):1591–1601. https://doi.org/10.1002/mds.26424
Prots I, Grosch J, Brazdis RM, Simmnacher K, Veber V, Havlicek S, Hannappel C, Krach F, Krumbiegel M, Schutz O, Reis A, Wrasidlo W, Galasko DR, Groemer TW, Masliah E, Schlotzer-Schrehardt U, Xiang W, Winkler J, Winner B (2018) alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc Natl Acad Sci USA 115(30):7813–7818. https://doi.org/10.1073/pnas.1713129115
Puzzo D, Bizzoca A, Privitera L, Furnari D, Giunta S, Girolamo F, Pinto M, Gennarini G, Palmeri A (2013) F3/Contactin promotes hippocampal neurogenesis, synaptic plasticity, and memory in adult mice. Hippocampus 23(12):1367–1382. https://doi.org/10.1002/hipo.22186
Quadalti C, Calandra-Buonaura G, Baiardi S, Mastrangelo A, Rossi M, Zenesini C, Giannini G, Candelise N, Sambati L, Polischi B, Plazzi G, Capellari S, Cortelli P, Parchi P (2021) Neurofilament light chain and alpha-synuclein RT-QuIC as differential diagnostic biomarkers in parkinsonisms and related syndromes. NPJ Parkinsons Dis 7(1):93. https://doi.org/10.1038/s41531-021-00232-4
Remnestal J, Just D, Mitsios N, Fredolini C, Mulder J, Schwenk JM, Uhlen M, Kultima K, Ingelsson M, Kilander L, Lannfelt L, Svenningsson P, Nellgard B, Zetterberg H, Blennow K, Nilsson P, Haggmark-Manberg A (2016) CSF profiling of the human brain enriched proteome reveals associations of neuromodulin and neurogranin to Alzheimer’s disease. Proteomics Clin Appl 10(12):1242–1253. https://doi.org/10.1002/prca.201500150
Rossi M, Baiardi S, Teunissen CE, Quadalti C, van de Beek M, Mammana A, Stanzani-Maserati M, Van der Flier WM, Sambati L, Zenesini C, Caughey B, Capellari S, Lemstra AW, Parchi P (2021) Diagnostic value of the CSF alpha-synuclein real-time quaking-induced conversion assay at the prodromal MCI stage of dementia with lewy bodies. Neurology 97(9):e930–e940. https://doi.org/10.1212/WNL.0000000000012438
Rudenko G (2017) Dynamic control of synaptic adhesion and organizing molecules in synaptic plasticity. Neural Plast 2017:6526151. https://doi.org/10.1155/2017/6526151
Ruiz A, Walker MC, Fabian-Fine R, Kullmann DM (2004) Endogenous zinc inhibits GABA(A) receptors in a hippocampal pathway. J Neurophysiol 91(2):1091–1096. https://doi.org/10.1152/jn.00755.2003
Rust MB, Maritzen T (2015) Relevance of presynaptic actin dynamics for synapse function and mouse behavior. Exp Cell Res 335(2):165–171. https://doi.org/10.1016/j.yexcr.2014.12.020
Sako W, Murakami N, Izumi Y, Kaji R (2014) Reduced alpha-synuclein in cerebrospinal fluid in synucleinopathies: evidence from a meta-analysis. Mov Disord 29(13):1599–1605. https://doi.org/10.1002/mds.26036
Sandelius A, Portelius E, Kallen A, Zetterberg H, Rot U, Olsson B, Toledo JB, Shaw LM, Lee VMY, Irwin DJ, Grossman M, Weintraub D, Chen-Plotkin A, Wolk DA, McCluskey L, Elman L, Kostanjevecki V, Vandijck M, McBride J, Trojanowski JQ, Blennow K (2019) Elevated CSF GAP-43 is Alzheimer’s disease specific and associated with tau and amyloid pathology. Alzheimers Dement 15(1):55–64. https://doi.org/10.1016/j.jalz.2018.08.006
Sauvola CW, Littleton JT (2021) SNARE regulatory proteins in synaptic vesicle fusion and recycling. Front Mol Neurosci 14:733138. https://doi.org/10.3389/fnmol.2021.733138
Selnes P, Stav AL, Johansen KK, Bjornerud A, Coello C, Auning E, Kalheim L, Almdahl IS, Hessen E, Zetterberg H, Blennow K, Aarsland D, Fladby T (2017) Impaired synaptic function is linked to cognition in Parkinson’s disease. Ann Clin Transl Neurol 4(10):700–713. https://doi.org/10.1002/acn3.446
Sensi SL, Paoletti P, Bush AI, Sekler I (2009) Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 10(11):780–791. https://doi.org/10.1038/nrn2734
Sevlever D, Zou F, Ma L, Carrasquillo S, Crump MG, Culley OJ, Hunter TA, Bisceglio GD, Younkin L, Allen M, Carrasquillo MM, Sando SB, Aasly JO, Dickson DW, Graff-Radford NR, Petersen RC, Deak F, Belbin O (2015) Genetically-controlled vesicle-associated membrane protein 1 expression may contribute to Alzheimer’s pathophysiology and susceptibility. Mol Neurodegener 10:18. https://doi.org/10.1186/s13024-015-0015-x
Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, Mollenhauer B, Soto C (2017) Development of a biochemical diagnosis of Parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 74(2):163–172. https://doi.org/10.1001/jamaneurol.2016.4547
Sikora J, Ouagazzal AM (2021) Synaptic zinc: an emerging player in Parkinson’s disease. Int J Mol Sci. https://doi.org/10.3390/ijms22094724
Singer W, Schmeichel AM, Shahnawaz M, Schmelzer JD, Boeve BF, Sletten DM, Gehrking TL, Gehrking JA, Olson AD, Savica R, Suarez MD, Soto C, Low PA (2020) Alpha-synuclein oligomers and neurofilament light chain in spinal fluid differentiate multiple system atrophy from lewy body synucleinopathies. Ann Neurol 88(3):503–512. https://doi.org/10.1002/ana.25824
Sjogren M, Minthon L, Davidsson P, Granerus AK, Clarberg A, Vanderstichele H, Vanmechelen E, Wallin A, Blennow K (2000) CSF levels of tau, beta-amyloid(1–42) and GAP-43 in frontotemporal dementia, other types of dementia and normal aging. J Neural Transm (vienna) 107(5):563–579. https://doi.org/10.1007/s007020070079
Soldan A, Moghekar A, Walker KA, Pettigrew C, Hou X, Lu H, Miller MI, Alfini A, Albert M, Xu D, Xiao MF, Worley P (2019) Resting-state functional connectivity is associated with cerebrospinal fluid levels of the synaptic protein NPTX2 in non-demented older adults. Front Aging Neurosci 11:132. https://doi.org/10.3389/fnagi.2019.00132
Sorrentino ZA, Giasson BI, Chakrabarty P (2019) alpha-Synuclein and astrocytes: tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol 138(1):1–21. https://doi.org/10.1007/s00401-019-01977-2
Sourty M, Thoraval L, Roquet D, Armspach JP, Foucher J, Blanc F (2016) Identifying dynamic functional connectivity changes in dementia with lewy bodies based on product hidden Markov models. Front Comput Neurosci 10:60. https://doi.org/10.3389/fncom.2016.00060
Sousa VL, Bellani S, Giannandrea M, Yousuf M, Valtorta F, Meldolesi J, Chieregatti E (2009) α-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol Biol Cell 20(16):3725–3739. https://doi.org/10.1091/mbc.e08-03-0302
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840. https://doi.org/10.1038/42166
Stoeckli ET (2010) Neural circuit formation in the cerebellum is controlled by cell adhesion molecules of the Contactin family. Cell Adh Migr 4(4):523–526. https://doi.org/10.4161/cam.4.4.12733
Taguchi K, Watanabe Y, Tsujimura A, Tanaka M (2016) Brain region-dependent differential expression of alpha-synuclein. J Comp Neurol 524(6):1236–1258. https://doi.org/10.1002/cne.23901
Tai CY, Mysore SP, Chiu C, Schuman EM (2007) Activity-regulated N-cadherin endocytosis. Neuron 54(5):771–785. https://doi.org/10.1016/j.neuron.2007.05.013
Trojanowski JQ, Revesz T (2007) Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 33(6):615–620. https://doi.org/10.1111/j.1365-2990.2007.00907.x
Tsui CC, Copeland NG, Gilbert DJ, Jenkins NA, Barnes C, Worley PF (1996) Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci 16(8):2463–2478
Ubhi K, Low P, Masliah E (2011) Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci 34(11):581–590. https://doi.org/10.1016/j.tins.2011.08.003
Vallortigara J, Rangarajan S, Whitfield D, Alghamdi A, Howlett D, Hortobagyi T, Johnson M, Attems J, Ballard C, Thomas A, O’Brien J, Aarsland D, Francis P (2014) Dynamin1 concentration in the prefrontal cortex is associated with cognitive impairment in Lewy body dementia. F1000Res 3:108. https://doi.org/10.12688/f1000research.3786.1
van Steenoven I, Majbour NK, Vaikath NN, Berendse HW, van der Flier WM, van de Berg WDJ, Teunissen CE, Lemstra AW, El-Agnaf OMA (2018) alpha-Synuclein species as potential cerebrospinal fluid biomarkers for dementia with lewy bodies. Mov Disord 33(11):1724–1733. https://doi.org/10.1002/mds.111
van Steenoven I, Noli B, Cocco C, Ferri GL, Oeckl P, Otto M, Koel-Simmelink MJA, Bridel C, van der Flier WM, Lemstra AW, Teunissen CE (2019) VGF peptides in cerebrospinal fluid of patients with dementia with lewy bodies. Int J Mol Sci. https://doi.org/10.3390/ijms20194674
van Steenoven I, Koel-Simmelink MJA, Vergouw LJM, Tijms BM, Piersma SR, Pham TV, Bridel C, Ferri GL, Cocco C, Noli B, Worley PF, Xiao MF, Xu D, Oeckl P, Otto M, van der Flier WM, de Jong FJ, Jimenez CR, Lemstra AW, Teunissen CE (2020) Identification of novel cerebrospinal fluid biomarker candidates for dementia with Lewy bodies: a proteomic approach. Mol Neurodegener 15(1):36. https://doi.org/10.1186/s13024-020-00388-2
Vasconcellos LF, Pereira JS (2015) Parkinson’s disease dementia: diagnostic criteria and risk factor review. J Clin Exp Neuropsychol 37(9):988–993. https://doi.org/10.1080/13803395.2015.1073227
Wallin A, Jennersjo C, Granerus AK (1999) Prevalence of dementia and regional brain syndromes in long-standing Parkinson’s disease. Parkinsonism Relat Disord 5(3):103–110. https://doi.org/10.1016/s1353-8020(99)00018-8
Wang Z, Danscher G, Kim YK, Dahlstrom A, Mook Jo S (2002) Inhibitory zinc-enriched terminals in the mouse cerebellum: double-immunohistochemistry for zinc transporter 3 and glutamate decarboxylase. Neurosci Lett 321(1–2):37–40. https://doi.org/10.1016/s0304-3940(01)02560-5
Wellington H, Paterson RW, Portelius E, Tornqvist U, Magdalinou N, Fox NC, Blennow K, Schott JM, Zetterberg H (2016) Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 86(9):829–835. https://doi.org/10.1212/WNL.0000000000002423
Wennstrom M, Surova Y, Hall S, Nilsson C, Minthon L, Bostrom F, Hansson O, Nielsen HM (2013) Low CSF levels of both alpha-synuclein and the alpha-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS ONE 8(1):e53250. https://doi.org/10.1371/journal.pone.0053250
Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O’Brien RJ, Worley P (2003) Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron 39(3):513–528
Yakovleva T, Bazov I, Cebers G, Marinova Z, Hara Y, Ahmed A, Vlaskovska M, Johansson B, Hochgeschwender U, Singh IN, Bruce-Keller AJ, Hurd YL, Kaneko T, Terenius L, Ekstrom TJ, Hauser KF, Pickel VM, Bakalkin G (2006) Prodynorphin storage and processing in axon terminals and dendrites. FASEB J 20(12):2124–2126. https://doi.org/10.1096/fj.06-6174fje
Yamada M, Hashimoto T, Hayashi N, Higuchi M, Murakami A, Nakashima T, Maekawa S, Miyata S (2007) Synaptic adhesion molecule OBCAM; synaptogenesis and dynamic internalization. Brain Res 1165:5–14. https://doi.org/10.1016/j.brainres.2007.04.062
Zhang Z, Zhang S, Fu P, Lin K, Ko JK, Yung KK (2019) Roles of glutamate receptors in Parkinson’s disease. Int J Mol Sci. https://doi.org/10.3390/ijms20184391
Zhou B, Wen M, Yu WF, Zhang CL, Jiao L (2015) The diagnostic and differential diagnosis utility of cerebrospinal fluid alpha -synuclein levels in Parkinson’s disease: a meta-analysis. Parkinsons Dis 2015:567386. https://doi.org/10.1155/2015/567386
Funding
This work was funded by the Fondos de Investigaciones Sanitarias (PI18/00327 to OB) from the “Fondo Europeo de Desarrollo Regional (FEDER)”, Unión Europea, “Una manera de hacer Europa” and the “Instituto de Salud Carlos III, Ministerio de Economia y Competitividad, Gobierno de España” (ISCIII) and also by the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR), Spain & Regional Development Funds, EU Knowledge Industry Valorisation Grants-AGAUR-PRODUCTE (2019 PROD 00088 to OB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr Belbin is inventor of a patent application (EP18382175) pending approval entitled “Markers of synaptopathy in neurodegenerative disease”.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cervantes González, A., Belbin, O. Fluid markers of synapse degeneration in synucleinopathies. J Neural Transm 129, 187–206 (2022). https://doi.org/10.1007/s00702-022-02467-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02467-8