
Abstract
Evaluating each patient and animal as its own control achieves personalized medicine, which honors the hippocratic philosophy, explaining that “it is far more important to know what person has the disease than what disease the person has.” Similarly, individualizing molecular signaling directly from the patient’s brain in real time is essential for providing prompt, patient-based treatment as dictated by the point of care. Fortunately, nanotechnology effectively treats many neurodegenerative diseases. In particular, the new medicinal frontier for the discovery of therapy for Parkinson’s disease is nanotechnology and nanobiotechnology. Indeed, the unique nanotechnology of neuromolecular imaging combined with the series of nanobiosensors enables continuous videotracking of molecular neurotransmitters in both the normal physiologic and disease states with long-term electrochemical operational stability. This nanobiotechnology is able to track a signal in real time with excellent temporal and spatial resolution directly from each patient’s brain to a computer as subjects are behaving during movement, normal and/or dysfunctional including prion-like Parkinson’s behavioral biometrics. Moreover, the molecular signaling performed by these nanobiosensors live streams directly online and originates from precise neuroanatomic brain sites such as, in this case, the dorsal striatum in basal ganglia. Thus, the nanobiotechnology studies discussed herein imaged neuromolecules with and without l-3,4-dihydroxyphenylalanine (l-DOPA) in dorsal striatal basal ganglia neurons. Parkinsonian and non-Parkinsonian animals were video-tracked, and images were readily seen on a laptop via a potentiostat using a semiderivative electrical circuit. Administered l-DOPA doses were 50 and 100 mg/kg intraperitoneally (ip); the same experimental paradigm was used to image and then contrast data. Results showed that the baseline release of biogenic amine molecules was significantly above detection limits in non-Parkinsonian animals. After administration of l-DOPA, biogenic amines significantly increased in these non-Parkinson’s animals. Nevertheless, it is intriguing to see that l-DOPA could not enable synaptic dopamine release in Parkinson’s animals, thereby demonstrating that biogenic amines are biomarkers for Parkinson’s disease. Biomarkers are biochemical, genetic, or molecular measures of biological reactions. Importantly, there were other significant biomarkers present in Parkinsonian animals and absent in non-Parkinsonian animals; these were peptide neurotransmitters that include dynorphin and somatostatin in the brain with detection limits of 40 nM for dynorphin and 37 nM for somatostatin (see Table 1). Furthermore, l-DOPA significantly increased these peptide biomarkers, dynorphin and somatostatin, in Parkinson’s animals. Targeting biomarkers enables new diagnostic devices and treatments for Parkinson’s disease through nanotechnology and nanobiotechnology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
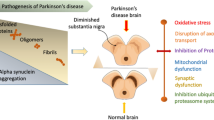
Parkinson’s disease is a neurodegenerative disease characterized by Lewy bodies and neurites comprised primarily of the protein, α-synuclein, a presynaptic protein in the cytomatrix of the neuronal active zone; the active zone forms a bouton at the synaptic cleft to affect the release of neurotransmitters. Lewy body dementia is another of these diseases associated with abnormal deposits of this protein called α-synuclein in the brain (Zarranz et al. 2004). Lewy body deposits affect brain neurotransmitters enabling a myriad of cognitive problems further associated with behavior and mood changes. The peptide, β-amyloid, is currently accepted to be one of the major pathomechanisms underlying Alzheimer’s disease given the caveat that the main species form of this peptide may not be fully characterized. Recent evidence indicates that soluble oligomers rather than plaques are the major cause of the synaptic dysfunction leading to neurodegeneration in the Alzheimer’s form (Danysz and Parsons 2012). Nonetheless, the pathomechanism of Parkinson’s and Alzheimer’s diseases bear a strong resemblance to one another as both brain disorders are similar not only in the presence of the presynaptic protein, α-synuclein, but also in the presence of the peptide, β-amyloid (Dugger et al. 2012; Forman et al. 2004; Vila and Przebdorski 2004). Kornhuber et al. (1994) showed the relationship among neurodegenerative interactive mechanisms for Parkinson’s, Alzheimer’s, epilepsy and ischemia via glutamatergic receptors N-methyl-d-aspartate and the treatment of such neurological disorders with NMDA-receptor channel antagonists.
Both Alzheimer’s and Parkinson’s patients exhibit α-synuclein, but there is a distinction in that in Parkinson’s, the α-synuclein protein may appear in more clump-like structures, allowing the destruction of the clumping as a therapeutic target (Wakabayashi et al. 2007). Since Parkinson’s and Alzheimer’s are similar in the common Lewy body protein excess, there is a new trend of thinking in pharmacotherapeutics for these two neurodegenerative diseases. This trend is critically important in pharmacotherapeutics, because it directly connects Alzheimer’s with Parkinson’s. Moreover, the consequences of Parkinson’s disease are known to be both non-motor and motor (Ford et al. 1996; Hely et al. 1994; Henry and Brotchie 1996; Matuja and Aris 2008). Thus, it makes sense that Parkinson’s patients suffer from non-motor symptoms such as cognitive defects and mood disorders as does the Alzheimer’s patient. Perhaps there are so many similarities between Parkinson’s and Alzheimer’s disease, because the condition of Parkinson’s disease acts as a reliable predictor of the onset of Alzheimer’s disease (Dugger et al. 2012).
Dementia with Lewy bodies in Parkinson’s disease affects about ten million people worldwide and contrary to what is sometimes expected, it is not the elderly person who is the victim. Indeed, the incidence of Parkinson’s does increase with age, but an estimated four percent of people with Parkinson’s are diagnosed before the age of 50 (Schrag and Schott 2006). When Parkinson’s disease is seen in individuals under the age of 40, it is called “young onset” Parkinson’s disease (YOPD), although the age range considered “young onset” varies. Although both environmental and genetic causes for YOPD have been implicated, it appears that genetics may play a role in its development via mutations in many genes including the PINK1 gene to create PARK6 and missense mutations of ATP13A2 (Di Fonzo et al. 2007; Healy et al. 2004; Rezak et al. 2008; Schrag and Schott 2006; Vila and Przebdorski 2004).
Parkinson’s disease is known to be caused by a loss of DA neurons in SN, the neuroanatomic site for DA somatodendritic cell bodies in basal ganglia. The basal ganglia circuitry is shown in Fig. 1.

Source: http://www.expertreviews.org
In Parkinson’s disease, the natural balance of the circuit is lost owing to the depletion of DA in the corpus striatum also called the putamen. Both the direct and indirect pathways operate through the GPi/SNr output nuclei and their influence is inhibitory on the thalamus. Thus, increased activity in the output nuclei leads to increased inhibition on the glutamatergic excitation of the motor cortex and a subsequent reduction in movement, observed in patients as bradykinesia. In addition, other structures such as the pedunculopontine nucleus (PPN) receive an abnormal input from the basal ganglia, which contributes to some of the clinical signs, such as gait disturbance. These changes in the Parkinson’s brain are shown here by the differing thickness of arrows, which represents the relative degree of activation in each projection. DA dopamine, Enk enkephalin, GABA gamma-aminobutyric acid, GPe globus pallidus, external segment, GPi globus pallidus, internal segment, SNc substantia nigra pars compacta, SNr substantia nigra pars reticulata, SP substance P, STN subthalamic nucleus (Expert Reviews, Cambridge University Press, 2003; Basal ganglia circuitry in Parkinson’s disease)
Pharmaceutical studies for Parkinson’s disease
l-DOPA is the current mainstay pharmaceutical therapy for Parkinson’s disease (Montastruc et al. 1994). Carbidopa is routinely used in combination with l-DOPA. The rationale is based on the finding that carbidopa inhibits aromatic-l-amino-acid decarboxylase (DOPA decarboxylase) (Sletzinger et al. 1963). The inhibitory action of the decarboxylase occurs in the biosynthesis of l-DOPA to DA, thus enabling increasingly available DA in the synaptic cleft to reach presynaptic DAergic sites in basal ganglia. Birkmayer and Mentasti (1967) were successful in using the decarboxylase inhibitor benserazide in Parkinson’s disease. Many, if not all, of the current medications for Parkinson’s disease are based on enhancing DA in the neuronal synapse either by acting as a DA agonist or by inhibiting the enzymatic metabolism of DA to increase the DA concentration in the neuronal synapse. In other words, the underlying mechanism of all Parkinson’s medications involves the facilitation of DA to reach presynaptic DAergic sites in basal ganglia.
Another enzyme in the DA metabolic pathway that can be blocked to enhance DA release and/or reuptake inhibition in the synapse is catechol-O-methyltransferase (COMT). Examples of this medication are entacapone, tolcapone, and opicapone. The active substance in both entacapone and opicapone works peripherally to restore the levels of DA in the parts of the brain that control movement and coordination known as the basal ganglia. This medication primarily alleviates symptoms of Parkinson’s only when it is taken with levodopa (Kurth et al. 1997). Johnston (1968) first developed the concept of MAO-A/B. Knoll and Magyar (1972) showed that selegiline, a preferentially monoamine oxidase inhibitor of DA metabolism, selectively inhibits the B-isoform of monoamine Oxidase, and later this monoamine oxidase B inhibitor proved useful in Parkinson’s treatment. In fact, the first clinical trials showed that selegiline reduced and, in many cases, abolished the ‘on–off’ periods produced by l-DOPA (Birkmayer et al. 1975). Even later studies showed that the dyskinesias produced by l-DOPA were blocked by this monoamine oxidase inhibitor B, selegiline (Gerlach et al. 2011).
Among other important pharmaceutical strategies for Parkinson’s, the drug, amantadine, plays another critical part in the pharmaceutical history of Parkinson’s therapy. The role that amantadine plays clinically can be deciphered by looking into its function as an N-methyl-d-aspartate-R-channel antagonist, because it was shown to antagonize tremor, balance and akinesia in Parkinson’s patients per se, as well as in experimental Parkinsonism modeling of HIV and AIDS-related parkinsonism-dementia complex (Czub et al. 2001; Kornhuber et al. 1989, 1991, 1994, 1995, 1999).
Some medications for Parkinson’s have receptor function at more than one DA receptor, exhibit receptor affinity at 5-HT receptors, and further demonstrate partial agonist and antagonist mechanisms of action. Indeed, most ergot derivatives with DA-R-activity show 5-HT2B-R activity. This may be causal for severe adverse reactions like fibrotic cardiopathology. In contrast, lisuride, a DA-R-agonist (Schechter 1984) is an antagonist at 5-HT2B-R sites and does not present fibrosis (Nomoto et al. 1998).
Finally, enter into the equation for Parkinson’s pharmacotherapy, the novel clinical properties of D3 receptors that underpin new interactions in Parkinson’s treatment despite the convention that Parkinson’s is known as a D2 disease. Pramipexole provides a good example of this type of receptor activity, particularly autoreceptor activity (Carlsson 1995). Pramipexole is a partial DAergic autoreceptor antagonist and DA agonist. The differences between the D2 and D3 receptor properties are discussed in Svensson et al. (1994). The non-ergoline and D3 properties of pramipexole introduce a notable contribution to enrich the pharmaceutical entourage of strategies to treat Parkinson’s. Pramipexole is used for early stages of Parkinson’s and, moreover, for advanced Parkinson’s treatment in patients presenting with mood disorders (Rektorová et al. 2003). Perhaps research into autoreceptor, as well as partial agonist and antagonist mechanisms, may lend an explanatory note to the treatment of affect disorders that frequently accompany Parkinson’s symptomatology. Indeed, studies demonstrating that autoreceptor antagonist, AJ 76, simultaneously upregulates accumbens 5-HT presynaptic release while increasing motility highlight the dual therapeutic nature of medications that target both physical and psychological manifestations of Parkinson’s (Broderick 2013; Broderick et al. 1989; Broderick and Piercey 1998; Eng et al. 1992; Piercey et al. 1990, 1993).
Pharmaceuticals based on anticholinergic agents for Parkinson’s
Anticholinergic agents for Parkinson’s treatment use imbalances between DA and the amino acid neurotransmitter, Ach, in the basal ganglia neuronal circuitry of the Parkinson’s patient (Girisha et al. 2009). Medications for Parkinson’s based on this anticholinergic hypothesis include benztropine. It is noteworthy that the glutamate, GABA, and glutamine neuronal interactions with intermediary neurochemicals are factored into an exceptionally delicate balance needed for Parkinson’s and Alzheimer’s. Such a mix is necessary for the natural, non-diseased brain-body connection within each prescient and non-prescient mammal (Fig. 2).

Schematic representation that describes the delicate balance between DA and ACH neuronal circuitry in basal ganglia. In Parkinson’s disease, a deficiency in DA may lead to ACH excess (Kvell et al. 2011). In addition, the glutamatergic system is highly involved in the mechanism of action of Parkinson’s disease, as mentioned above in the description for Fig. 1
Clinical studies of gene therapy in Parkinson’s disease
Clinical studies of gene therapy for Parkinson’s disease have been reported (Bohn and Choi-Lundberg 1998; Horellou and Mallet 1997; Ridet et al. 1999). Freed et al. (2001) demonstrated that transplantation of human embryonic DA neurons in human patients with severe Parkinson’s disease effectively alleviated Parkinsonian symptoms, especially in younger patients. Similarly, Ma et al. (2005) transplanted embryonic DA cells into the human brain to successfully reinnervate neurons along the nigrostriatal pathway in Parkinson’s patients. In 2007, surgeons at New York-Presbyterian Hospital implanted GAD, the gene responsible for making GABA, with an adeno-associated virus into the subthalamic nucleus to alleviate Parkinson’s symptoms in humans (Kaplitt et al. 2007).
Research focusing on Caspases (Cas) revealed the link between this family of programmable cell death enzymes and Parkinson’s disease (Smith et al. 2005). To understand how endoplasmic reticulum stress and mitochondrial dysfunction contribute to A53T α-synuclein-induced cell death, Smith et al. (2005) used the Tet-off regulatory system to create A53T mutant α-synuclein-inducible PC12 cell lines. Upon inducing expression of these PC12 cell lines, proteasome activity decreased, ROS intracellular levels increased, and 40 % of cells died with a concurrent release of mitochondrial cytochrome C and elevations of Cas9 and Cas3 activity. Thus, this study highlighted the connection between elevated Cas9 activity and the development of Parkinsonian cellular toxicity.
Jinek et al. (2012) demonstrated that Cas9 had the potential to be used as a therapeutic agent by cutting specific sites in DNA; Cong et al. 2013 demonstrated that Cas9 could be used with clustered regularly interspaced short palindromic repeats (CRISPR) to target specific sites in mammalian cells. These research discoveries culminated in the development of RNA-programmable CRISPR–Cas 9 biotechnology that could be used as “molecular scissors” in the surgical removal of mutated gene sites for subsequent replacement with physiological gene parts (Doudna and Charpentier 2014). This technology was derived from bacterial type II CRISPR–Cas systems that provided these single-cell organisms with an adapted immunity to plasmids and viruses. To model itself after the ancient immune defenses of bacteria, the CRISPR–Cas system is based on an RNA component such as crRNAs and a protein component such as Cas proteins. Together, the RNA and protein components form ribonucleoprotein complexes to oppose pathological nucleic acids via two phases. In the adaptation phase, the endonuclease Cas9 may be used to induce a double-strand break in DNA target sequences using its two catalytic centers as blades to cleave each strand of a DNA target site. The Cas protein then inserts a portion of this target sequence into a CRISPR array in the bacterial genome to memorize this genetic element. In the interference phase, the re-offending genetic element is recognized and destroyed to prevent re-infection.
CRISPR–Cas9 has been used to create knock-in mutations to generate loss-of-function phenotypes in animal models of disease (Tu et al. 2015). Similarly, the Whitehead Institute in Cambridge, MA used CRISPR–Cas9 to induce mutations into the isotonic human pluripotent stem cells of only one chromosome, leaving the other chromosome as an internal control. This mutation caused a nucleotide to change from adenine to guanine, which prevented transcription factor from effectively binding and, thereby, decreasing alpha-synuclein gene (SNCA) expression. In this manner, the CRISPR–Cas9-induced mutations revealed a genetic mechanism for high risk alleles causing sporadic Parkinson’s disease via reduced transcription factor binding, increased SNCA expression, and increased Parkinson’s risk (Soldner et al. 2016). Given the fact that this biotechnology has already treated plants and animals, as well as elucidated human mechanisms of neurodegenerative disease, perhaps CRISPR–Cas9 may be implemented as the future gene therapy for cancer, infectious disease such as HIV, and devastating genetic disorders such as Parkinson’s disease in humans (Charpentier 2015).
Clinical studies of gene therapy in Parkinson’s disease animal models
In addition to the advances made in these important human studies of Parkinson’s disease, animal research continues to provide critical new knowledge on gene therapies for Parkinson’s. Bergman et al. (1990) demonstrated that lesions to the subthalamic nucleus reduced akinesia, rigidity, tremor, and major motor impairments in contralateral limbs of monkeys. Choi-Lundberg et al. (1997) found that GDNF gene therapy prevented degeneration of DAergic neurons. In 2001, Kordower and Aebischer also determined that sustained delivery of GDNF to the nigrostriatial system provided neuroprotection and functional recovery in a primate model of Parkinson’s. Not only did GDNF provide symptomatic relief by enhancing function of remaining DAergic neurons, but GDNF also counteracted the degeneration of nigrostriatal neurons (Kordower and Aebischer 2001). Furthermore, in animal studies, Shen et al. (2000) found that GCH, in addition to TH and AADC, is important for effective gene therapy of Parkinson’s disease. Animal studies have also provided important preliminary data regarding the most effective vector use (Bemelmans et al. 1999; Fan et al. 1998; Kordower et al. 2000). However, the optimal vector remains uncertain.
Animal studies have further elucidated the role of the BDNF gene in Parkinson’s. Blum et al. (2001) found that BDNF caused a limited behavioral recovery in striatal DA systems made toxic by 6-OHDA in aged animals. Other animal research found that BDNF might possess a dual role in Parkinson’s disease. Indeed, BDNF is both a neuroprotective molecule, as its inhibition leads to loss of nigral DAergic neurons, and a neuromodulator, as its enhanced expression ameliorates cognitive processes (Bhave et al. 1999). In 2002, the Kolodny laboratory found that BDNF protected DAergic neurons from apoptosis induced by 6-OHDA and significantly increased the long-term survival of TH-positive astrocytes in the striatum (Wang et al. 2002). After normal parental neonatal rat astrocytes were transfected with AAVBDNF using the FuGENE 6 transfection kit, TH-positive status was determined using mRNA slot blotting and Western blotting. In mRNA slot blotting, the TH cDNA probes from pCDM8-TH were labeled with [K-32P]dCTP by random primer extension, and used to assay expression of TH mRNA in striatal astrocytes. In Western blotting, protein obtained from transfected cell extracts was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis, electroblotted onto nitrocellulose membranes, preincubated with 5 % non-fat milk, and incubated with monoclonal mouse anti-rat TH (0.4 ng ml−1) with subsequent incubation by either goat F(abP)2 anti-mouse or goat F(abP)2 anti-rabbit antibodies conjugated with horseradish peroxidase (1:5000). Finally, the Amersham ECL system was used to visualize and determine TH+ status (Wang et al. 2002).
This is significant as Hirsch et al. (1999) demonstrated that the main mechanism of DAergic neuron degeneration in Parkinson’s is apoptosis. Indeed, the Kolodny group found that the combined use of TH and BDNF has a synergistic therapeutic effect, and this combined use of gene therapy is more efficient for the treatment of Parkinson’s disease than a single gene therapy using TH or BDNF alone (Wang et al. 2002). Perhaps the mechanism of this therapeutic effect is partially explained by BDNF’s rapid upregulation of synaptophysin and tau proteins via the neurotrophin receptor TrkB as demonstrated by Coffey et al. (1997) in murine cerebellar granule cells. Finally, Levivier et al. (1995) revealed the neuroprotective effects of BDNF after they implanted fibroblasts genetically engineered to produce BDNF that successfully prevented DAergic neuron degeneration in striatum of the non-prescient mammalian brain. More recently, an array approach was used to reveal upregulation of a noncoding, neurodegenerative, and proinflammatory gene regulator called microRNA-155 in a mouse model of Parkinson’s disease created via adeno-associated-virus-mediated expression of α-synuclein (Thome et al. 2016). Conversely, in a Parkinson’s mouse model with full deletion of microRNA-155, the proinflammatory response to α-synuclein was reduced, as well as the consequent α-synuclein-mediated neurodegeneration; and the treatment of this mouse with microRNA-155 restored the inflammatory reaction to α-synuclein. Thus, microRNA is a promising genetic target for inflammatory downregulation in Parkinson’s disease (Thome et al. 2016). Based on the knowledge gained from both human and animal studies of Parkinson’s disease, the purpose of our nanotechnology studies herein is to share new data on l-DOPA to further the advances already accomplished in the field of Parkinson’s nanomedicine with the novel brain sensing nanobiotechnology of NMI and the series of nanobiosensors. Indeed, nanomedicine is a novel branch of medical treatment encompassing therapeutics such as nanopharmacology to treat disease on the molecular scale. Thus, the objective of this paper is to look directly inside the Parkinson’s brain and compare Parkinsonian striatal neurotransmitters with the milieu existing in the non-Parkinson’s brain. In this manner, a closer cause and effect relationship between therapy and outcome is presented.
Methods
Both lesioned and non-lesioned animals were implanted with miniature series of nanobiosensors minimally inserted into the dura mater and then advanced by microns into the dorsal striatum under sodium pentobarbital anesthesia (50 mg kg−1 ip at 6 % solution) in the Broderick laboratory. Stereotaxic coordinates were chosen as described by Pellegrino et al. (1979). Subsequently, Parkinson’s and non-Parkinson’s animals underwent neuromolecular imaging (NMI) of striatal DA neurons with nerve terminals for the somatodendritic substantia nigra. NMI enabled the same animal to be imaged as its own control, enabling minimized animal variability and further reducing the number of animals needed for accurate statistical analysis. Recordings were taken every 2 min, allowing a 2-min cell deposition period. Each scan selectively imaged seven neurotransmitters in less than 1 min. Endogenous neurochemical profiles for Parkinsonian compared with non-Parkinsonian animals were studied in Part A. In Part B, l-DOPA was cumulatively administered at 50 and 100 mg kg−1 ip.
Nanobiotechnology model
The nanobiotechnology used to study neural transmission in the Parkinson’s versus non-Parkinson’s brain is described schematically in Fig. 3. The nanobiotechnology involved herein includes the series of nanobiosensors and all related technologies.

Schematic drawing of just one example of one of the several designs of the BRODERICK PROBE® series of nanobiosensors
The series of nanobiosensors is comprised of several formulations of the carbon-based biosensors, patented by City University of New York and assigned in part to New York University, and all are assigned to Eazysense Nanotechnologies Incorporated in New York State (Broderick 1989a, b, 1995, 1999, 2015; Broderick and Pacia 2006, 2011). These formulations were tested in controlled studies. Details for the manufacture of these nanobiosensors, which include description of specific components of each formulation in addition to the use, design, and applications for these nanobiosensors are published. The NMI biotechnology, such as detector/potentiostat electrical circuits, is also published (Broderick 1988, 1989a, b, 1995, 1999, 2008; Broderick et al. 2000, 2008; Broderick and Pacia 2005, 2006). Pending patents, also listed in these latter references, show novel inventive constructions and formulations of our nanobiosensor (Broderick 2015; Broderick and Pacia 2011). This miniature device uses electron transfer kinetics to select an image for a specific neurochemical at an electroactive oxidation/half wave potential. An electroactive signature for each neurochemical is detected in subunits of volts and amperes, dependent on the electronic circuitry chosen for use in the detector/potentiostat. In the studies presented here, a semidifferential electrical circuit was chosen, because several neurotransmitters and neurochemicals can be separately imaged within 1 min with each neurochemical imaged within seconds, and recordings can be repeated continuously for hours or longer periods of time, for weeks and months without the occurrence of gliosis (scar tissue). Using the semidifferential/semiderivative electrical circuit, millivolts are shown on the x-axis and current in nano- and picoamperes is shown on the y-axis. Current is derived from electron transfer kinetics determined by specific biosensor properties, such as hydrophobicity and hydrophilicity of biosensor formulation within the context of specific interactions with neurotransmitters and neuromolecules. In these studies, a series of laurate nanobiosensors was used to image neurotransmitter profiles. We have determined that we have indeed imaged release mechanisms at the presynapse via the use of γ-butyrolactone investigations. γ-Butyrolactone was used to block neuronal depolarization in specific neuroanatomic sites (Broderick 1991). It is best though to provide the caveat that reuptake inhibitory actions at the presynapse can be factored as well, albeit reuptake inhibitory mechanisms may not be the primary mode of action (Fig. 4).

This figure shows how the BRODERICK PROBE® works in concert with NMI nanobiotechnologies. Simply, in one example, apply a voltage to the BRODERICK PROBE® nanobiosensor, voltage is converted into current via a potentiostat and a software application displays the NMI signals on a computer monitor or laptop. When a semiderivative electrical circuit is used, the voltage is usually in the unit of millivolts
NMI has made several advances in the field of electrochemistry. One of these significant advances is the fact that NMI biosensors do not form gliosis (i.e., scar tissue), which impedes detection of neurotransmitters by causing electroactive signals to decay. This property improves the sensitivity, selectivity and operational stability of the biosensors, allowing the detection of reliable electroactive signals for long periods of time; this enables their use not only for diagnosis and treatment for PD, which we discuss here, but also for cardiac disease such as acute ischemic stroke (Broderick et al. 2009a, b, c) and hypoxia (Bekker et al. 2007; Haile et al. 2008, 2009), as well as for peripheral body disorders such as human uterine cervical cancer (Broderick 1989a, b, 1995, 1999; Broderick et al. 2009a). Another significant advance of NMI in concert with the series of nanobiosensors is unique calibration curves for neurochemicals delineated in situ at water and phosphatidyl interfaces in both human epilepsy patients and mammalian animals. In this manner, NMI has discovered a lipid amplification factor in brain matrix and body neurochemicals using a neuroprobe.
The series of nanobiosensors has successfully made the critical advance to clinical use by imaging neurotransmitters, neurochemicals, and peptides in the neocortex of epilepsy patients on line, in real time and in vivo during intraoperative surgery. It is important to note that the pathology reports from epilepsy patients show that the series of nanobiosensors do not produce gliosis/sclerosis/scar tissue in human studies; moreover, these nanobiosensors do not produce bacterial growth in either animals or humans (Broderick et al. 2009a) as presented at the American Epilepsy Society (Broderick et al. 2009a, b, c) (Table 1).
Results
Live neurotransmitter profiles in the dorsal striatum of the Parkinson’s intact brain in the natural state on line, in vivo and in real time without treatment and data are compared with neurotransmitter profiles in the dorsal striatum of a normal, non-Parkinson’s intact brain. These data are shown in Fig. 5a, b.

a, b A representative example of neurotransmitter profiles imaged directly by NMI and the BRODERICK PROBE® nanobiosensor in the intact dorsal striatum of the murine Parkinson’s brain (a). This Parkinson brain image is then compared with neurotransmitter profiles imaged directly in the intact dorsal striatum of the murine non-Parkinson’s brain, that is, the non-Parkinson’s brain image (b). NMI endogenous (baseline, control) neurotransmitter chemical signature profiles in the dorsal striatum of the Parkinson’s subject (a) and the endogenous (baseline, control) non-Parkinson’s animal (b) are displayed in vivo, on line and in real time, within milliseconds. BRODERICK PROBE® nanobiosensors exhibit excellent properties for reliable operational stability and superior temporal and spatial resolution over periods of months. Gliosis (brain scarring, sclerosis) using the BRODERICK PROBE® is not significant as reported using post-operative histological studies by NYU Pathology studies. Moreover, these nanobiosensors do not promote bacterial growth, neither in sterilized nor in unsterilized forms, as reported by NYU Immunology studies. Neurotransmitter profiles are drawn directly from original in vivo data as videotracking of live signals simultaneously appear on the computer monitor. Abbreviations for a: 5-HT Serotonin, L-trp L-tryptophan, Dyn Dynorphin A 1-17, SRIF Somatotropin Release-Inhibiting Factor. Abbreviations for b: DA Dopamine, 5-HT Serotonin, HVA Homovanillic acid, L-trp L-tryptophan
This is the first report of an in vivo, live neurochemical comparison between the Parkinson’s and the non-Parkinson’s brain in the natural state and with and without l-DOPA. In addition, it is the first report of l-DOPA’s impact upon both Parkinson’s and non-Parkinson’s neuronal circuitry. The data are derived from six Parkinson’s animal models and another additional six non-Parkinson’s animal models. In this research, we examined the DAergic motor neurons of the dorsal striatal brain with and without administration of l-DOPA, the current pharmacotherapeutic mainstay for Parkinson’s. A previous in vivo report of live neurochemical comparisons between Parkinson’s versus non-Parkinson’s motor neurons in the striatal brain was also conducted by the Broderick Lab. Moreover, in this past study, we investigated the activity of motor neurons with and without administration of the DA agonist, bromocriptine (Broderick and Kolodny 2009).
Both the present report and the Broderick and Kolodny (2009) research are critical to the process of describing exactly what happens to live neurochemistry during the progressive neurodegeneration of the Parkinson’s brain. The dramatic deficiency of some neurochemicals versus the striking excess of others in both the Parkinson’s and non-Parkinson’s brain is fascinating. Indeed, neuronal brain differences endogenously present in the Parkinson’s brain were demonstrated by the fact that some Parkinson’s animals imaged 5-HT release, whereas others did not produce detectable concentrations of 5-HT.
Thus, the biomarkers are (a) the absence of presynaptic DA release; (b) the presence or absence of catecholamine, 5-HT, release; (c) the upregulation or downregulation of L-TP release; (d) the presence of Dyn A (1–17) and SRIF release; and (e) the presence of presynaptic release of the biomarker occurring at redox peak 0.83 V. It is noteworthy that substantia nigra lesioning was performed by expert animal surgeons at the Charles River Facility in North Carolina, USA using the exact same stereotaxic coordinates in all animals for both bromocriptine and l-DOPA studies with weight and age kept constant. Yet, both Parkinson’s animals receiving bromocriptine and l-DOPA showed different degrees of neurodegeneration a priori. This clearly demonstrates that precise differences in the milieu of brain neurotransmitters occur within each Parkinson’s patient. Thus, the need for Personalized Medicine is emphasized.
Live neurotransmitter profiles in the dorsal striatum of the Parkinson’s intact brain after l-DOPA treatment are shown in histogram form in Fig. 6.

A histogram is shown, illustrating the effects of l-DOPA administration on the Parkinson’s intact murine brain image (n = 6). It is noteworthy that only a small sample size is needed for accurate data as well as data at a statistically significant level. Statistical significance at the alpha level of p < 0.05 was used, and statistical significance is clear at the level of p < 0.05 to p < 0.001. Post-hoc analysis was conducted using Tukey’s test. The biomarkers L-TP, DYN A, and SRIF reach statistical significant over its own control at both the 50 and 100 mg kg−1 ip. Moreover, the effect of l-DOPA at the 50 and the 100 mg kg−1 dose in the Parkinson’s brain are essentially equivalent. Thus, a non-linear dose–response relationship for the effect of l-DOPA on the Parkinson’s brain image is clear. DA, 5-HT, and HVA profiles were below detectable limits in dorsal striatal Parkinson’s brain
Live neurotransmitter profiles in the dorsal striatum of the non-Parkinson’s intact brain after l-DOPA treatment are shown in histogram form in Fig. 7.

A histogram is shown, illustrating the effects of l-DOPA administration on the non-Parkinson’s intact murine brain image (n = 6). It is noteworthy that only a small sample size is needed for accurate data as well as data at a statistically significant level. Statistical significance at the alpha level of p < 0.05 was used, and statistical significance is clear at the level of p < 0.05 to p < 0.001. Post-hoc analysis was conducted using Tukey’s test. The biogenic biomarkers, metabolites and precursors reach statistical significance over its own control at both the 50 and 100 mg kg−1 ip. Moreover, the effect of l-DOPA at the 50 and 100 mg kg−1 dose in the murine non-Parkinson’s brain is dependent on and proportional to the dose of l-DOPA administered. Thus, a linear dose–response curve for the effect of l-DOPA on neurotransmitters is clear in the non-Parkinson’s brain image
This is the second report from the Broderick Lab showing an in vivo, live neurochemical comparison between the Parkinson’s and the non-Parkinson’s live and intact brain in the natural state uniquely enabled by the nanobiotechnology of nanobiosensors; each group was comprised of six study subjects, allowing a lesser number of animals to be studied with a reliable and accurate outcome. Thus, these data directly applicable to the clinic and to Parkinson’s disease are shown to be repeatable over time. In addition, it is the first report of l-DOPA’s impact upon both Parkinson’s and non-Parkinson’s neuronal circuitry, wherein we examined the DAergic motor neurons of the dorsal striatal brain with and without administration of l-DOPA. A previous in vivo report of live neurochemical comparisons between Parkinson’s versus non-Parkinson’s motor neurons in the striatal brain was also conducted by the Broderick Lab, wherein we investigated the DA agonist, bromocriptine (Broderick and Kolodny 2009).
Comparison of results from another pharmaceutical treatment, bromocriptine
Bromocriptine is a biphasic DA receptor agonist that induces excitatory effects at a low dose and inhibitory effects at a high dose. The biphasic property exhibited by bromocriptine presented here and previously presented by Broderick and Kolodny (2009) supports the findings of other investigators including (Monti et al. 1988). The data presented here are reprinted with permission from the publisher MDPI in Basel, Switzerland and Beijing, China. It is suggested insofar as bromocriptine is concerned that 5-HTergic function may be compensating for the lack of endogenous DA and HVA in the Parkinsonian dorsal striatal brain in addition to compensating for the decreased DA and HVA biphasic effect of the high-dose bromocriptine in the non-Parkinsonian dorsal striatal brain. Moreover, we also agree with other researchers such as Monti et al. (1988) who state that the DA agonist activity is possibly derived from activation of presynaptic D2 axons in the non-Parkinson’s animal model.
Temporal course data showing live neurotransmitter profiles in the dorsal striatum of the Parkinson’s intact brain after low-dose bromocriptine treatment are shown in line graph form in Fig. 8a.

a, b Temporal course line graphs for the effects of the DA agonist, bromocriptine, on live neurotransmitter image profiles in the murine Parkinson’s intact brain (a) compared with the murine non-Parkinson’s intact brain (b) at the 5 mg kg−1 dose (low dose) of bromocriptine. Reprinted with permission from the publisher, MDPI, Basel, Switzerland with Beijing, China
Temporal course data showing live neurotransmitter profiles in the dorsal striatum of the non-Parkinson’s intact brain after low-dose bromocriptine treatment are shown in line graph form in Fig. 8b.
Figure 8a, b shows the first in vivo neurochemical profile study of Parkinson’s disease in any neuroanatomic substrate of animal after a low dose of bromocriptine. The present data are derived from ten NMI studies in the dorsal striatum of Parkinson’s animals and are compared with another ten studies in non-Parkinson’s animals. The results showed that DA and its metabolite HVA were not imaged in the Parkinsonian striatal brain. What was readily apparent in the Parkinsonian striatal brain was the selective imaging of the neurotransmitter, 5-HT, and its precursor, L-TP. Moreover, two neurotransmitter peptides, Dyn A and SRIF, were also separately imaged in the Parkinsonian striatal brain. Repeatedly imaged to a significant degree was a peptide at an oxidation/half-wave potential of about 0.83 V, which this laboratory is in the process of defining. These peptide neurotransmitters were not imaged in the non-Parkinsonian dorsal striatal brain. In the non-Parkinson’s dorsal striatum, selective electroactive signals for DA, 5-HT, HVA, and L-TP were repeatedly imaged.
Temporal course data showing live neurotransmitter profiles in the dorsal striatum of the Parkinson’s intact brain after low- and after high-dose bromocriptine treatments are shown in line graph form in Fig. 9a.

a, b Temporal course line graphs for the effects of the DA agonist, bromocriptine, on live neurotransmitter image profiles in the murine Parkinson’s intact brain (a) compared with the murine non-Parkinson’s intact brain (b) at the 5 mg kg−1 dose (low dose) followed by the 10 mg kg−1 ip dose (high dose) of bromocriptine. Reprinted with permission from the publisher, MDPI, Basel, Switzerland with Beijing, China
Temporal course data showing live neurotransmitter profiles in the dorsal striatum of the non-Parkinson’s intact brain after low- and high-dose bromocriptine treatments are shown in line graph form in Fig. 9b.
In Fig. 9b, high-dose bromocriptine showed significantly greater effects on 5-HT and L-TP in Parkinson’s versus non-Parkinson’s animals (ANOVA; p < 0.0001). It is suggested, as previously mentioned, that 5-HTergic function may be compensating for the lack of endogenous DA and HVA in the Parkinsonian dorsal striatal brain in addition to compensating for the decreased DA and HVA biphasic effect of the high-dose bromocriptine in the non-Parkinsonian dorsal striatal brain.
It is important to reiterate that the high dose of bromocriptine exhibits its inhibitory action on DAergic function in the non-Parkinsonian dorsal striatal brain. Both DA and HVA are decreased in non-Parkinson’s animals as expected due to the biphasic dose-dependent property of bromocriptine. In contrast, the dorsal striatal brain of Parkinson’s animals did not exhibit any DAergic function, as expected.
An interrogative perspective on bromocriptine: a bromocriptine assay test may be used to rule out a clinical diagnosis of Parkinson’s disease
Our research demonstrated that bromocriptine exhibited a surprising effect in non-Parkinson’s animals in that the biogenic amine release, although significant, was dramatically less than that observed after l-DOPA treatment. Perhaps this finding could be translated into a clinical test for Parkinson’s disease in animals and/or humans. Given the caveat of the “gut to brain” phenomenon and that 5-hydroxyindolacetic acid (5-HIAA) is a metabolite of 5-HT, serum 5-HIAA could be measured after bromocriptine administration to assess Parkinson’s neurodegeneration.
Discussion
The series of nanobiosensors with NMI presents the uniquely interrelated marriage between the brain and nanotechnology. These nanobiosensors were used to image neurotransmitters in vivo, on line, in real time in milliseconds both with and without l-DOPA. In particular, NMI enabled the selective and separate imaging of each neuromolecule, such as DA, 5-HT, DA metabolite HVA, 5-HT, L-TP, a precursor to 5-HT, as well as peptide neurotransmitters such as Dyn A (1–17) and SRIF in the natural state, which is unique to in vivo studies. Indeed, NMI allows studies in real time in the diseased or dysfunctional state even during behavioral activities. These studies allowed for the first time, the establishment of a closer cause and effect relationship among the natural condition of neurons, brain disorder and therapy. Each neurotransmitter produced a characteristic experimentally derived electrochemical signature. Imaging was performed in the dorsal striatal brain of Parkinsonian and non-Parkinsonian murine models. In Parkinson’s animals, results demonstrated that neither presynaptic release of DA nor 5-HT was above detectable limits, while the precursor to 5-HT, L-TP, in addition to the peptide neurotransmitters Dyn A (1–17) and SRIF were significantly above detectable limits. In contrast, non-Parkinson’s animals imaged presynaptic release of DA, 5-HT, HVA and L-TP, while the peptide biomarkers Dyn A (1–17) and SRIF were not above detectable limits. Thus, our data show that Parkinson’s biomarkers are consistently detected as the absence of the biogenic amine, DA, the presence or absence of 5-HT release, as well as the clear images of the peptides Dyn A, SRIF and a 0.83 V neuromolecule. Indeed, biogenic amines and peptide neurotransmitters likely signal stages of neurodegeneration. Furthermore, as presented at the American Epilepsy Foundation, Dec 2009, we hypothesized that Dyn A (1–17) is excitatory, because it contains glutamate in its amino acid structure (Sigma Aldrich Inc., St. Louis Missouri), whereas SRIF is neuroprotective as demonstrated by enhanced motor function after the administration of somatostatin in the Parkinsonian mammal (Lu and Stoessel 2002).
Nanotechnology platforms for electrochemical detection of DA, 5-HT, AA, UA, and α-synuclein in Parkinson’s disease
Before the advent of nanotechnology and nanobiotechnology to assist in Parkinson’s diagnosis and treatment, medical protocols relied primarily on clinical acumen, symptomatology and behavioral biometrics. We now realize that the detection of DA itself is a biomarker for Parkinson’s disease (Adhikary et al. 2015).
DA biomarkers
DA is a known biomarker for Parkinson’s disease (Cipriani et al. 2010; Hass et al. 2012; Kish et al. 1988). Tashkhourian et al. (2009) reported in vitro work on a silver/carbon electrode in the nanotube structure. Silver nanoparticles were embedded into the carbon, so that the charge transfer kinetics allowed further electrocatalytic conversion of the AA/DA interaction for detection of both molecules in one signal. The result is an electrochemical signal that oxidizes AA and DA each in the same signal and at the same high uM concentrations. This leads to an efficient electrochemical catalysis; yet, it is catalysis with less selectivity for each molecule but higher sensitivity for molecules when these are oxidized in combination. Such a phenomenon may be useful for many applications but insofar as in vivo work is concerned, this appears to be a less optimal option.
In yet another modification for detection of the DA biomarker for Parkinson’s, Kurzatkowska et al. (2009) developed an electrode consisting of ion-channel mimetic self-assembled monolayers of macrocyclic polyamines deposited onto gold electrodes for electrochemical determination of DA. The corrole molecules covalently absorbed onto the metal surface making the electrode surface semi-permeable to a redox marker. When DA, in the form of a corrole DA complex, is added, a positive charge forms on the monolayer and allows detection of the cation, DA. The different compositions of the modification solution consist of a corrole-SH, a thiol derivative. Some of the background compounds included 1-dodecanethiol (DDT), 6-mercapto-1-hexanol [HS(CH(2))(6)OH], or 11-mercapto-1-undecanol [HS(CH(2))(11)OH]. The most effective modification for DA detection was the self-assembled monolayer (SAM). Among them, the mixed SAM comprised of corroles with the –SH group and 6-mercapto-1-hexanol [HS(CH(2))(6)OH] in the molar ratio 1:10 was the most sensitive. The signals, generated by the formation of a complex between the corrole host and the DA guest, were measured using a square-wave voltammetric circuit according to Osteryoung (OSWV), and electrochemical impedance spectroscopy (EIS) with [Ru(NH(3))(6)](3+) was used as an electroactive marker. Lowest detection limits were in the pM range.
UA and AA are biomarkers for Parkinson’s disease
A study by Yue et al. (2014) reported a DA electrochemical signal detected along with separate electrochemical signals for UA and AA. The investigators utilized ZnO assembled with graphene nanowire arrays comprised of individual nanowires that were less than 100 nm in diameter and a couple of microns in height; a differential pulse voltammetric electrical circuit was used to apply the potential to the electrode. The addition of nanowires can lead to high electrical conductivity of electrochemical biosensors, thus making this nanotechnology amenable to study the serum concentrations of DA and UA in Parkinson’s patients. The peak near 0.15 V corresponds to DA oxidation but DA overlaps with signals from NE, epinephrine, and l-DOPA excluding the possibility of its use as a DA biomarker for Parkinson’s. The peak near 0.29 V originates from UA. The field testing of this array led to discoveries in Parkinson’s disease. The results of the study showed a reduced UA level in the serum of seven Parkinson’s patients compared to healthy individuals who can make UA. Therefore, UA is a potential biomarker in the diagnosis of Parkinson’s, and this ZnO NWA/GF electrode may be a promising candidate for electroanalysis applications in Parkinson’s.
The biomarker α-synuclein is detected via H2O2
An et al. (2010) developed highly ordered microfabricated arrays using gold-doped TiO2 nanotubes for photoelectrochemical detection of α-synuclein. This is an immunosensor that employs TiO2 nanotubes as a substrate for the determination of proteins in photoelectrochemical bioanalysis. The highly ordered TiO2 nanotube arrays were fabricated by the electrochemical anodic oxidation technique. The arrays were effective platforms for the immobilization of primary antibodies while retaining their stability and α-synuclein binding (An et al. 2010). Then, the attachment of a secondary antibody and gold nanoparticle-conjugated glucose oxidase allowed higher sensitivity by signal amplification. Glucose oxidase catalyzed the conversion of glucose into gluconic acid and H2O2. Upon irradiating the other side of the titanium foil, the holes that were formed within the valence band of the nanotubes could be scavenged by H2O2 leading to a photocurrent proportional to concentrations of α-synuclein with a detection concentration in the range of pg/ml. The peroxide acted as a sacrificial electron donor to scavenge the holes generated in the valence band of TiO2 nanotubes upon irradiation of the other side of the Ti foil, thereby improving the accumulation of electrons and leading to a remarkable photocurrent. The α-synuclein concentration, which was proportional to that of the GOx labels linked to Au nanoparticles, could be readily examined through measurement of the photocurrent derived from the photoelectrochemical reaction of H2O2. Under optimal conditions, the linear relationship between the photocurrent and α-synuclein concentration was obtained in the range of 50 pg mL−1 to 100 ng mL−1. The detection limit was estimated to be 34 pg mL−1, and α-synuclein concentration was obtained from the photoelectrochemical reaction ending in the byproduct, H2O2. The peroxide byproduct is proportional to the α-synuclein concentration, and α-synuclein per se is not detected at all.
DA and AA are biomarkers for Parkinson’s disease
The Broderick Laboratory has discovered that DA is a biomarker for Parkinson’s disease in vivo and on line by looking directly at the Parkinson’s brain with a mini-implantable nanobiosensor, smaller than one human hair, in conjunction with NMI techniques at a p value ≤0.0001. When looking at the Parkinson’s brain as electrochemical redox reactions occurring at the very neuronal site for Parkinson’s inception, DA is clearly absent. DA is below detectable limits in each of the 16 Parkinson's animal models studied previously by Broderick and Kolodny (2009) and in the work described herein in the present paper. Therefore, the Broderick Laboratory data supports and confirms the conventional data from so many other investigators that DA or lack thereof is essential to study Parkinson’s syndrome, although these investigators did not perform studies in vivo. The Broderick Lab has not seen AA as a biomarker for Parkinson’s in these 16 animals but has interestingly identified an association between DA and AA in resected tissue from neocortical temporal lobe epilepsy patients versus mesial temporal lobe epilepsy patients. Therefore, this team has seen AA to be a reliable biomarker for epilepsy, another neurodegenerative disease, and has actually imaged AA as a biomarker in epilepsy patients, especially in the subtypes of temporal lobe epilepsy that is primarily neocortical rather than mesial (Broderick et al. 2000, 2009a, b, c; Broderick and Pacia 2005). As demonstrated, the use of NMI improves the detection of DA, AA, UA, and NE. In particular, NMI and a series of nanobiosensors have demonstrated a 0.50 V difference in the oxidation potential of NE versus DA (Broderick 1988).
5-HT and L-TP are biomarkers for Parkinson’s disease: the Yin Yang hypothesis
The Broderick team is the first to identify 5-HT and L-TP as reliable biomarkers responsible for the etiology of Parkinson’s disease. This is clear as some, but not all, of the murine Parkinson’s animals imaged the 5-HT indoleamine neurotransmitter as it was released in the dorsal striatum above detectable limits. Moreover, increased 5-HT release is concomitant with decreased L-TP release and vice versa only when Parkinson’s disease is present. The inverse relationship between 5-HT and L-TP demonstrates the Yin and Yang hypothesis, which highlights the opposite yet complementary relationship between the biomarkers 5-HT and L-TP. Indeed, due to the interconnection between 5-HT and L-TP, the presence or absence of 5-HT and L-TP is highly specific at the p ≤ 0.0001 level, because NMI is a technology that uniquely images the baseline state of subjects before a drug is administered or a disease state occurs. In the same subject, with no gliosis, the drug or diseased condition is imaged. Then, in the next phase, the treatment is imaged in the same subject and baseline profile returns to control values. Such imaging is available only with NMI nanobiosensor technology.
In fact, with this new NMI nanobiosensor technology, specificity is high because of the ability of NMI to compare baselines in thousands of studies in non-prescient mammalians in vivo. This ability to compare baselines during drug and disease studies clearly reports that Parkinson’s is the only disease in which 5-HT release is absent during baseline. Indeed, after examining neurodegenerative diseases, such as stroke and hypoxia, as well as psychiatric diseases, including anxiety, bipolar disorder, depression, drug addiction, drug reinforcement, and schizophrenia in the animal model, none of these brain disorders display an absence of 5-HT during baseline. Furthermore, animals with Parkinson’s that have no 5-HT release also exhibit more dramatic movement disorders. In either intraoperative human models or human tissue resections, 5-HT release is absent during later stages of neurodegeneration. Conversely, 5-HT is above detectable limits in human tissue resections that are harvested during early stages of neurodegeneration. Thus, the degree of immobility with absence of 5-HT release may reflect the degree of neurodegenerative progress.
Thus, the 5-HT biosynthesis pathway may be seriously considered as a therapeutic target. Contrary to the single-step DA biosynthetic pathway needed for Parkinson’s treatment, the anabolic 5-HT pathway involves two steps mediated by two enzymes. These 5-HT anabolic enzymes are tryptophan hydroxylase and aromatic-l-amino acid decarboxylase (AAAD).
Thus, the two-step anabolic 5-HT pathway initially involves the enzyme tryptophan hydroxylase. Tryptophan hydroxylase is the enzyme responsible for transforming tryptophan to 5-hydroxytryptophan and is the rate-limiting enzyme in the 5-HT biosynthesis pathway. Tryptophan hydroxylase is activated by phosphorylation via protein kinase A and calcium/calmodulin kinase (Kuhn et al. 1997). Phosphorylating conditions are defined as an ATP-Mg2+-rich environment that triggers a calcium-dependent, cyclic AMP-independent mechanism of phosphorylation and thereby may be a target for Parkinson’s treatment.
The second enzyme in the two-step anabolic 5-HT pathway is AAAD. AAAD is the enzyme responsible for converting 5-hydroxytryptophan to 5-HT. AAAD activity is regulated by activation and induction by second messengers, and interestingly, l-DOPA decarboxylase activity reflects AAAD activity as well. Thus, drugs that antagonize and enhance AAAD activity can be used to indirectly modulate l-DOPA decarboxylation in parallel and, thus, may effectively treat Parkinson’s by increasing 5-HT production. Indeed, based on a review conducted by Hadjiconstantinou and Neff (2008), l-DOPA adjuvants amantadine, budipine, and memantine effectively augment AAAD activity and l-DOPA decarboxylation to decrease Parkinsonian symptoms and motor impairments in animal studies. Similarly, the atypical antipsychotic clozapine increases AAAD activity and l-DOPA decarboxylation in Parkinson’s models (Hadjiconstantinou and Neff 2008). Moreover, L-TP dehydrogenase and monoamine oxidase are two catabolic enzymes used in the 5-HT pathway. Thus, these two enzymes may be used as therapeutic targets in simili modo to the treatments for Parkinson’s that use the DA catabolic pathway.
Another important therapeutic target is the 5-HT autoreceptor. An autoreceptor is a presynaptic inhibitor, a priori, an antagonist. Consequently, autoreceptors, particularly antagonists, may provide treatment for Parkinson’s, and this phenomenon may be, again, in simili modo to established treatment for DAergic Parkinson’s pharmacotherapeutics. Based on this hypothetical paradigm, Parkinson’s disease may actually stem from an autoreceptor defect as a presynaptic 5-HT release mechanism.
Also in the Broderick Laboratory, peptide neurotransmitter biomarkers, such as dynorphin and somatostatin, were identified, as well as another intriguing biomarker at oxidation potential 0.83 V that has yet to be fully characterized. A myriad of neurotransmitters, including DA, AA, and 5-HT, was selectively imaged using a polymeric carbon-based nanobiosensor anion–cation exchange mechanism with a semiderivative reduction circuit in vivo, in vitro and in situ. Furthermore, the Broderick lab has also identified critical peptide neurotransmitters such as dynorphin and somatostatin in the basal ganglia of Parkinson’s animals. Due to the identification of common biomarkers for both epilepsy and Parkinson’s, we posit that these diseases share common pathological pathways akin to Parkinson’s and Alzheimer’s that may share a common pathogenesis.
Nanotechnology for peptide Biomarkers
Stress proteins either incite neuronal damage or are a by-product and, thus, a biomarker of neuronal pathology. The neurotransmitter peptide, dynorphin, is a stress protein, and much more research is needed to clarify exactly which forms of dynorphin are neuroprotective versus neurotoxic, and this particular point cannot be stressed enough. In the present paper and also in a previous paper from the Broderick Laboratory (Broderick and Kolodny 2009), we determined that Dyn A (1–17) was excitatory, because it contains glutamate and is, thus, neurotoxic. Consequently, it is clear that Dyn A (1–17) is a biomarker for Parkinson’s disease (Broderick and Kolodny 2009).
The clinical usage of dynorphin as a stress protein biomarker of functional DA neurons is supported by numerous studies. In 1991, Engber et al. reported dynorphin downregulation in the striatal neurons of rats made Parkinsonian via 6-OHDA lesioning. In 1994, Xu et al. discovered that D1 receptor mutant mice exhibiting hyperactive locomotion and failure to respond to D1 receptor agonists and antagonists had greatly reduced expression of dynorphin in the striatum and basal ganglia. Similarly, based on their previous in vitro research demonstrating that exogenous dynorphin is a potent neuroprotective agent against DAergic neurodegeneration incited by inflammation, Wang et al. (2012) administered neurotoxins MPTP and methamphetamine to Parkinsonian rats with either endogenous prodynorphin, the precursor to dynorphin, or without endogenous prodynorphin. Neurotoxin administration caused behavioral impairments, substantia nigral and striatal DAergic neuronal loss, depletion of DA and DA metabolites, and increased microglial activation in rats with and without endogenous dynorphin (Wang et al. 2012). However, rats deficient in endogenous dynorphin suffered more severe neurotoxin-induced damage with concurrent exacerbation of methamphetamine neurotoxicity compared to their endogenous prodynorphin counterparts. Thus, these in vivo results demonstrated that endogenous dynorphin inhibits inflammation and promotes DA neuron function (Wang et al. 2012).
However, despite current research supporting the neuroprotective effects of dynorphin, we would like to reiterate that older research revealed certain forms of dynorphin to be neurotoxic. Faden and Jacobs (1984) reported that intrathecal administration of dynorphin 1–17, dynorphin 1–13, dynorphin 1–8, des-Tyr-dynorphin (DYN 2–17), and α-neo-endorphin produced dose-related, non-opiate-mediated flaccid, hindlimb paralysis in non-prescient mammals that was unresponsive to blocking or reversal by the opiate receptor antagonist naloxone. Moreover, Hugonin et al. (2008) conducted a structural analysis of dynorphin neuropeptides after sodium dodecyl sulfate administration and found that “big dynorphin” protein is neurotoxic due to its dynorphin A and B composite secondary structure which synergistically aids “big dynorphin” in cell membrane disruption (Hugonin et al. 2008). Thus, more research is needed to clarify exactly which forms of dynorphin are neuroprotective versus neurotoxic. Once again, this particular point cannot be stressed enough.
Somatostatin
Another biomarker, somatostatin, simply does not co-localize with α-synuclein positive cells in Parkinson’s disease. Indeed, in the human anterior olfactory nucleus, olfactory bulb, and olfactory cortices in Parkinson’s patients, somatostatin rarely co-localized with cells containing α-synuclein and, thus, were unaffected by neurodegeneration (Ubeda-Bañon et al. 2010). Similarly, mouse hippocampal neuronal cultures comprised of inhibitory neurons naturally had either weak or absent expression of α-synuclein, which produced positive somatostatin immunostaining. Conversely, excitatory mouse hippocampal neuronal cultures expressed high levels of α-synuclein, which produced negative somatostatin immunostaining. In vitro double immunofluorescence with antibodies against α-synuclein and somatostatin confirmed that somatostatin-positive inhibitory neurons have minimal expression of α-synuclein (Taguchi et al. 2014). Interestingly, Rubio et al. (2012) report that β-amyloid in Alzheimer’s disease actually increases somatostatin and cortistatin gene expression via increased histone 3 lysine 4 methylation with transcriptional activation. In this manner, somatostatin and cortistatin in primary cortical neurons partially decreased β-amyloid toxicity in vitro, suggesting that somatostatin and cortistatin are neuroprotective agents released by neurons to shield against β-amyloid insult (Rubio et al. 2012; Sharrad et al. 2013).
Nanotechnology for neuroblastoma that may be extrapolated to Parkinson’s disease via mitochondrial, electrochemical redox mechanics
Ma et al. (2013) have discovered a QD biosensor, ten thousand times smaller than one human hair, which has powerful optical fluorescence sensing ability and with results displayed as wavelength frequency. These scientists have manufactured a QD crystal biosensor to monitor the progression of Parkinson’s disease. Thus, QD biosensors may be particularly useful in early diagnosis of Parkinson’s disease during early stages 3–5. The authors based their idea of QDs on SY5Y neuroblastoma cells. According to Xie et al. (2010), the SH-SY5Y cell line is increasingly used in Parkinson’s research as it shares many similarities with DAergic neurons such as DA-β-hydroxylase expression, TH expression, and DA transporter use. Moreover, differentiating agents may be used on SH-SY5Y cells to prime them for Parkinson’s studies examining neurotoxicity and neuroprotection (Xie et al. 2010). Thus, given the caveat that Ma et al. (2013) did not directly study Parkinson’s disease, their QD biosensors effectively measured neurodegeneration via redox reactions in mitochondria in situ. Correspondingly, their research is directly applicable to the study of SH-SY5Y neuroblastoma cells and may be indirectly related to the discovery of redox mechanisms in Parkinson’s disease.
The prion protein connection
Parkinson’s disease may have a prion-like pathogenesis as α-synuclein protein is a principal component of Lewy bodies and neurites (Olanow and Brundin 2013). Prion is a composite term derived by Prusiner (1982) from the words “proteinaceous” and infectious with “-on” added to the end of the word by homology to “virion.” The derivation of this word is fitting as Parkinson’s disease may be conceptualized as a protein infection. Indeed, α-synuclein gene point mutations, duplications, triplications, and single nucleotide polymorphisms are sufficient to cause Parkinson’s disease (Olanow and Brundin 2013) as demonstrated by a single intracerebral injection of human wild-type α-synuclein into transgenic and wild-type mice inducing the prion-like spread of α-synuclein in both Parkinson’s and multiple system atrophy animal models (Bernis et al. 2015).
To further elucidate the pathological mechanism of α-synuclein, Iljina et al. (2016) used single-molecule fluorescence and kinetic analysis to determine that α-synuclein self-assembles into small oligomers which compact into fibrils and lengthen via monomer addition. These fibrils are more effective at seeding the brain than their oligomer counterparts. Thus, the “prion hypothesis” of Parkinson’s disease involves α-synuclein’s conformational change to α-helical fibrils and oligomers that form β-sheet rich fibrils with resultant protein aggregation that migrates from affected to unaffected neurons causing further misfolding (Chu and Kordower 2015; Iljina et al. 2016; Olanow and Brundin 2013).
Another protein that incites proteopathy is P123H β-synuclein, a pathologic protein that receives less attention despite its association with numerous α-synucleinopathies, such as familial Lewy body dementia. P123H β-synuclein works in concert with α-synuclein to stimulate neurodegeneration in mouse models (Fujita et al. 2010). Indeed, the self-propagating seeding of neurons by α-synuclein with cooperation from β-synuclein is tantamount to the prion mechanism of neurodegeneration (Fujita et al. 2010; Luna and Luk 2015; Tyson et al. 2015; Rey et al. 2015). More importantly, the ability of α-synuclein to produce β-sheet amyloid plaques in a prion-like manner demonstrates that Parkinson’s disease shares a similar pathophysiology with prion disease, Alzheimer’s disease, and multiple system atrophy (Milisav et al. 2015; Prusiner et al. 2015; Rey et al. 2015). Fortunately, α-synuclein phosphorylated at serine 129 may be used as a pathological biomarker to identify neurodegeneration for the development of specific therapies (Stewart et al. 2015).
After α-synucleinopathy identification, many treatment modalities targeting the prion-like spread of neurodegeneration are available (Cooper et al. 2006). Milisav et al. (2015) suggest that the pathological unfolded protein response and macroautophagy involved in the pathogenesis of neuronal disease may be targeted by pharmacological treatments. More importantly, both Bieschke et al. (2012) and Lam et al. (2016) demonstrated in vitro that the orcein-related small molecule, O4, binds directly to hydrophobic amino acids in amyloid-β fibrils, stabilizes them, and accelerates mature, nontoxic amyloid-β fibrillogenesis to prevent prefibrillar α-synuclein substrates from catalyzing the prion-like spread of toxic amyloid-β oligomers via seeding. Thus, fibril stabilization is a potential therapeutic strategy for the treatment of neurodegenerative disease. More research is needed to determine the efficacy of utilizing P123H β-synuclein as a therapeutic target. Similarly, the structure and function of γ-synuclein, a disordered, small protein found in the peripheral nervous system that is overexpressed in the late stage breast cancer (Yerramilli et al. 2013), requires further study to determine its potential as a therapeutic target.
Finally, in prion disease itself, nanotechnology may detect prion protein to diagnose this form of neurodegeneration. Nanotechnology may also distinguish the infectious β-structure insoluble conformer prion protein from the less infectious α-helix rich prion protein via two aptamers capable of recognizing these two distinct epitopes in serum and brain homogenate (Xiao et al. 2010). Similarly, specific sites of prion protein expression on cell surfaces may be labeled in vitro using site-specific labeling of prion protein ex PEG-interspersed nitrilotriacetic acid-functionalized QDs (Xie et al. 2010). To treat prion disease, in vitro studies demonstrate that polyamine dendrimers remove pathogenic β-structure insoluble conformer prion protein from infected cells by transporting them to lysosomal degradation (Lim et al. 2010; Ai Tran et al. 2010; Sousa et al. 2010). In vivo studies of nanotechnology targeting prion protein are needed.
Nanotechnologies for the treatment of Alzheimer’s disease
Engineered materials between 1 and 100 nm allow diverse therapeutic modalities to cross the blood brain barrier via nanoparticles that diagnose and treat neurodegeneration without disrupting normal barrier function (Badilescu and Packirisamy 2012; Karnik et al. 2008; Muthu and Singh 2009; Re et al. 2012). Indeed, the blood brain barrier is a major obstacle to treating neurodegenerative disorders such as Alzheimer’s disease and ALS. Fortunately, nanobiotechnology may measure pathogenic markers in human cerebrospinal fluid such as soluble amyloid-β oligomers and tau protein to diagnose Alzheimer’s in vitro, as well as detect amyloid-β deposits in the brain in vivo (Georganopoulou et al. 2005). To treat Alzheimer’s disease, administration of peripheral nanoparticles with high Aβ affinity sequesters peptides to the plasma, thereby exploiting the “sink effect” to prevent or reduce brain amyloidosis (Matsuoka et al. 2003; Robinson et al. 2015). Similarly, hydrated fullerene C60 nanoparticles may centrally interfere with Aβ aggregation in the Alzheimer’s murine model (Matsuoka et al. 2003; Podolski et al. 2007). Nanoparticles may also prevent neuronal cell oxidative damage to alleviate Alzheimer’s symptoms (Re et al. 2012). In the murine model of ALS, solid lipid nanoparticles carrying riluzole successfully crossed the blood brain barrier to treat neurodegeneration (Bondi et al. 2010). However, further research on nanobiotechnology for ALS diagnosis and treatment is needed.
Conclusion
Nanobiotechnology surmounts the medical challenges in the diagnosis and treatment of Parkinson’s with a penchant for detail that was previously unattainable. Inroads to the underlying brain mechanics responsible for neurodegenerative disease are not only possible but probable given the fact that research in this area has been met with success. In particular, insights into Parkinson’s, Alzheimer’s, and the truly elusive Lou Gehrig’s disease have provided a novel therapeutic arsenal against these disruptive disorders as described herein. The series of nanobiosensors with NMI, a particularly effective component of the nanobiotechnology repository, is not just one sensor or nanobiosensor; rather, it is comprised of thousands of tested formulas for subtypes of sensors and sensor platforms that are already available for use in multifunctional assays for diverse purposes in the living patient and animal. It is likely that intimately related genes and neurosignals could be an answer to some therapeutic problems. Indeed, NMI deciphers molecular biomarkers for such treatment challenges and targets these neuromolecules in real time to craft “cause and effect” roadmaps enabling optimal outcomes in Personalized Medicine.
Abbreviations
- Ach:
-
Acetylcholine
- ALS:
-
Amyotrophic lateral sclerosis
- AA:
-
Ascorbic acid
- BDNF:
-
Brain-derived neurotrophic factor
- DA:
-
Dopamine
- Dyn A:
-
Dynorphin A 1-17
- Enk:
-
Enkephalin
- GABA:
-
Gamma-aminobutyric acid
- GDNF:
-
Glial cell line-derived neurotrophic factor
- GPe:
-
Globus pallidus, external circuit
- GPi:
-
Globus pallidus, internal circuit
- GAD:
-
Glutamic acid decarboxylase
- Au:
-
Gold
- GF:
-
Graphene foam
- GCH:
-
GTP cyclohydrolase
- HVA:
-
Homovanillic acid
- H2O2 :
-
Hydrogen peroxide
- 6-OHDA:
-
6-Hydroxydopamine
- i.p.:
-
Intraperitoneal
- AADC:
-
l-Amino-acid decarboxylase
- Levodopa, l-DOPA:
-
l-3,4-Dihydroxyphenylalanine
- l-TP:
-
l-Tryptophan
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NWA:
-
Nanowire Arrays
- NWA/GF (ZnO):
-
Nanowire arrays/graphene foam/zinc oxide
- NMI:
-
Neuromolecular Imaging
- NE:
-
Norepinephrine
- PEG:
-
Polyethylene glycol
- 5-HT:
-
Serotonin
- SRIF:
-
Somatostatin releasing inhibitory factor
- SNc:
-
Substantia nigra pars compacta
- SNr:
-
Substantia nigra pars reticulata
- SP:
-
Substance P
- STN:
-
Subthalamic nucleus
- Ti:
-
Titanium
- TiO2 :
-
Titanium oxide
- TH:
-
Tyrosine hydroxylase
- UA:
-
Urate/uric acid
- QD:
-
Quantum dot
- ZnO:
-
Zinc oxide
References
Adhikary RR, Sandbhor P, Banerjee R (2015) Nanotechnology platforms in Parkinson’s disease. ADMET DMPK 3(3):155–181
Ai Tran HN, Sousa F, Moda F, Mandal S, Chanana M, Vimercati C, Morbin M, Krol S, Tagliavini F, Legname G (2010) A novel class of potential prion drugs: preliminary in vitro and in vivo data for multilayer coated gold nanoparticles. Nanoscale 2(12):2724–2732
An Y, Tang L, Jiang X, Chen H, Yang M, Jin L, Zhang S, Wang C, Zhang W (2010) A photoelectrochemical immunosensor based on au-doped TiO2 nanotube arrays for the detection of α-synuclein. Chem Eur J 16(48):14439–14446
Badilescu S, Packirisamy M (2012) Microfluidic-nano-integration for synthesis and sensing. Polymers 4(2):1278–1310
Bekker A, Haile M, Gingrich K, Wenning L, Gorny A, Quartermain D, Blanck T (2007) Physostigmine reverses cognitive dysfunction caused by moderate hypoxia in adult mice. Anesth Analg 105(3):739–743
Bemelmans AP, Horellou P, Pradier L, Brunet I, Colin P, Mallet J (1999) Brain-derived neurotrophic factor-mediated protection of striatal neurons in an excitotoxic rat model of Huntington’s disease, as demonstrated by adenoviral gene transfer. Hum Gene Ther 10(18):2987–2997
Bergman H, Wichmann T, Delong MR (1990) Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249(4975):1436–1438
Bernis ME, Babila JT, Breid S, Wüsten KA, Wüllner U, Tamgüney G (2015) Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun 3(1):75
Bhave SV, Ghoda L, Hoffman PL (1999) Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci 19(9):3277–3286
Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, del Amo JML, Grüning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fändrich M, Frank RF, Reif B, Günther S, Walsh DM, Wanker EE (2012) Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat Chem Biol 8:93–101
Biomarkers Definitions Working Group (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 69(3):89–95
Birkmayer W, Riederer P, Youdim MBH, Linauer W (1975) The potentiation of the anti akinetic effect after l-Dopa treatment by an inhibitor of Mao-B, deprenil. J Neural Transm 36(3):303–326
Blum D, Torch S, Lambeng N, Nissou MF, Benabid AL, Sadoul R, Verna JM (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol 65(2):135–172
Bohn MC, Choi-Lundberg DL (1998) Gene therapies for Parkinson’s disease. In: Chiocca EA, Breakefield XO (eds) Gene therapy for neurological disorders and brain tumors. Human Press, New Jersey, pp 377–395
Bondi ML, Craparo EF, Giammona G, Drago F (2010) Brain-targeted solid lipid nanoparticles containing riluzole: preparation, characterization and biodistribution. Nanomedicine 5(1):25–32
Broderick PA (1988) Distinguishing in vitro electrochemical signatures for norepinephrine and dopamine. Neurosci Lett 95:275–280
Broderick PA (1989a) Cathodic electrochemical current arrangement with telemetric application. US Patent # 4, 883,057, Issued
Broderick PA (1989b) Characterizing stearate probes in vitro for the electrochemical detection of dopamine and serotonin. Brain Res 495:115–121
Broderick PA (1991) In vivo voltammetric studies on release mechanisms for cocaine with gammabutyrolactone. Pharmacol Biochem Behav 40:969–975
Broderick PA (1995) Microelectrodes and their use in cathodic electrochemical current arrangement with telemetric application. US Patent # 5,433,710, Issued
Broderick PA (1999) Microelectrodes and their use in an electrochemical arrangement with telemetric application. US Patent # 5,938,903, Issued
Broderick PA (2008) Studies of oxidative stress mechanism using a morphine/ascorbate animal model and novel N-stearoyl cerebroside and laurate sensors. J Neural Transm 115(1):7–17
Broderick PA (2013) Neuromolecular imaging shows temporal synchrony patters between serotonin and movement within neuronal motor circuits in the brain. Brain Sci 3(2):992–1012
Broderick PA (2015) Noninvasive photonic sensor with polymer memory transduction using organic and inorganic elements as platforms. USPTO Patent Pending
Broderick PA, Kolodny EH (2009) Real time imaging of biomarkers in the Parkinson’s brain using mini-implantable biosensors. II. Pharmaceutical therapy with bromocriptine. Pharmaceuticals 2:236–249
Broderick PA, Pacia SV (2005) Imaging white matter signals in epilepsy patients: a unique sensor technology. In: Broderick PA, Rahni DN, Kolodny EH (eds) Bioimaging in neurodegeneration. Humana Press Inc, New Jersey, pp 199–206
Broderick PA, Pacia SV (2006) Identification, diagnosis, and treatment of neuropathologies, neurotoxicities, tumors, and brain and spinal cord injuries using microelectrodes with microvoltammetry. USPTO_US Patent #7,112,319, Issued
Broderick PA, Pacia SV (2011) Identification, diagnosis, and treatment of neuropathologies, neurotoxicities, tumors, and brain and spinal cord injuries using microelectrodes with microvoltammetry. USPTO USSN Patent #2011/13/083,810, Pending
Broderick PA, Piercey MF (1998) Neurochemical and behavioral evidence supporting (+)-AJ 76 as a potential pharmacotherapy for cocaine abuse. J Neural Transm 105(10):1307–1324
Broderick PA, Phelan FT, Lum JT, Hoffmann WE, Piercey MF (1989) Buspirone: effects on serotonin/norepinephrine cell firing rates and release mechanisms. Soc Neurosci Abstr 15:1097
Broderick PA, Pacia SV, Doyle WK, Devinsky O (2000) Monoamine neurotransmitters in resected hippocampal subparcellations from neocortical and mesial temporal lobe epilepsy patients: in situ microvoltammetric studies. Brain Res 878:49–63
Broderick PA, Ho H, Wat K, Murthy V (2008) Laurate biosensors image brain neurotransmitters in vivo: can an antihypertensive medication alter psychostimulant behavior? Sensors 8:4033–4061
Broderick PA, Doyle WK, Pacia SV, Kuzniecky RI, Devinsky O, Kolodny EH (2009a) A clinical trial of an advanced diagnostic biomedical device for epilepsy patients. J Long-Term Eff Med Implants 18:50
Broderick PA, Doyle WK, Pacia SV, Kuzniecky RI, Devinsky O, Kolodny EH (2009b) Intraoperative neuromolecular (NMI) in neocortex of epilepsy patients: comparison with resected neocortical epileptogenic tissue. Poster presentation at The American Epilepsy Society, Boston, MA, USA, December 4–8
Broderick PA, Ho H, Li YS, Kolodny EH (2009c) Lovenox® affects striatal neurotransmitters on line with laser Doppler flowmetry: neuromolecular imaging (NMI) in an animal model of acute ischemic stroke. Oral Presentation in Society for Neuroscence, Chicago
Carlsson A (1995) Towards a new understanding of dopamine receptors. Symposium: Dopamine receptor subtypes in neurological and psychiatric diseases. Clin Neuropharmacol 18(Suppl.):6S–13S
Charpentier E (2015) CRISPR-Cas9: how research on a bacterial RNA-guided mechanism opened new perspectives in biotechnology and biomedicine. EMBO Mol Med. doi:10.15252/emmm.201504847
Choi-Lundberg DL, Lin Q, Chang YN, Chiang YL, Hay CM, Mohajeri H, Davidson BL, Bohn MC (1997) Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science 275(5301):838–841
Chu Y, Kordower JH (2015) The prion hypothesis of Parkinson’s disease. Curr Neurol Neurosci Rep 15(5):28
Cipriani S, Chen X, Schwarzschild MA (2010) Urate: a novel Biomarker of Parkinson’s disease risk, diagnosis and prognosis. Biomark Med 4:701–712
Coffey ET, Åkerman KEO, Courtney MJ (1997) Brain derived neurotrophic factor induces a rapid upregulation of synaptophysin and tau proteins via the neurotrophin receptor TrkB in rat cerebellar granule cells. Neurosci Lett 227(3):177–180
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, LaBaer J, Rochet JC, Bonini NM, Lindquist S (2006) α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313(5785):324–328
Czub S, Koutsilieri E, Sopper S, Czub M, Stahl-Hennig C, Müller JG, Pedersen V, Gsell W, Heeney JL, Gerlach M, Gosztonyi G, Riederer P, ter Meulen V (2001) Enhancement of central nervous system pathology in early simian immunodeficiency virus infection by dopaminergic drugs. Acta Neuropathol 101(2):85–91
Danysz W, Parsons CG (2012) Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine—searching for the connections. Br J Pharmacol 167(2):324–352
Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, Marini P, Squitieri F, Horstink MW, Montagna P, Dalla Libera A, Stocchi F, Goldwurm S, Ferreira JJ, Meco G, Martignoni E, Lopiano L, Jardim LB, Oostra BA, Barbosa ER, Bonifati V (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68(19):1557–1562
Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science. doi:10.1126/science.1258096
Dugger BN, Serrano GE, Sue LI, Walker DG, Adler CH, Shill HA, Sabbagh MN, Caviness JN, Hidalgo J, Saxon-LaBelle M, Chiarolanza G, Mariner M, Henry-Watson J, Beach TG, Arizona Parkinson’s Disease Consortium (2012) Presence of striatal amyloid plaques in Parkinson’s disease dementia predicts concomitant Alzheimer’s disease: usefulness for amyloid imaging. J Parkinsons Dis 2(1):57–65
Eng F, Phelan FT, Wechsler RT, Piercey MF, Broderick PA (1992) The D3 antagonist, AJ76, increases accumbens dopamine and serotonin release: simultaneous detection on line with behavior. Soc Neurosci Abstr 18:1172
Engber TM, Susel Z, Kuo S, Gerfen CR, Chase TN (1991) Levodopa replacement therapy alters enzyme activities in striatum and neuropeptide content in striatal output regions of 6-hydroxydopamine lesioned rats. Brain Res 552:113–118
Faden AI, Jacobs TP (1984) Dynorphin-related peptides cause motor dysfunction in the rat through a non-opiate action. Br J Pharmacol 81(2):271–276
Fan DS, Ogawa M, Fujimoto KI, Ikeguchi K, Ogasawara Y, Urabe M, Nishizawa M, Nakano I, Yoshida M, Nagatsu I, Ichinose H, Nagatsu T, Kurtzman GJ, Ozawa K (1998) Behavioral recovery in 6-hydroxydopamine-lesioned rats by cotransduction of striatum with tyrosine hydroxylase and aromatic l-amino acid decarboxylase genes using two separate adeno-associated virus vectors. Hum Gene Ther 9(17):2527–2535
Ford B, Greene P, Louis ED, Petzinger G, Bressman SB, Goodman R, Brin MF, Sadiq S, Fahn S (1996) Use of intrathecal baclofen in the treatment of patients with dystonia. Arch Neurol 53(12):1241–1246
Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, Eidelberg D, Fahn S (2001) Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med 344(10):710–719
Fujita M, Sugama S, Sekiyama K, Sekigawa A, Tsukui T, Nakai M, Waragai M, Takenouchi T, Takamatsu Y, Wei J, Rockenstein E, LaSpada AR, Masliah E, Inoue S, Hashimoto M (2010) A β-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat Commun 1(110):1–9
Georganopoulou DG, Chang L, Nam JM, Thaxton S, Mufson EJ, Klein WL, Mirkin CA (2005) Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc Natl Acad Sci USA 102(7):2273–2276
Gerlach M, Riederer P, Scheller D (2011) Mechanisms underlying and medical management of l-Dopa-associated motor complications. J Neural Transm 118(12):1659–1660
Girisha HR, Narendra Sharath Chandra JN, Boppana S, Malviya M, Sadashiva CT, Rangappa KS (2009) Active site directed docking studies: synthesis and pharmacological evaluation of cis-2,6-dimethyl piperidine sulfonamides as inhibitors of acetylcholinesterase. Eur J Med Chem 44(10):4057–4062
Hadjiconstantinou M, Neff NH (2008) Enhancing aromatic l-amino acid decarboxylase activity: implications for l-DOPA treatment in Parkinson’s disease. CNS Neurosci Ther 14:340–351
Haile MM, Broderick PA, Li Y-S, Quartermain D, Blanck T, Bekker AY (2008) Nimodipine reverses the hypoxia-induced elevation of dopamine and serotonin in striatum of adult rats. Poster Presentation at American Society of Anesthesiologists, Orlando
Haile MM, Broderick PA, Li Y-S, Quartermain D, Blanck T, Bekker AY (2009) Nimodipine reverses the hypoxic elevation of tryptophan and serotonin in the striatum of adult rats. Poster Presentation at the International Anesthesia Research Society, San Diego
Hass BR, Stewart TH, Zhang J (2012) Premotor biomarkers for Parkinson’s disease: a promising direction of research. Transl Neurodegener 1:11
Healy DG, Abou-Sleiman PM, Ahmadi KR, Muqit MMK, Bhatia KP, Quinn NP, Lees AJ, Latchmann DS, Goldstein DB, Wood NW (2004) The gene responsible for PARK6 Parkinson’s disease, PINK1, does not influence common forms of parkinsonism. Ann Neurol 56(3):329–335
Hely MA, Morris JG, Reid WG, O’Sullivan DJ, Williamson PM, Rail D, Broe GA, Margrie S (1994) The Sydney Multicentre Study of Parkinson’s disease: a randomized, prospective five year study comparing low dose bromocriptine with low dose levodsopa-carbidopa. J Neuro Neurosurg Psychiatry 57:903–910
Henry B, Brotchie JM (1996) Potential of opioid antagonists in the treatment of levo-dopa-induced dyskinesias in Parkinson’s disease. Drugs Aging 9:149–158
Hirsch EC, Hunot S, Faucheux B, Agid Y, Mizuno Y, Mochizuki H, Tatton WG, Tatton N, Olanow WC (1999) Dopaminergic neurons degenerate by apoptosis in Parkinson’s disease. Mov Disord 14(2):383–386
Horellou P, Mallet J (1997) Gene therapy for Parkinson’s disease. Mol Neurobiol 15(2):241–256
Hugonin L, Barth A, Gräslund A, Perálvarez-Marín A (2008) Secondary structure transitions and aggregation induced in dynorphin neuropeptides by the detergent sodium dodecyl sulfate. Biochim Biophys Acta Biomembr 1778(11):2580–2587
Iljina M, Garcia GA, Horrocks MH, Tosatto L, Choi ML, Ganzinger KA, Abramov AY, Gandhi S, Wood NW, Cremades N, Dobson CM, Knowles TP, Klenerman D (2016) Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc Natl Acad Sci 113(9):E1206–E1215
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA—guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821
Johnston JP (1968) Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem Pharmacol 17(7):1285–1297
Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, During MJ (2007) Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet 369(9579):2097–2105
Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W, Langer R, Farokhzad OC (2008) Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett 8(9):2906–2912
Kish SJ, Shannak K, Hornykiewicz O (1988) Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N Engl J Med 318:876–880
Knoll J, Magyar K (1972) Some puzzling pharmacological effects of monoamine oxidase inhibitors. Adv Biochem Psychopharmacol 5:393–408
Kordower JH, Aebischer P (2001) Gene therapy to the rescue in Parkinson's disease. Response from Kordower and Aebischer. Trends Pharmacol Sci 22(3):105–106
Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, Pyzalski R, Taylor MD, Carvey P, Ling ZD, Trono D, Hantraye P, Deglon N, Aebischer P (2000) Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 290(5492):767–773
Kornhuber J, Bormann J, Retz W, Hübers M, Riederer P (1989) Memantine displaces [3H]MK-801 at therapeutic concentrations in postmortem human frontal cortex. Eur J Pharmacol 166(3):589–590
Kornhuber J, Bormann J, Hübers M, Rusche K, Riederer P (1991) Effects of the 1-amino-adamantanes at the MK-801-binding site of the NMDA-receptor-gated ion channel: a human postmortem brain study. Eur J Pharmacol 206(4):297–300
Kornhuber J, Weller M, Schoppmeyer K, Reiderer P (1994) Amantadine and memantine are NMDA receptor antagonists with neuroprotective properties. J Neural Transm 43(suppl.):91–104
Kornhuber J, Quack G, Danysz W, Jellinger K, Danielczyk W, Gsell W, Riederer P (1995) Therapeutic brain concentration of the NMDA receptor antagonist amantadine. Neuropharmacology 34(7):713–721
Kornhuber J, Jellinger K, Wiltfang J, Leblhuber F, Riederer P (1999) The N-methyl-d-aspartate receptor channel blocker amantadine does not cause histopathological alterations in human brain tissue. Acta Neuropathol 98(1):85–90
Kuhn DM, Arthur R, States JC (1997) Phosphorylation and Activation of Brain Tryptophan Hydroxylase: Identification of Serine-58 as a Substrate Site for Protein Kinase A. J Neurochem 68(5):2220–2223
Kurth MC, Adler CH, St. Hilaire M, Singer C, Waters C, LeWitt P, Chernik DA, Dorflinger EE, Yoo K (1997) tolcapone improves motor function and reduces levodopa requirement in patients with Parkinson’s disease experiencing motor fluctuations a multicenter, double-blind, randomized, placebo-controlled trial. Neurology 48:81–87
Kurzatkowska K, Dolusic E, Dehaen W, Sieroń-Stołtny K, Sieroń A, Radecka H (2009) Gold electrode incorporating corrole as an ion-channel mimetic sensor for determination of dopamine. Anal Chem 81(17):7397–7405
Kvell K, Pongrácz J, Székely M, Balaskó M, Pétervári E, Bakó G (2011) Figure I. 10-2 neurotransmitter imbalance in Parkinson’s disease in molecular in neurological and psychological disorders in the elderly. In: Kvell et al (eds) Molecular and clinical basics of gerontology. University of Pécs, Hungary, p 94
Lam HT, Graber MC, Gentry KA, Bieschke J (2016) Stabilization of α-synuclein fibril clusters prevents fragmentation and reduces seeding activity and toxicity. Biochemistry 55(4):675–685
Levivier M, Przedborski S, Bencsics C, Kang UJ (1995) Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson’s disease. J Neuroscience 15(12):7810–7820
Lim Y, Mays CE, Kim Y, Titlow WB, Ryou C (2010) The inhibition of prions through blocking prion conversion by permanently charged branched polyamines of low cytotoxicity. Biomaterials 31(8):2025–2033
Lu JQ, Stoessel AJ (2002) Somatostatin modulates the behavioral effects of dopamine receptor activation in parkinsonian rats. Neuroscience 112:261–266
Luna E, Luk KC (2015) Bent out of shape: α-Synuclein misfolding and the convergence of pathogenic pathways in Parkinson’s disease. FEBS Lett 589(24):3749–3759
Ma Y, Dhawan V, Freed C, Fahn S, Eidelberg D (2005) Positron emission tomography and embryonic dopamine cell transplantation in Parkinson’s disease. In: Rahni DN, Kolodny EH (eds) Broderick PA. Bioimaging in neurodegeneration, Springer, pp 45–57
Ma W, Qin L-X, Liu F-T, Gu Z, Wang J, Pan ZG, James TD, Longa Y-T (2013) Ubiquinone-quantum dot bioconjugates for in vitro and intracellular complex I sensing. Sci Rep 3:1537
Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K (2003) Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. J Neurosci 23(1):29–33
Matuja WB, Aris EA (2008) Motor and non-motor features of Parkinson’s disease. E Afr Med J 85:3–9
Milisav I, Šuput D, Ribarič S (2015) Unfolded protein response and macroautophagy in Alzheimer’s, Parkinson’s and prion diseases. Molecules 20(12):22718–22756
Montastruc JL, Rascol O, Senard JM, Rascol A (1994) A randomised controlled study comparing bromocriptine to which levodopa was later added, with levodopa alone in previously untreated patients with Parkinson’s disease: a five year follow up. J Neurol Neurosurg Psychiatry 57:1034–1038
Monti JM, Hawkins M, Jantos H, D’Angelo L, Fernández M (1988) Biphasic effects of dopamine D-2 receptor agonists on sleep and wakefulness in the rat. Psychopharmacology 95(3):395–400
Muthu MS, Singh S (2009) Targeted nanomedicines: effective treatment modalities for cancer, AIDS and brain disorders. Nanomedicine 4(1):105–118
Nomoto M, Iwata S, Irifune M, Kaseda S, Osame M, Fukuda T (1998) Dermal application of lisuride on parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in the common marmoset and on cases with Parkinson’s disease. Nihon Shinkei Seishin Yakurigaku Zasshi 18(3):81–87
Olanow CW, Brundin P (2013) Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord Spec Issue Vatican Conf Neuroprot Parkinson’s Dis 28(1):31–40
Pellegrino LJ, Pellegrino AS, Cushman AJ (1979) A stereotaxic atlas of the rat brain. Plenum Press, New York
Piercey MF, Broderick PA, Hoffman WE, Vogelsang GD (1990) Neurotransmitter release and impulse frequency studies of dopaminergic autoreceptor agonists. In: Kalsner S, Westfall TC (eds) presynaptic receptors and the question of autoregulation of neurotransmitter release. Ann NY Acad Sci 604:637–638
Piercey MF, Svensson KA, Lin CH, Haadsman-Svensson SR, McCall RB, Smith MW, Broderick PA, Carlsson A (1993) U-93385, a high intrinsic activity 5-HT1A agonist with good oral activity. Soc Neurosci Abstr 19:1866
Podolski IY, Podlubnaya ZA, Kosenko EA, Mugantseva EA, Makarova EG, Marsagishvili LG, Shpagina MD, Kaminsky YG, Andrievsky GV, Klochkov VK (2007) Effects of hydrated forms of C 60 fullerene on amyloid β-peptide fibrillization in vitro and performance of the cognitive task. J Nanosci Nanotechnol 7(4–5):1479–1485
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216(4542):136–144
Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K (2015) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA 112(38):E5308–E5317
Re F, Gregori M, Masserini M (2012) Nanotechnology for neurodegenerative disorders. Nanomedicine: nanotechology. Biol Med 8:S51–S58
Rektorová I, Rektor I, Bareš M, Dostál V, Ehler E, Fanfrdlová Z, Fiedler J, Klajblová H, Kulišt’ák P, Ressner P, Svátová J, Urbánek K, Velísková J (2003) Pramipexole and pergolide in the treatment of depression in Parkinson’s disease: a national multicentre prospective randomized study. Eur J Neurol 10(4):399–406
Rey NL, George S, Brundin P (2015) Spreading the word: precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol Appl Neurobiol. doi:10.1111/nan.12299
Rezak M, Reese S, Sacks J (2008) Young Parkinson’s handbook: a guide for patients and their families. American Parkinson Disease Association Inc, USA
Ridet JL, Corti O, Pencalet P, Hanoun N, Hamon M, Philippon J, Mallet J (1999) Toward autologous ex vivo gene therapy for the central nervous system with human adult astrocytes. Hum Gene Ther 10(2):271–280
Robinson M, Lee BY, Leonenko Z (2015) Drugs and drug delivery systems targeting amyloid-ß in Alzheimer’s disease. Mol Sci 2(3):332–358
Rubio A, Sánchez-Mut JV, García E, Velasquez ZD, Oliver J, Esteller M, Avila J (2012) Epigenetic control of somatostatin and cortistatin expression by β amyloid peptide. J Neurosci Res 90(1):13–20
Schechter MD (1984) Evidence for a direct dopaminergic effect of lisuride. Pharmacol Biochem Behav 21(2):185–189
Schrag A, Schott JM (2006) Epidemiological, clinical, and genetic characteristics of early-onset parkonsinism. Lancet Neurol 5(4):355–363
Sharrad DF, Gai WP, Brookes SJ (2013) Selective coexpression of synaptic proteins, α-synuclein, cysteine string protein-α, synaptophysin, synaptotagmin-1, and synaptobrevin-2 in vesicular acetylcholine transporter-immunoreactive axons in the guinea pig ileum. J Comp Neurol. 521(11):2523–2537
Shen Y, Muramatsu SI, Ikeguchi K, Fujimoto KI, Fan DS, Ogawa M, Mizukami H, Urabe M, Kume A, Nagatsu I, Urano F, Suzuki T, Ichinose H, Nagatsu T, Monahan J, Nakano I, Ozawa K (2000) Triple transduction with adeno-associated virus vectors expressing tyrosine hydroxylase, aromatic-l-amino-acid decarboxylase, and GTP cyclohydrolase I for gene therapy of Parkinson’s disease. Hum Gen Ther 11(11):1509–1519
Sletzinger M, Chemerda JM, Bollinger FW (1963) Potent decarboxylase inhibitors. Analogs of methyldopa1. J Med Chem 6(2):101–103
Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA (2005) Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Human Molecular Genetics 14(24):3801–3811
Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533:95–99
Sousa F, Mandal C, Garrovo C, Astolfo A, Bonifacio A, Latawiec D, Menk RH, Arfelli F, Huewel S, Legname G, Galla H, Krol S (2010) Functionalized gold nanoparticles: a detailed in vivo multimodal microscopic brain distribution study. Nanoscale 2(12):2826–2834
Stewart T, Sossi V, Aasly JO, Wszolek ZK, Uitti RJ, Hasegawa K, Yokoyama T, Zabetian CP, Leverenz JB, Stoessl AJ, Wang Y, Ginghina C, Liu C, Cain KC, Auinger P, Kang UJ, Jensen PH, Shi M, Zhang J (2015) Phosphorylated α-synuclein in Parkinson’s disease: correlation depends on disease severity. Acta Neuropathol Commun 3:7
Svensson K, Carlsson A, Huff RM, Kling-Petersen T, Waters N (1994) Behavioral and neurochemical data suggest functional differences between dopamine D2 and D3 receptors. Eur J Pharmacol 263(3):235–243
Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, Mizuno T, Tanaka M (2014) Differential expression of alpha-synuclein in hippocampal neurons. PLoS One 9(2):e89327
Tashkhourian J, Nezhad MRH, Khodavesi J, Javadi S (2009) Silver nanoparticles modified carbon nanotube paste electrode for simultaneous determination of dopamine and ascorbic acid. J Electroanal Chem 633(1):85–91
Thome AD, Harms AS, Volpicelli-Daley LA, Standaert DG (2016) MicroRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J Neurosci 36(8):2383–2390
Tu Z, Yang W, Yan S, Guo X, Li X-J (2015) CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol Neurodegener. doi:10.1186/s13024-015-0031-x
Tyson T, Steiner JA, Brundin P (2015) Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. J Neurochem. doi:10.1111/jnc.13449
Ubeda-Bañon I, Saiz-Sanchez D, de la Rosa-Prieto C, Argandoña-Palacios L, Garcia-Muñozguren S, Martinez-Marcos A (2010) α-Synucleinopathy in the human olfactory system in Parkinson’s disease: involvement of calcium-binding protein- and substance P-positive cells. Acta Neuropathol 119(6):723–735
Vila M, Przebdorski S (2004) Genetic clues to the pathogenesis of Parkinson’s disease. Nat Med 10(Suppl):S58–S62
Wakabayashi K, Tanji K, Mori F, Takahashi H (2007) The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 27(5):494–506
Wang ZH, Ji Y, Shan W, Zeng B, Raksadawan N, Pastores GM, Wisniewski T, Kolodny EH (2002) Therapeutic effects of astrocytes expressing both tyrosine hydroxylase and brain-derived neurotrophic factor on a rat model of Parkinson’s disease. Neuroscience 113(3):629–640
Wang Q, Shin EJ, Nguyen X-KT, Li Q, Bach JH, Bing G, Kim W-K, Kim HC, Hong JS (2012) Endogenous dynorphin protects against neurotoxin-elicited nigrostriatal dopaminergic neuron damage and motor deficits in mice. J Neuroinflamm 9:124
Xiao SJ, Hu PP, Wu XD, Zou YL, Chen LQ, Peng L, Ling J, Zhen SJ, Zhan L, Li YF, Huang CZ (2010) Sensitive discrimination and detection of prion disease-associated isoform with a dual-aptamer strategy by developing a sandwich structure of magnetic microparticles and quantum dots. Anal Chem 82(23):9736–9742
Xie M, Luo K, Huang BH, Liu SL, Hu J, Cui D, Zhang ZL, Xiao GF, Pang DW (2010) PEG-interspersed nitrilotriacetic acid-functionalized quantum dots for site-specific labeling of prion proteins expressed on cell surfaces. Biomaterials 31:8362–8370
Xu M, Moratalla R, Gold LH, Hiroi N, Koob GF, Graybiel AM, Tonegawa T (1994) Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell 79:729–742
Yerramilli VS, Golebiewska UP, Scarlata S (2013) Characterizing interactions between γ-synuclein and PLCβ2 in breast cancer cells. Biophys J 104(2-Suppl 1):p51a
Yue HY, Huang S, Chang J, Heo C, Yao F, Adhikari S, Gunes F, Liu LC, Lee TH, Oh ES, Li B, Zhang JJ, Huy TQ, Luan NV, Lee YH (2014) ZnO nanowire arrays on 3D heirachical grapheme foam biomarker: detection of Parkinson’s disease. ACS Nano 8(4):1639–1646
Zarranz JJ, Alegre J, Gómez-Esteban J, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Tortosa EG, del Ser T, Muñoz DG, de Yebenes JG (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55(2):164–173
Acknowledgments
The author wishes to thank the Broderick Brain Foundation, the F.M. Kirby Foundation, the Center for Advanced Technology, CUNY, and the MacKenzie Foundation for partial support of our laboratory and students during these studies. Other grants, including the National Institute of Health, National Institute on Drug Abuse, The Lowenstein Foundation, and the FACES and PACE Foundations for Epilepsy in addition to the former Pharmacia and Upjohn Company in Kalamazoo, Michigan, deserve honorable mention. It is important to note that the figures and the table in this manuscript are the result of detailed high-resolution work by graphics expert, Paul Sethi. It is noteworthy that the development and pioneering of Neuromolecular Imaging and this series of nanobiosensors (BRODERICK PROBE®) is named for Paddy Broderick, father of the corresponding author.
Author information
Authors and Affiliations
Corresponding author
Additional information
Definition of biomarkers
According to the National Institutes of Health (NIH) working group, a biomarker is “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes or pharmacological responses to a therapeutic intervention” (Biomarkers Definitions Working Group 2001).
Rights and permissions
About this article

Cite this article
Broderick, P.A., Wenning, L. & Li, YS. Neuromolecular imaging, a nanobiotechnology for Parkinson’s disease: advancing pharmacotherapy for personalized medicine. J Neural Transm 124, 57–78 (2017). https://doi.org/10.1007/s00702-016-1633-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-016-1633-3