
Abstract
The prevalence of both Alzheimer’s disease (AD) and vascular dementia (VaD) is increasing with the aging of the population. Studies from the last several years have shown that people with diabetes have an increased risk for dementia and cognitive impairment. Therefore, the authors of this consensus review tried to elaborate on the role of diabetes, especially diabetes type 2 (T2DM) in both AD and VaD. Based on the clinical and experimental work of scientists from 18 countries participating in the International Congress on Vascular Disorders and on literature search using PUBMED, it can be concluded that T2DM is a risk factor for both, AD and VaD, based on a pathology of glucose utilization. This pathology is the consequence of a disturbance of insulin-related mechanisms leading to brain insulin resistance. Although the underlying pathological mechanisms for AD and VaD are different in many aspects, the contribution of T2DM and insulin resistant brain state (IRBS) to cerebrovascular disturbances in both disorders cannot be neglected. Therefore, early diagnosis of metabolic parameters including those relevant for T2DM is required. Moreover, it is possible that therapeutic options utilized today for diabetes treatment may also have an effect on the risk for dementia. T2DM/IRBS contribute to pathological processes in AD and VaD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A causative association between diabetes mellitus (DM) and cognitive impairment has been suggested based on clinical, epidemiological, and experimental studies (Alafuzoff et al. 2009; Bitel et al. 2012; Vagelatos and Eslick 2013; Carvalho et al. 2015; Feinkohl et al. 2015; Jellinger 2015a).
In fact, recent studies demonstrate a pathophysiological link between diabetes mellitus type II (T2DM) and cognitive decline (Jellinger 2015b). This is demonstrated in persons with DM showing that a higher risk of developing Alzheimer’s disease (AD), vascular dementia (VaD) and mixed-type dementia (AD plus cerebrovascular disease), and comorbidity, in particular cerebrovascular disease, hypertension, hypercholesterolemia, etc. increases this risk (Jellinger 2015b; Haroon et al. 2015; Kuo et al. 2015). Insulin resistance predicts medial temporal hypermetabolism in Mild Cognitive Impairment (MCI) conversion to AD (Willette et al. 2015b). In addition, changes in glucose uptake in medial temporal regions in AD predict worse memory performance (Willette et al. 2015a). Moreover, DM facilitates cognitive decline in patients with mild AD compared to those without comorbid DM (Jellinger 2015a; Ascher-Svanum et al. 2015). However, the precise mechanisms involved in the development of AD in diabetics are not yet fully understood, and several pathogenic pathways have been discussed (Feinkohl et al. 2015; Abner et al. 2016; Hao et al. 2015; Chiu et al. 2015; Verdile et al. 2015; Bedse et al. 2015; De Felice et al. 2014), including vascular brain disease, insulin resistance, and other metabolic effects on the brain.
In a meta-analysis, Chatterjee et al. (2016) estimated the sex-specific relationship between women and men with DM with incident dementia. Fourteen studies with 2310.330 individuals and 102.174 dementia cases were included. T2DM showed a ~60% greater risk for the development of dementia compared with those without DM. For VaD but not for non-vascular dementia, the additional risk is greater in women (Chatterjee et al. 2016).
In the study of Marseglia et al. (2016), the authors aimed to identify the cognitive domains initially impaired by diabetes and the factors that play a role in this first stage. There were 2.305 cognitively intact participants aged ≥60 years. A variety of memory tests were assessed. Diabetes (controlled and uncontrolled) as well as prediabetes were ascertained by clinicians. Information on vascular disorders and vascular risk factors has been recorded. Mainly uncontrolled diabetes in APOEɛ4 non-carriers was related to lower performance in perceptual speed, category fluency, and digit scan forward, and this association was present only among participants with vascular disorders or vascular risk factors (Marseglia et al. 2016).
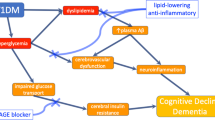
One-fifth of dementia cases are caused by VaD, a disorder with heterogenous spectrum of cerebrovascular pathologies (Nizam and Hyer 2007). VaD is one of the most prevalent dementia disorders after AD (Ozbabalik et al. 2012). The prevalence of VaD rises rapidly between ages 65–85. People with DM as compared to those without DM have a higher risk for developing VaD [pooled RR 2.27 [95% CI 1.94–2.66] (Gudala et al. 2013) and 2.2 (95% CI 1.7–2.8)] (Ninomiya 2014). They are 2–4 times more likely to develop AD and have a 1.5–2-fold greater risk for an accelerated rate of age-related cognitive decline (Cukierman et al. 2005). This has been demonstrated utilizing both neuropsychological instruments and surrogates such as change in MRI volumes (van den Berg et al. 2010; van Harten et al. 2006; Reijmer et al. 2011). Individuals with elevated blood glucose levels are at an increased risk to develop dementia, and those with elevated blood glucose levels have a more pronounced conversion from MCI to AD, suggesting that disrupted glucose homeostasis could play a more causal role in AD pathogenesis (Macauley et al. 2015).
Observational studies have also shown an increase in the incidence of other types of dementia than AD or VaD in DM (Gudala et al. 2013; Macauley et al. 2015; Irie et al. 2008; Ahtiluoto et al. 2010). Therefore, the precise mechanisms involved in the development of cognitive impairment in diabetic patients are not yet fully understood (Alafuzoff et al. 2009; Feinkohl et al. 2015).
Advances in the management of T2DM have enhanced preventive and medical services and have diminished its macro- and microvascular complications. This has led to an increase in life expectancy of people with diabetes, however, that has increased the population at risk for cognitive impairment and dementia (Ninomiya 2014).
Given all these aspects, the group concluded that disrupted glucose homeostasis is of risk for developing dementia. This includes diabetes-dependent cerebrovascular pathology. Therefore, the cascade of pathological events in AD may show first onset of non-vascular pathology followed by cerebrovascular changes, while for VaD, cerebrovascular pathology is of primary importance.
Possible mechanisms for the relationship between diabetes and cognitive impairment
Is cognitive impairment in patients with diabetes mellitus type 1 (T1DM) a consequence of vascular impairment or a separate process?
Modest cognitive impairment in patients with T1DM does not follow any dementia pattern. Compared to healthy controls, patients with T1DM were slower in information-processing, but had better scores on visuospatial tests (Brands et al. 2006). It was shown that patients with T1DM have an increased risk of lacunar stroke (Luitse et al. 2012) and those with additional microangiopathy had decreased structural connectivity in posterior brain regions (van Duinkerken et al. 2012a) and impaired function in the ventral attention network (Van Duinkerken et al. 2012b). However, the effect of vascular lesions on the cognitive decline in T1DM patients is not entirely clear (Brands et al. 2006; Nunley et al. 2015; Biessels and Reijmer 2014; Huang et al. 2014). In contrast to T2DM, T1DM begins earlier in life and may influence brain development (Biessels et al. 2008; de Felice and Benedict 2015) via insulin receptors in the hypothalamus, which play a key role in the memory system (De Felice et al. 2014; de Felice and Benedict 2015). In a recent small study, patients with T1DM had partly altered CSF AD biomarkers (Ouwens et al. 2014). Levels of p-Tau were elevated similar to those in AD patients. Another biomarker is soluble low-density lipoprotein receptor-related protein 1 (sLRP1) protein, which regulates efflux of beta-amyloid (Aβ) from the brain to the blood and is impaired in patients with AD (Ramanathan et al. 2015). T1DM patients who had elevated levels of sLRP1 in the CSF, performed better on the cognitive tests (Ouwens et al. 2014).
Hyperglycemia, which is a primary impairment in T1DM, can cause permanent cognitive impairment, thus contrasting the situation with hypoglycemia. In the brain of streptozotocin (STZ)–T1DM rats and mice decreased neurogenesis (Alvarez et al. 2009), mitochondrial dysfunction due to decreased activity of respiratory chain complex I (Taurino et al. 2012), lower release of adenosine triphosphate (ATP) and downregulation of synaptic purinergic receptors in the hippocampus (Duarte et al. 2007), a region involved in learning and memory (Duarte et al. 2007), have been reported. Moreover, STZ–T1DM animals performed poorly on cognitive tasks (Alvarez et al. 2009).
Taken together, it is likely that vascular risk factors together with metabolic causes may facilitate neurodegeneration and contribute to cognitive impairment in T1DM patients.
Atherosclerosis, stroke, and insulin resistance
Several pathogenic routes have been suggested for this relationship. First, chronic hyperglycemia may cause cognitive impairments and abnormalities in synaptic plasticity (Jacobson et al. 2007). Tight glycemic control significantly reduced the rate of brain atrophy over a period of 40 months in STZ-induced diabetic rats compared with the standard glucose treatment (Biessels et al. 1996). Second, relative insulin deficiency (also termed “insulin resistance”) may be of importance. The Hisayama study reported an increase in the presence of neuritic plaques with higher postprandial glycemic (PG) levels, fasting insulin level, and insulin resistance in AD (Doi et al. 2010), which might be also relevant for mixed forms of dementia with VaD involvement. It is reasonable to postulate a close association between 2-h PG levels and the risk of VaD, because increased 2-h PG levels are associated with the development of stroke (Thacker et al. 2011; Doi et al. 2010). Insulin resistance is associated with VaD through atherosclerosis (De Felice et al. 2014; Fitzpatrick et al. 2009). Obesity in T2DM contributes to hyperinsulinemia and insulin resistance. Insulin also regulates acetylcholine synthesis (Kimura et al. 2016), thus possibly affecting cognitive functions in dementia. Insulin resistance reduces the amount of insulin that crosses the blood–brain barrier (BBB), which hinders its role in the brain (see details in glycemic control). It has been found that prolonged hyperinsulinemia induces an impaired response to insulin through decreased expression of insulin receptors at the BBB and brain and consequently inhibits the insulin transport into cerebrospinal fluid (CSF) and brain tissues (Neumann et al. 2008). These changes could cause deficits in learning and memory formation, probably due to a neuroglial energy crisis (Kimura 2001, 2016; Craft et al. 1998). Higher levels of plasma insulin provoke amyloid accumulation by limiting the degradation of Aβ by direct competition for the insulin degrading enzyme (IDE), which degrades both insulin and Aβ (Neumann et al. 2008). However, lower insulin levels in CSF and the impaired response to insulin and insulin-like growth factor-1 inhibit the transportation of these carrier proteins and decrease the clearance of Aβ (Craft and Watson 2004). Third, chronic exposure to hyperglycemia in DM also induces abnormalities in the cerebral capillaries (termed “vasculopenia”) (Serlin et al. 2011). Recent human study in asymptomatic, late middle-aged adults (N = 186) from the Registry for Alzheimer’s Prevention who underwent [C-11]Pittsburgh compound B (PiB) position emission tomography as an indicator of amyloid deposition in the brain tested the interaction between insulin resistance and glycemic status on PiB distribution volume in the cerebral cortex (Willette et al. 2015a). The results of that study demonstrated that in normoglycemia, higher peripheral insulin resistance corresponded to higher PiB uptake in frontal and temporal areas, indicating that in individuals at risk for AD, peripheral insulin resistance may contribute to and predicts brain amyloid deposition in brain regions affected by AD. Since this association was not confirmed in a much smaller study on 47 participants (Thambisetty et al. 2013), further studies are needed to resolve the nature of the link between insulin resistance/T2DM and amyloid load. On the other hand, peripheral insulin resistance has been found to predict MCI progression to AD, as shown by the study of the Alzheimer’s Disease Neuroimaging Initiative which included 194 MCI, 60 AD, and 26 cognitively normal subjects (Willette et al. 2015b). The results suggested that during the MCI stage, the homeostatic model assessment of insulin resistance (HOMA-IR) as an index of peripheral insulin resistance is differently associated with either hypo- or hyper- glucose (FDG-PET) metabolism in different brain areas, depending on whether participants progress to develop clinical AD. Therefore, evidence accumulated showing that peripheral insulin resistance, which is often associated with a metabolic syndrome and T2DM, has a role in prediction of AD pathology development, but its most specific AD correlates have not been clearly defined yet.
Finally, severe hypoglycemia may be also a risk factor for cognitive impairments in patients with DM. It has been reported that patients with recurrent severe hypoglycemic episodes have a 1.5–2.0 times greater risk of the development or deterioration of cognitive impairment (Thacker et al. 2011). These are, however, prospective studies, and as cognitive impairment is a long process, it is hard to delineate the direction of the relationship, i.e., does cognitive impairment cause severe hypoglycemia or does severe hypoglycemia cause dementia. Older patients are thought to have less brain reserve or brain plasticity than younger patients (Artola et al. 2002). Therefore, it is plausible that hypoglycemia could cause neurologic changes that render an older patient more susceptible to dementia.
Glycemic control
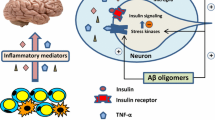
Peripheral insulin of pancreatic origin crosses the BBB in a tightly controlled manner, as the BBB expresses insulin receptors, which may decrease in number in response to specific conditions associated with chronic hyperinsulinemia and insulin resistance (Banks 2004; Banks 2006; Unger et al. 1991). At the level of the BBB, there is a tight relationship between the presence of insulin receptors and the topographic expression of glucose transporters particularly abundant in medial temporal lobe and diencephalic structures, which notably are related to neurocognitive functions (White 2002; Zhou et al. 2001), suggesting an important role of insulin in modulation of glucose uptake and utilization (Banks 2004; Baker et al. 2011; Craft et al. 2012; Hertz et al. 1981). Insulin stimulation of glucose transporter-4 (GLUT4) seems to be critical to the regulation of neuronal metabolism and the generation of energy needed for memory and other neurocognitive functions. The presence and functional activity of major insulin signal transduction molecules in human primary astrocytes has also been demonstrated, including glycogen formation and cell proliferation, thus supporting neurons with energy, since neurons cannot store glycogen for their own activity (Heni et al. 2011).
A growing body of evidence points to the importance of a condition of the insulin inability to serve its physiological function in the brain, in literature described by two alternative terms, “brain insulin resistance” (BIR) (Su et al. 2017; Talbot et al. 2012; de la Monte et al. 2012) or “insulin resistant brain state” (IRBS) (Correia et al. 2013; Frisardi et al. 2010; Plaschke et al. 2010a, b; Salkovic-Petrisic et al. 2009). At the molecular level, BIR is characterized by a reduced response to insulin signalling generally downstream the insulin receptor (IR)—insulin receptor substrate (IRS)—phosphatidyl inositol kinase-3 (PI-3) pathway in the brain, which, particularly considering the neurotrophic, neuroprotective, and neuromodulatory roles of brain insulin, may lead to neurodegeneration and cognitive impairment as seen in AD as well as metabolic alterations in hypothalamic functions, as seen in obesity and T2DM (Kullmann et al. 2016). Although some authors proposed that it might be considered as type 3 diabetes (de la Monte and Tong 2014), others strongly disagree (Talbot 2014; Talbot and Wang 2014). BIR actually represents a brain-related metabolic syndrome associated with metabolic and oxidative stresses and neuroinflammation in the brain, which may or may not be accompanied by alterations in peripheral metabolic homeostasis, since T2DM increases the risk for AD (and vice versa), but neither all T2DM patients develop AD (and vice versa) nor AD is necessarily associated with hyperglycemia (Talbot 2014; Talbot and Wang 2014; Blázquez et al. 2014).
A clinical study on 30 normal, 29 MCI, and 30 AD patients (Talbot et al. 2012) demonstrated that cognition was negatively associated with levels of candidate biomarker of BIR serin-phosphorylated insulin receptor substrate-1 (IRS-1 pS616) in the hippocampus, and that association of episodic memory and IRS-1 pS616 was statistically independent of Aβ plaques, suggesting that BIR is mechanistically closer than the plaques to the molecular causes of cognitive decline in AD. A very recent longitudinal, 35-month study in 57 MCI and 64 cognitively unimpaired controls confirmed the existence of the interaction between insulin resistance-related genetic polymorphisms (AKT2, PIK3CB, IGF1R, PIK3CD, MTOR, IDE, AKT1S1, and AKT1) and cognitive impairments in MCI subjects, providing in vivo evidence that pathway of BIR modifies cognitive performance, further showing that the influence occurred in the absence of diabetes (Su et al. 2017).
Insulin resistance impairs the normal activity of the brain; both experimental, imaginistic, and clinical non-interventional studies have identified correlations between insulin and cognitive functions—in particular impaired memory and AD but also increased insulin resistance in a significant number of patients with other neurodegenerative diseases (de Felice et al. 2014; Craft et al. 2012; de la Monte et al. 2012; Craft and Christen 2010; Rönnemaa et al. 2008; de la Monte et al. 2009). These implications could be related to the role of insulin in the normal APP and Aβ cellular synthesis and processing, but also in the brain-liver metabolic axis (de la Monte et al. 2012; de la Monte 2009; Banks et al. 2012; Craft et al. 2013; Gasparini and Xu 2003; Lin et al. 2000; Matsuzaki et al. 2010; Passafaro et al. 2001; Sagare et al. 2012; Tamaki et al. 2007; Lopez et al. 2011). The role of brain insulin in the control of the turn-over of Aβ is also important for mixed and vascular cognitive impairment as there is a tight interference between the brain vascular risk factors and Aβ (as recently stated by AHA/ASA based on a significant number of published research data) (Gorelick et al. 2011).
In addition, insulin has been shown to regulate the phosphorylation of tau proteins (Rudolph et al. 2016). Hyperphosphorylated tau contributes to the formation of neurofibrillary tangles (Kimura 2016). It is also reported that there are genomic/transcriptomic links between AD and DM by meta-analysis study (Mirza et al. 2014).
A neuropathological evaluation of glucose/insulin-related molecules in AD, DM and controls is presented in Table 1. These molecular post-mortem brain data agree with histological and clinical studies underlying the importance of glucose/insulin pathology as risk factors for cognitive dysfunction. These results point to a concomitant occurrence of alterations in the energy metabolism pathways.
Considering the dysfunction of the brain insulin system found in AD patients post-mortem (Luchsinger 2012), an experimental rat model, the STZ-ICV model, which mirrors an insulin resistant brain state, seems to be an appropriate animal model for AD (Salkovic-Petrisic et al. 2009; Luchsinger 2012; Hoyer 1998, 2004; Israili 2011; Grünblatt et al. 2007; de la Monte 2009).
Brain insulin resistance (BIR) as a shared pathological feature in obesity, cardiovascular disease, T2DM, and dementia
Evidence has gathered suggesting that BIR seems to be a shared pathological feature of metabolic and cognitive disturbances in T2DM, obesity, cardiovascular disease, and dementia patients (Kullmann et al. 2016; Lutski et al. 2017), which may provide the missing link between these disorders. Indeed, recent evidence suggests that insulin resistance is related to subsequent poorer cognitive performance and greater cognitive decline among patients with cardiovascular disease with and without diabetes (Lutski et al. 2017). Clinical investigation of the link between the obesity and BIR showed that obese men respond to cognitive but not to catabolic brain insulin signalling (Hallschmid et al. 2008), indicating that not all insulin activities in the brain have been equally affected by BIR and that insulin resistance in metabolic disorders does not uniformly affect all target cells and intracellular signaling pathways in the brain (Könner and Brüning 2012). Whereas dementia predominately affects cognitive target regions of insulin action, T2DM- and obesity-associated BIR predominately targets hypothalamic insulin action, but there is overlap of these three disorders in impairment of functional connectivity in prefrontal and lateral temporal cortices and hippocampus as reviewed by Kullmann et al. (2016). Thus, numerous clinical phenotypes may arise from selective insulin resistance, leading to inhibition of defined intracellular signaling pathways in some tissues, while in other cell types, insulin action is maintained or even overactivated (Könner and Brüning 2012). Furthermore, magnetoencephalographic studies on carriers of obesity- and diabetes-risk genes (fat-mass and obesity-associated gene/FTO/and IRS-1, respectively) showed an attenuated insulin-mediated response in the brain (Tschritter et al. 2006, 2007). In lean humans, insulin infusion modulates cerebrocortical activity as demonstrated by magnetoencephalography, while these effects are suppressed in obese individuals, indicating lower cerebrocortical response to insulin, i.e., BIR in this particular region, found in individuals with the Gly972Arg polymorphism in IRS-1, a T2DM risk gene (Tschritter et al. 2006). The same group demonstrated also that variation in the FTO gene locus (obesity-risk gene) is associated with cerebrocortical insulin resistance, but in these subjects, the effect of FTO polymorphism was independent of the Gly972Arg polymorphism in IRS-1 (Tschritter et al. 2007). These studies clearly indicate that each genetic determinant for BIR involves different neuronal systems (Kullmann et al. 2016), which may provide an explanation why AD is associated with T2DM in some, but not all demented patients, and vice versa, why T2DM is associated with AD in some but not all diabetic patients.
BIR is not necessarily a secondary pathological event as mentioned earlier in the text (references Neumann et al. 2008; Kimura 2001, 2016; Craft et al. 1998; Craft and Watson 2004). Considering the BIR as a shared feature in obesity, T2DM, and dementia, etiology of BIR as a primary pathological event could be related to the maternal environment during pregnancy and its influence on the fetus, according to the studies showing that the change of insulin action in fetuses of diabetic mothers influences the fetal brain (Sobngwi et al. 2003). Intrauterine exposure of fetuses to a non-physiological concentration of insulin during critical periods of early development can lead to a permanent malprogramming of fundamental regulatory systems including those in hypothalamus, as demonstrated for elevated insulin level during perinatal life which programmed the development of obesity and diabetes (Plagemann 2008). Recent meta-analysis of 19 studies including 2260 subjects has confirmed a strong support for the fetal programming hypothesis (Pearson et al. 2015). It is not only stress that might be a confounding factor, as the effects of chronic exposure to stress hormones on cognition at different stages in life including the prenatal age, depend on the brain areas that are developing or declining at the time of exposure (Lupien et al. 2009). Therefore, environmental factors and epigenetic mechanisms operating during pregnancy and postnatally may affect particular susceptibility genes and stress factors, consequently affecting brain development and causing respective diseases like AD and/or T2DM that manifest late in life when aging takes place and may become a trigger of desynchronization of biological systems (Salkovic-Petrisic et al. 2009).
Cerebral blood flow
Age-related dysfunction based on reduced capillary function declines in uptake of energy metabolites, amino acids, trophic factors, and other metabolic constituents, is of eminent importance in a variety of brain-related disorders (Kang et al. 2017; Bellou et al. 2017). T2DM favours such age-dependent dysfunction and potentiates energy loss in brain tissue. Therefore, aging eventually combined with stress, which per se exerts negative effects on T2DM, is both potential risk factors for AD (de Matos et al. 2017).
While under physiological conditions, compensatory mechanisms are able to keep the homeostasis of brain nutrition for a long time, chronic dysfunction finally will overcome compensatory functions leading to neuronal death.
Glucose-6-phosphate dehydrogenase plays a pivotal role in homeostatic redox control by providing reducing equivalents to glutathione, the major non-enzymatic cellular antioxidant. As OS plays an important role in the pathogenesis of AD, it is noteworthy that both glucose-6-phosphate dehydrogenase and sulfhydryl concentrations are upregulated in AD, showing compensatory regulation.
According to an alternative two-hit vascular hypothesis, Aβ accumulation in the brain is a second pathology (hit 2) initiated by vascular damage (hit 1; Fig. 1). Neurovascular dysfunction and hypoperfusion/hypoxia can reduce Aβ vascular clearance across the BBB and increase Aβ production from Aβ precursor protein (APP), respectively, causing Aβ accumulation in the brain. Elevated levels of Aβ in the brain may in turn accelerate neurovascular and neuronal dysfunction and promote self-propagation, leading to cerebral β-amyloidosis (Sagare et al. 2012).

Modified from Sagare et al. (2012) (This copyright agreement is admitted by describing Cold Spring Herb Perspect Med 2012;2:a011452)
Vascular hypothesis of Alzheimer disease. HIT 1 Vascular damage as primary pathological event. HIT 2 Aβ accumulation as secondary pathological event.
Chronic brain hypoperfusion (CBH) can be present for many years without eliciting mental symptoms, creating instead an insidious neuronal energy crisis that is finally expressed by progressive cognitive deficits in affected individuals.
In this scenario, the presence of advanced aging plus vascular risk factors can lower cerebral perfusion by inducing any of number of abnormal hemodynamic mechanisms affecting blood pressure, vessel patency, vascular wall shear stress, blood flow resistance, blood viscosity, and chemical blood flow regulators (Blennow et al. 1990).
As neurons have no energy reserves, the performance of cognitive tasks is critically dependent on the steady delivery of adequate oxygen and glucose to produce adenosine triphosphate (ATP). This nutrient delivery is inadequate in the aging brain.
Diminished CBF, neurovascular dysfunction, and impaired vascular clearance of Aβ from brain support an essential role in linking DM and AD pathogenesis (de la Torre 2010).
The glymphatic system mediates clearance of the interstitial solutes in the brain by exchange of cerebrospinal and interstitial fluids (CSF and ISF). The glymphatic system consists of CSF influx from the paravascular space of cerebral arteries, ISF clearance along the para-venous space and the astroglial water channel AQP4 that partially mediates transparenchymal changes of CSF and ISF (Iliff et al. 2013; Yang et al. 2013). Impairment of the glymphatic system induces accumulation of Aβ (Iliff et al. 2013; Yang et al. 2013). Using a rat model of DM induced by nicotinamide and STZ, Jiang et al. (2016) showed that compared to age matched non-diabetic rats, middle-aged DM rats exhibited spatial learning deficits. An odour recognition test which detects non-spatial memory deficits showed that DM rats failed to form new memories. In vivo dynamic Gd-DTPA contrast-enhanced MRI analysis confirmed by ex vivo confocal image analysis indicated that DM impairs the glymphatic system that mediates clearance of the interstitial solutes in the brain (Jiang et al. 2017). Cognitive deficits were highly and inversely correlated to the impairment of the glymphatic system. Immunohistological analysis showed the presence of microvascular leakage and loss of AQP4, axons, and oligodendrocytes in the hippocampi of DM rats (Hamed 2017).
Inflammation in the diabetic brain
It has been shown clinically that disturbances of the BBB play a role in the development of AD, especially in elderly patients (Blennow et al. 1990). Therefore, peripheral inflammatory factors from DM could leak to the brain parenchyma and induce activation of microglial cells to release inflammatory molecules (Breteler 2000), thus contributing to the pathophysiology of AD and VaD. There have been studies which have pointed out that inflammatory pathways may be acting as a possible mechanistic link between the two disorders. Takeda et al. (2010, AD and VaD) crossed transgenic mice (APP23) with diabetic mice (ob/ob) and looked at the metabolism and pathology of the brains in those double mutant mice (APP+-ob/ob). AD-like cognitive impairment was observed in APP+-ob/ob mice. Cerebrovascular inflammation, severe cerebral amyloid angiopathy, and up-regulation of RAGEs were observed in those double mutant mice even before the appearance of cerebral amyloid angiopathy, suggesting their role in cognitive impairment (Takeda et al. 2010). These findings agree with pathology of the cerebral vasculature in AD and DM (Blennow et al. 1990; Breteler 2000).
Oxidative stress (OS) caused by chronic hyperglycemia in chronic experimental diabetic neuropathy has been shown to cause oxidative injury of dorsal root ganglion neurons, specifically damaging the mitochondrial function and neuronal cell death (Schmeichel et al. 2003).
Prolonged metabolic stress conditions could be activated by various cell stressors, as hypoxia, oxidative stress, viral infections, and trophic withdrawal or various insults unveil deleterious effects of p53-evoked insulin resistance in neurons; enhancement of transcription of pro-oxidant factors, accumulation of toxic metabolites (AGE and ROS)-modified cellular components, together with activation of proapoptotic genes, could finally move a suicide death program of autophagy/apoptosis in neurons. The important role of p53 driving insulin resistance in AD brains validates attempts to inhibit p53 activity in neurons, since it could promise an improvement of the disease therapy. Recent studies reveal the impact of p53 on expression and processing of several microRNA (miRs) under DNA damage-inducing conditions. In addition, the role of miRs in promotion of insulin resistances and in T2DM has been well documented. Detailed recognition of the role of p53/miRs crosstalk in driving insulin resistance in AD brains could improve the disease diagnostics and future therapy (Vousden 2010).
The kynurenine pathway, the main metabolic route of tryptophan degradation, produces several neuroactive molecules [such as the excitotoxin antagonist kynurenic acid (KYNAC) and the excitotoxin quinolinic acid (QA)]. Alterations in the kynurenine pathway may promote glutamate-mediated excitotoxic neuronal damage and inflammatory processes (Vecsei et al. 2013). Recently, it was shown that STZ-induced experimental T1DM increases hippocampal content of KYNAC (Chmiel-Perzynska et al. 2014). The increased KYNAC level may have negative impact on cognition. KYNAC in the course of DM could be associated with an enhanced ketone body formation. In cortical slices and glial cultures, beta-hydroxybutyrate (BHB) augments KYNAC production by stimulating KATs activity in the protein kinase-A dependent way, thus explaining the neuroprotective actions of BHB (Chmiel-Perzynska et al. 2011).
Potential role of butyrylcholinesterase in linking diabetes and cognitive dysfunction
In AD, the brain levels of AChE go down, while those of BChE (the protein butyrilcholinesterase) go up, resulting in a dysregulation causing cholinergic deficit. As levels of that enzyme are altered in T2DM too, those authors suggest a synergistic negative interaction of T2DM and AD on cholinergic neurotransmission (Mushtaq et al. 2014). Among common pathogenic factors between DM and AD, BChE has been studied in vitro and in plasma (Sridhar et al. 2006; Rao et al. 2007; Shaikh et al. 2014). Alterations in the level of plasma BChE occur in DM; variant forms of the plasma enzyme occur in both DM and AD (Sridhar et al. 2010; Raygani et al. 2004). In vitro studies demonstrate a common pathogenic mechanism (Sridhar et al. 2006; Diamant et al. 2006). Whereas brain hyperglycemia mediates hippocampal neuron responses (Macauley et al. 2015), BCHE levels also correlate with cerebral glucose metabolism and cerebral Aβ load (Darreh-Shori et al. 2011). BChE associates particularly with the malignant form of Aβ plaques, suggesting its role in transforming non-fibrillar to the malignant fibrillar form (Reid and Darvesh 2015). To account for a gender difference, a gene–gene interaction between BChE and estrogen-associated genes was proposed (Reid and Darvesh 2015). However, the relation between BChE and AD is not settled yet. While an earlier meta-analysis of the K-Variant of BChE suggested that it was related to development of AD in Asians (Want et al. 2015), but a more comprehensive meta-analysis failed to confirm the relation (Ji et al. 2015).
T2DM is not only associated with an increased risk of cognitive decline and different types of dementia but also with cerebrovascular and peripheral vascular disease (Hoyer et al. 1999; Hoyer 1998, 2004; Israili 2011). Moreover, cerebrovascular disease may contribute to the severity of cognitive decline in AD (Last et al. 2007).
For the group, it is evident that disruption of glucose metabolism in both AD and VaD is based on multiple triggers. However, there is no agreement on follow-up and time-course of pathological cascade.
Imaging the diabetes–cognitive impairment relationship
Rapid advances in neuroimaging have confirmed a link between cognitive impairment and poor metabolic control in DM, mediated by the structural and functional brain changes (van Bussel et al. 2017). Whole-brain analysis revealed a consistent link between DM and brain atrophy and this atrophy is often more pronounced within the hippocampus (Gold et al. 2007). However, a pooled analysis of three cohort studies showed that the degree of hippocampal atrophy in T2DM is comparable to the degree of total brain atrophy (Biessels et al. 2006a, b). Brain atrophy in T2DM is associated with poor cognition, predominantly attention and executive function, and information-processing speed and memory (Moran et al. 2013; van Elderen et al. 2010; Manschot et al. 2006).
Whole-brain grey matter (GM) atrophy may be associated with T2DM; the association is more convincing for regional GM atrophy (Gold et al. 2007; Last et al. 2007). Similarly, the association of global and regional white matter (WM) atrophy and WM hyperintensities with DM was not consistently reported (Friedman et al. 2014). T2DM is clearly associated with the occurrence of lacunes (van Harten et al. 2007).
Functional magnetic resonance imaging (fMRI) demonstrated reduced synchronized activity within default mode network in cognitively normal T2DM patients (Musen et al. 2012). Regional basal cerebral blood flow (CBF) and cerebrovascular reactivity (CVR) have been shown to be decreased in T2DM patients (Zlokovic 2008). Longitudinal studies have confirmed the association of CBF and CVR with cognitive function and total brain volume in T2DM at baseline. However, both indexes of cerebral hemodynamics have not been predictive for atrophy and cognitive decline, and seem to be secondary phenomena (Brundel et al. 2012).
Neuroimaging studies may serve as early biomarkers and as monitors of progression of cognitive impairment in subjects with DM (Moran et al. 2015). Several methods have been attempted to identify anatomical and biomolecular markers linking accelerated cognitive decline with insulin resistance. First, MRI studies have consistently shown that chronic hyperglycemia is associated with brain atrophy and cerebrovascular lesions (Moran et al. 2013; van Bussel et al. 2017), which are hallmarks of attention deficits and impaired executive functioning (McCrimmon et al. 2012). There is no consensus on the exact mechanism of neurodegeneration leading to accelerated cognitive decline in DM and whether it is mediated by neuronal atrophy or/and cerebrovascular lesions (Biessels 2013). Such uncertainty undermines MRI as an early predictive tool for the transformation potential of MCI into AD in normal as well as DM subjects. For example, the AD Neuroimaging Initiative (http://www.adni-info.org/) has been validating the use of MRI/PET imaging for the prediction of MCI-to-dementia conversion within 18 months of diagnosis. Patients who converted to dementia showed changes in GM volume, amyloid deposition, and glucose metabolism in multiple regions compared with those who did not develop dementia. In a recent analysis of the data collected using structural MRI, amyloid-PET and 18F-FDG-PET scans, investigators could predict the transition with maximum accuracy of 72% (Teipel et al. 2015). In addition to its low predictive potential, this approach can only provide circumstantial clues on the underlying mechanism of accelerated cognitive decline leading to AD and dementia in DM patients.
In addition, DM triggers molecular alterations that elicit deranged microvascular and mitochondrial functions, increased inflammation, and elevated levels of advanced glycation end products (AGEs) (Kim et al. 2012; Goldin et al. 2006). All these diverse pathways converge at a nodal point where positive feedback loops exacerbate OS which is invariably implicated in neurotoxicity, neurodegeneration, and cognitive deficits. It can be suggested, therefore, that biomarkers of OS may provide early predictive probes for cognitive decline in DM subjects (Praticò et al. 2000, 2002; Keller et al. 2005; Aluise et al. 2011; Baldeiras et al. 2010; Thomas et al. 1996).
The group concluded that imaging studies (MRI, PET) contribute to an early diagnosis of AD and VaD. However, specificity and selectivity do not reach sufficient levels to be used solely for a precise clinical diagnosis.
Pathology
Both T1DM and T2DM induce regional microstructural changes in cortical and subcortical brain structures that are associated with impairment of neurocognitive functions (Seaquist 2015). Some autopsy studies stated that patients with DM have significantly less AD pathology but more frequent cerebrovascular lesions including microvascular changes (Alafuzoff et al. 2009; Beeri et al. 2005; Nelson et al. 2009; Ahtiluoto et al. 2010) or both types of cerebral pathology (Alafuzoff et al. 2009; Vagelatos and Eslick 2013; Ahtiluoto et al. 2010; Takeda et al. 2011; Verdile et al. 2015), and white matter lesions (Jellinger 2015a, b). The increased risk of cognitive decline in elderly subjects with DM is due to dual pathology, involving both the CVD and cortical atrophy (Biessels et al. 2006a, b; Umegaki 2012). Two different patterns of cerebral injury were seen in patients with dementia depending on DM status: greater amyloid plaque load in untreated DM patients but more frequent deep microvascular infarcts in those with treated DM (Sonnen et al. 2009). Central vascular disease and exacerbated pathology were seen in a mixed model of DM and AD by crossing APP/PS1 mice (AD model) with db/db mice (DM model) that show an age-dependent synergistic effect between DM and AD, including brain atrophy, senile plaques, hemorrhagic burden, and increase of microglia activation (Ramos-Rodriguez et al. 2015). Insulin resistance, hyperinsulinemia, and hyperglycemia can promote the onset of AD (Rönnemaa et al. 2008; de Oliveira Lanna et al. 2014) by accelerating tau phosphorylation and neuritic plaque formation (Bitel et al. 2012; Matsuzaki et al. 2010) and, overlapping with AD pathology, aggravate the progression of neurodegeneration due to OS, mitochondrial dysfunction, neuroinflammation, etc. as a common background (Carvalho et al. 2015; Kraska et al. 2012; Roriz-Filho et al. 2009; Rosales-Corral et al. 2015). Thus, impaired insulin signaling may be a possible link between AD and DM (Jellinger 2015a, b; Sato et al. 2011). Although insulin mitigates Aβ deposition and phosphorylation of tau (Bedse et al. 2015), DM in combination with APOEε4 may lead to excessive hyperphosphorylation of tau (Matsuzaki et al. 2010) and exacerbation of AD pathology (Malek-Ahmadi et al. 2013). However, a very recent publication (Abner et al. 2016) concludes that diabetes is associated with cerebrovascular but not AD pathology.
An extensive literature search reviewing 275 publications reporting post-mortem brain analyses of AD and VaD and published between 1980 and 1994 was performed (Gsell et al. 1996). In comparison to AD, in VaD, human brain neurotransmitter alterations are mild, e.g., for choline acetyltransferase activity, muscarinic receptor density, serotonin, dopamine, homovanillic acid, dopamine D1-and D2-receptor density, noradrenaline, and gamma aminobutyric acid (GABA), while 5-hydroxyindoleacetic acid (5-HIAA) shows a more pronounced deficiency. This data summarized here agree in principle with more recent conclusions of post-mortem human brain studies and experimental models (Ohara et al. 1994; Pimlott et al. 2004; Jia et al. 2004; Tohgi et al. 1996; Chen et al. 2013; Lee et al. 2014; Niwa et al. 2002; Pedrós et al. 2014; Knezovic et al. 2015; Barilar et al. 2015). CSF concentrations of choline were significantly higher in VaD patients compared to AD and controls but did no correlate with mini-mental state examination (MMSE) scores (Jia et al. 2004; Tohgi et al. 1996).
It is evident for the group that the pathology of AD and VaD shows multiple alterations at both neuropathological and neurochemical levels. In addition, mixed-type dementia pathology is frequent.
Animal models
There are few literature data on brain insulin resistance (BIR) and glucose hypometabolism in widely exploited transgenic AD mice models in which amyloid/tau-related gene manipulation is an inevitable starting point as antecedent to BIR allowing thus no clear conclusion on BIR–cognition relationship (Chen et al. 2013; Lee et al. 2014; Niwa et al. 2002; Pedrós et al. 2014), which contrasts animal treated intracerebroventricularly with STZ (non-transgenic STZ-icv model). STZ-icv administration induces dysfunctional insulin receptor signalling and mirrors the etiology of AD and in part that of cerebrovascular diseases (Table 2). Two long-term follow-up studies of STZ-icv rat model which provided the first staging of cognitive, structural/ultrastructural, neuropathological, and BIR markers in the STZ-icv rat model showed that cognitive deficit correlated well with GSK-3β activity (larger deficits–higher activity) and IDE protein expression (larger deficits–lower expression), in a bi-phasic time-dependent manner, with cognitive deficits becoming manifested later than dysfunctions in brain insulin system (Knezovic et al. 2015; Barilar et al. 2015). AD-like structural pathology seen in STZ-icv rat model in the form of early neurofibrillary changes and Aβ accumulation becomes manifested later than insulin- and memory-related changes and follows slow, graduating progression (Knezovic et al. 2015). Findings in this non-transgenic sAD animal model strongly support clinical data indicating BIR as a possible primary pathological event in AD development. Furthermore, recently developed and thus far less explored STZ-icv mokey model demonstrates BIR (Lee et al. 2014) accompanied by Aβ deposition and tauopathy (Yeo et al. 2015), while BIR induced by STZ-icv treatment aggravates cognitive deficits and increases the formation of pathomorphological AD hallmarks, particularly Aβ accumulation in APP overexpressing (Plaschke et al. 2010a, b) and Presenilin-1-Val97Leu mutant (Lin et al. 2014) transgenic mice AD models.
Long-term drug testing polygon considering the preliminary data of the therapeutic role of icv insulin in the STZ-icv model (Shingo et al. 2013) and of intranasal insulin in AD patients (Claxton et al. 2015) should be performed to elucidate the importance of glucose/insulin pathology as risk factor for both, AD and VaD.
The representative experiments (≥3 mg/kg STZ-icv; rat) demonstrate the following order of AD-like pathology appearance: IRBS = oxidative stress = neuroinflammation > glucose hypometabolism = tau pathology = cognitive deficits > amyloid β1-42 accumulation > amyloid angiopathy > amyloid plaques. These data support possible causal role of IRBS in sAD etiopathogenesis (Chen et al. 2014; de la Monte et al. 2014), confirmed by therapeutic effect of icv insulin in this model (Shingo et al. 2013) and intranasal insulin in AD patients (Claxton et al. 2015), and contributing role of vascular pathology in progression of cognitive decline as demonstrated in 9-month follow-up studies of this model (Knezovic et al. 2015; Salkovic-Petrisic et al. 2011).
IRBS is a condition characterized at the molecular level by reduced response to insulin signaling downstream the insulin receptor (IR)–insulin receptor substrate (IRS)–phosphatidyl inositol kinase-3 (PI-3) pathway in the brain, which, particularly considering the neurotrophic, neuroprotective and neuromodulatory role of brain insulin (Craft and Christen 2010; Gasparini and Xu 2003; Sato et al. 2011), may lead to neurodegeneration and cognitive impairment as seen in AD. Although sometimes termed “type 3 diabetes” (de la Monte and Tong 2014), it actually represents a brain-related metabolic syndrome associated with metabolic and oxidative stress and neuroinflammation in the brain, which may or may not be accompanied by alterations in peripheral metabolic homeostasis, since T2DM increases the risk for AD (and vice versa), but neither all T2DM patients develop AD (and vice versa) nor AD is necessarily associated with hyperglycemia (Talbot and Wang 2014; Talbot and Wang 2014; Blázquez et al. 2014).
While animal models are referred to in other consensus BMC-related manuscript, the group focused here on modelling the IRBS. It is concluded that the icv STZ rodent model mirrors AD and VaD pathologies in many respects. The model should be used with or without combination of transgenic mouse models.
Treatment of diabetes-related cognitive impairment
Currently little is known regarding the effect of diabetes interventions on diabetes-related cognitive impairment. The ACCORD-MIND study conducted among ~3000 individuals with DM demonstrated that tight glycemic control significantly reduced the rate of brain atrophy over a period of 20–40 months compared with the standard glucose treatment; however, there was no difference between the groups in the rate of cognitive decline as measured by 4 cognitive instruments (Cukierman-Yaffe et al. 2009).
Analogue compounds for the incretin hormone GLP-1 (glucagon-like peptide-1), which facilitate endogenous insulin release and are used to treat T2DM, reduce Aβ accumulation, and rescue impairments in hippocampal synaptic plasticity and spatial learning memory in transgenic mouse models of AD (Gengler et al. 2012).
Many studies suggest that adding more insulin to the brain would improve memory and prevent cell damage (Shingo et al. 2013; Claxton et al. 2015). In individuals without DM, it has been shown amongst cognitively intact and cognitively impaired individuals that a form of insulin that enters the brain selectively has beneficial effects on some cognitive domains (Shemesh et al. 2012). In the ORIGIN cognitive sub-study, treatment of people with DM and prediabetes for 6.5 years with basal insulin had a neutral effect on cognitive function (Cukierman-Yaffe et al. 2014).
There is one interesting report on the use of bacteriophage as a common divergent therapeutic approach for treating AD and T2DM (Sohrab et al. 2014). Invokana (Canagliflozin), which has dual inhibitory effect on acetylcholinesterase as well as on SGLT2, represents advancement in the parallel management of AD and T2DM (Rizvi et al. 2014). Galangin (a novel natural ligand) has inhibition characteristics on human brain acetylcholinesterase, butyrylcholinesterase, and 5-lipoxygenase (Shaikh et al. 2014). Molecular interaction of human brain acetylcholinesterase (target enzyme in AD therapy) has also been studied with a natural inhibitor, Huperzine-B (Alam et al. 2014a).
Elements such as magnesium play an important role in the normal functioning of many enzymatic activities. There has been some evidence for the role of magnesium in the prevention and therapy of AD and T2DM (Gröber et al. 2015), and there are some recent nanotechnological approaches in the management of AD and T2DM (Alam et al. 2014b).
Pantethine has beneficial effects in vascular disease, is able to decrease the hyperlipidemia, moderates the platelet function, and prevents lipid-peroxidation (Horváth and Vécsei 2009). The disulfide group (oxidized form of pantethine) is necessary to lower the platelet response to activation by thrombin and collagen (Penet et al. 2008). It was found that orally active multi-functional antioxidants including pantethine delay cataract formation in streptozotocin T1DM and gamma-irradiated rats (Randazzo et al. 2011). Pantethine should be considered for the treatment of lipid abnormalities also in patients at risk such as those with DM and other dementia disorders.
The possible implications of the relationship between dementia/cognitive impairment and diabetes on the care of the older individual with diabetes
Current guidelines for treatment of individuals with DM include extensive life style changes in diet, physical activity, smoking cessation, medication, and routine medical follow-up (Powers et al. 2015). To successfully manage self-care of such changes, the individual with DM is required to have intact cognitive function; i.e., to understand and learn new information, memorize it, apply new behaviors and procedures, and make complex decisions in a changing environment. However, current DM treatment and surveillance do not include routine assessment of cognitive function and the cognitive function of the individual is not taken into consideration when devising a treatment plan. This is especially important when treating older people with DM, since DM and aging are both independent risk factors for cognitive dysfunction. In the face of increasing numbers of older people with DM the fact that cognitive impairment is another complication of DM has two important implications. One is that it is pivotal that the effect of currently used glucose lowering agents on this complication be understood. Second, cognitive assessment, i.e., screening and surveillance should be part of the routine care of the older person with DM.
Cognitive dysfunction can potentially present new barriers to self-care and to achieving glycemic control. Indeed, population studies have shown that among people with DM lower cognitive function was associated with worse efficacy of treatment indices such as glucose control (Cukierman-Yaffe et al. 2009) and a greater risk for incident hypoglycemia (Punthakee et al. 2012). Reciprocal associations are assumed between DM self-care, glycemic control, micro and macro vascular outcomes, and cognitive impairment. Indeed, in a sample of 1398 older community-dwelling adults with DM, as cognitive impairment worsened, so did participants’ adherence to each diabetes self-care task with incremental increases in DM comorbidity (Esmaeili et al. 2016). In a population-based study, amongst ~3000 middle-aged individuals with diabetes, those with lower cognitive scores had a higher risk for hypoglycemia events that required the help of another (a possible sequel of poor self-care management skills as it requires the patient to be self-alert and active in the management of the disease) (Punthakee et al. 2012). Another study reported that providing memory strategies improved adherence to medication amongst elderly DM patients (Vedhara et al. 2010). Finally, a recent study reported that in individuals with DM, lower executive function was associated with higher need of outpatient care.
Current guidelines for treating older people with DM recommend routine screening for cognitive dysfunction (Kirkman et al. 2012; Sinclair et al. 2015). However, cognitive dysfunction is only the tip of the iceberg of a continuum of cognitive decline which is accelerated in people with DM. Thus, it is also important to characterize the cognitive profile of the intact individual with DM enabling care takers to adapt the treatment plan according to the individual’s cognitive capacities. Indeed, in the past 3 years at the Center for Successful aging with Diabetes at the Sheba Medical Center, Israel, we have been conducting multi-disciplinary evaluation that include extensive neuropsychological testing and evaluation of the medical, functional, and physical status of the older person with DM, followed by cognitively adapted tailor-made recommendations (including the use of cognitive rehabilitation strategies) and a follow-up plan that takes into consideration the cognitive profile of the individual. Participants have reported a significant improvement in quality of life. We hypothesize that this type of approach which includes cognitive screening, surveillance, and rehabilitation will improve the self-care capacity of the older individual with DM, thus improving glucose control and reducing the risk for DM complications and possibly reducing the accelerated rate of cognitive decline this population experiences.
When deciding which drug to add to the regimen of an older individual with DM, the potential risk for hypoglycemia with this agent should be evaluated. Hypoglycemia unawareness is very common in elderly DM patients. A study involving T2DM patients over 65 years of age and using continuous glucose monitoring revealed hypoglycemic episodes in as many as 80% of patients, including 56% with severe hypoglycemic episodes (<40 mg/dl), and none of these episodes were actually “felt” by a patient (Kagansky et al. 2003; Zoungas et al. 2010; Whitmer et al. 2009; Sinclair et al. 2011; Kasiukiewicz et al. 2015; Tseng et al. 2014). Thus, the type of glucose lowering agent chosen should take into consideration hypoglycemia unawareness as well as the cognitive profile of the older person with diabetes choosing therapy that is safer in this respect such as metformin, alpha-glucosidase inhibitors, thiazolidinediones, GLP-1 receptor agonists, DPP-4 inhibitors, SGLT-2 as opposed to insulin, sulfonylureas, or glinides (Chamberlain et al. 2016).
Differences between treated and not treated DM2 patients
T2DM patients may be treated with oral antidiabetic agents [such as sulfonylurea, metformin and dipeptidyl peptidase 4 inhibitor (DPP-4I)], insulin, or by diet control only. Although all treatments may reduce glucose level, which type of treatment is helpful in reducing the cognitive impairment, is not clear.
Early studies suggest that cognitive decline may be slower, if T2DM patients are treated with insulin instead than oral antidiabetic agents. Therapy with insulin may lower neuritic plaque density in hippocampus and other brain areas (Beeri et al. 2008). In addition, Plastino et al. demonstrated slower cognitive decline in T2DM patients with AD treated with insulin in comparison to patients treated with oral antidiabetic agents (Plastino et al. 2010). However, this relationship may not be straight forward. In a recent study, Herath et al. followed T2DM patients for 4 years and found no significant differences between patients from diet only, oral antidiabetic agents, and insulin groups (Herath et al. 2016).
New research shows that treatment with DPP-4 inhibitors (DPP4I) is helpful. Rizzo et al. demonstrated in a prospective 2-year study that patients treated with DPP4I and metformin had better cognitive functioning in comparison to patients treated only with sulphonyl urea and metformin (Rizzo et al. 2014). Similar results were found in a shorter study, where they followed patients for 6 months (Isik et al. 2017). T2DM patients with AD and without AD treated with DPP-4I performed better on MMSE 6 months after they started with the treatment than patients taking metformin (Isik et al. 2017). In addition, patients treated with insulin or DPP-4I also had better glucose control and lower HbA1c at the end visit.
Future strategies
Increasing evidence suggests that the production of new neurons in the adult hippocampus (adult neurogenesis; AN) plays an important role in different subtypes of learning processes and memory formations (Deng et al. 2009a, b) and seems also to contribute to cognitive flexibility (Burghardt et al. 2012). Indeed, hippocampal AN was demonstrated to be diminished in these icv STZ rats after 3 months (Sun et al. 2015).
Therefore, it has been suggested that altered AN in the hippocampus plays a role in the etiopathology of neurodegenerative disorders such as AD (for review, see Winner and Winkler 2015) and vascular dementia (Ekonomou et al. 2011). Cerebrovascular functions and AN both decline during aging (Kalaria 2009; Kempermann 2015) which stands to reason as AN occurs within an angiogenic niche (Palmer et al. 2000). Improved energy supply in experimental AN studies show improved AN when using DM related therapeutic strategies (Luitse et al. 2012; Biessels 2013; Sonnen et al. 2009; Jia et al. 2004; Tohgi et al. 1996; Chen et al. 2013; Lee et al. 2014; Niwa et al. 2002), indicating an important role of glucose/energy supply for the proper integrity of AN physiology.
Conclusion
In many individuals, AD and VaD show an underlying pathology of glucose utilization based on a disturbance of insulin-related pathology, leading to a brain insulin resistance state. However, the risk factors rather induce a mechanisms independent from pathological mechanisms underlying AD and VaD. In fact, these probably have different causality, which is reflected in the prevalence/incidence of AD (about 60%) and VaD (about 20%) of all dementia disorders. Considering the brain insulin resistance as a shared pathological feature of T2DM and dementia which, most probably are a consequence of environmental factors and epigenetic mechanisms operating during pregnancy and postnatally may be manifested as one or the other disorder, T2DM contributes to disease onset and progression of both AD and VaD. Therefore, therapeutic strategies focusing on DM should be considered already in early stages of AD and VaD. Future strategies including AN may enlarge the therapeutic armamentarium. More focus should be put on delaying dementia onset in people with diabetes and on the challenges cognitive impairment imposes on the self-care capacity of this population.
Abbreviations
- Aβ:
-
Beta-amyloid-protein
- AChE:
-
Acetylcholinesterase
- AD:
-
Alzheimer’s disease
- AGEs:
-
Advanced glycation end products
- AKT1s1:
-
Proline-rich AKT1 substrate 1
- AKT-1:
-
RAC-alpha serine/threonine-protein kinase
- AKT-2:
-
RAC-beta serine/threonine-protein kinase
- APP:
-
Beta-amyloid precursor protein
- APOE ϵ4:
-
Apolipoprotein E ϵ4
- AQP4:
-
Aquaporin-4
- ATP:
-
Adenosine triphospate
- BBB:
-
Blood brain barrier
- BChE:
-
Butyrylcholinesterase
- BHB:
-
Beta-hydroxybutyrate
- BIR:
-
Brain insulin resistance
- CBF:
-
Cerebral blood flow
- CBH:
-
Chronic brain hypoperfusion
- CSF:
-
Cerebrospinal fluid
- Ct:
-
Control
- CVR:
-
Cerebrovascular reactivity
- DM:
-
Diabetes mellitus
- DNA:
-
Desoxyribonucleic acid
- FDG:
-
Fluorodeoxyglucose
- FTO:
-
Fat-mass and obesity-associated gene
- Gd-DTPA:
-
Gadolinium-based MRI contrast agent
- GLP-1:
-
Glucagon-like peptide 1
- GLUT3:
-
Glucose transporter 3
- GM:
-
Grey matter
- GSK3β:
-
Glycogen synthase kinase 3 β
- HOMA-IR:
-
Homeostatic model assessment of insulin resistance
- HNE:
-
4-Hydroxynonenal
- IDE:
-
Insulin degrading enzyme
- ICV:
-
Intracerebroventricular
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- IR:
-
Insulin receptor
- IRBS:
-
Insulin resistant brain state
- IRβ:
-
Insulin receptor subunit β
- IRS1:
-
Insulin receptor substrate-1
- IRS-1pS616:
-
Serin-phosphorylated insulin receptor substrate-1
- IRS2:
-
Insulin receptor substrate-2
- ISF:
-
Interstitial fluid
- KAT:
-
Kynurenine aminotransferase
- KYNAC:
-
Kynurenic acid
- MCI:
-
Mild cognitive impairment
- MRI:
-
Magnet resonance imaging
- mTOR:
-
Mechanistic target of rapamycin
- OS:
-
Oxidative stress
- PCAD:
-
Pre-clinical AD
- PET:
-
Positron emission tomography
- PG:
-
Postprandial glycemia
- PIK3CB:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta isoform
- PIK3CD:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta
- PI3:
-
Phosphatidylinositol-3-kinase
- PI3K:
-
Phosphoinositid-3-kinase
- PIP3:
-
Phosphatidylinositol (3,4,5)-triphosphate
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- P-Tau:
-
Phospho-Tau-Protein
- PYY:
-
Peptide YY
- P53:
-
Phosphoprotein p53
- QA:
-
Quinolinic acid
- RAGE:
-
Receptor for AGEs
- RNA:
-
Ribonucleic acid
- ROS:
-
Reactive oxygen species
- sAD:
-
Sporadic Alzheimer’s disease
- SGLT2:
-
Sodium/glucose cotransporter 2
- STZ:
-
Streptozotocin
- T2DM:
-
Type 2 diabetes mellitus
- T1DM:
-
Type 1 diabetes mellitus
- VaD:
-
Vascular dementia
- WM:
-
White matter
References
Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL, Cairns NJ, Yu L, Dodge HH, Xiong C, Masaki K, Tyas SL, Bennett DA, Schneider JA, Arvanitakis Z (2016) Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12(8):882–889
Agrawal R, Tyagi E, Shukla R, Nath C (2011) Insulin receptor signaling in rat hippocampus: a study in STZ (ICV) induced memory deficit model. Eur Neuropsychopharmacol 21:261–273
Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, Sulkava R, Kivipelto M (2010) Diabetes, Alzheimer disease, and vascular dementia. A population based neuropathologic study. Neurology 75:1195–1202
Alafuzoff I, Aho L, Helisalmi S, Mannermaa A, Soininen H (2009) Beta-amyloid deposition in brains of subjects with diabetes. Neuropathol Appl Neurobiol 35:60–68
Alam A, Shaikh S, Ahmad SS, Ansari MA, Shakil S, Rizvi SM, Shakil S, Imran M, Haneef M, Abuzenadah AM, Kamal MA (2014a) Molecular interaction of human brain acetylcholinesterase with a natural inhibitor Huperzine-B: an enzoinformatics approach. CNS Neurol Disord Drug Targets 13(3):487–490
Alam Q, ZubairAlam M, Karim S, Gan SH, Kamal MA, Jiman-Fatani A, Damanhouri GA, Abuzenadah AM, Chaudhary AG, Haque AA (2014b) Nanotechnological approach in management of Alzheimer’s diseases and type 2 diabetes. CNS Neurol Disord Drug Targets 13(3):478–486
Aluise CD, Robinson RA, Cai J, Pierce WM, Markesbery WR, Butterfield DA (2011) Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: insights into memory loss in MCI. J Alzheimers Dis 23:257–269
Alvarez EO, Beauquis J, Revsin Y, Banzan AM, Roig P, De Nicola AF, Saravia F (2009) Cognitive dysfunction and hippocampal changes in experimental type 1 diabetes. Behav Brain Res 198(1):224–230
Artola A, Kamal A, Ramakers GM, Gardoni F, Di Luca M, Biessels GJ, Cattabeni F, Gispen WH (2002) Synaptic plasticity in the diabetic brain: advanced aging? Prog Brain Res 138:305–314
Ascher-Svanum H, Chen YF, Hake A, Kahle-Wrobleski K, Schuster D, Kendall D, Heine RJ (2015) Cognitive and functional decline in patients with mild alzheimer dementia with or without comorbid diabetes. Clin Ther 37(6):1195–1205
Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S (2011) Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol 68(1):51–57
Baldeiras I, Santana I, Proença MT, Garrucho MH, Pascoal R, Rodrigues A, Duro D, Oliveira CR (2010) Oxidative damage and progression to Alzheimer’s disease in patients with mild cognitive impairment. J Alzheimers Dis 21:1165–1177
Banks WA (2004) The source of cerebral insulin. Eur J Pharmacol 490(1–3):5–12
Banks WA (2006) The blood–brain barrier as a regulatory interface in the gut–brain axes. Physiol Behav 89(4):472–476
Banks WA, Owen JB, Erickson MA (2012) Insulin in the brain: there and back again. Pharmacol Ther 136(1):82–93
Barilar JO, Knezovic A, Grünblatt E, Riederer P, Salkovic-Petrisic M (2015) Nine-month follow-up of the insulin receptor signalling cascade in the brain of streptozotocin rat model of sporadic Alzheimer’s disease. J Neural Transm 122:565–576
Bartl J, Monoranu CM, Wagner AK, Kolter J, Riederer P, Grünblatt E (2012) Alzheimer’s disease and type 2 diabetes: two diseases, one common link? World J Biol Psychiatry 14:233–240
Bedse G, Di Domenico F, Serviddio G, Cassano T (2015) Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci 9:204
Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J, Purohit DP, Perl DP, Davidson M, Mohs RC, Haroutunian V (2005) Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol A Biol Sci Med Sci 60:471–475
Beeri MS, Schmeidler J, Silverman JM, Gandy S, Wysocki M, Hannigan CM et al (2008) Insulin in combination with other diabetes medication is associated with less Alzheimer neuropathology. Neurology 71:750–757
Bellou V, Belbasis L, Tzoulaki I, Middleton LT, Ioannidis JP, Evangelou E (2017) Systematic evaluation of the associations between environmental risk factors and dementia: an umbrella review of systematic reviews and meta-analyses. Alzheimers Dement 13(4):406–418
Biessels GJ (2013) Brain MRI correlates of cognitive dysfunction in type 2 diabetes: the needle recovered from the haystack? Diabetes Care 36:3855–3856
Biessels GJ, Reijmer YD (2014) Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes 63(7):2244–2252
Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW et al (1996) Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes 45:1259–1266
Biessels GJ, De Leeuw F-E, Lindeboom J, Barkhof F, Scheltens P (2006a) Increased cortical atrophy in patients with Alzheimer’s disease and type 2 diabetes mellitus. J Neurol Neurosurg Psychiatry 3:304–307
Biessels GJ, Koffeman A, Scheltens P (2006b) Diabetes and cognitive impairment. Clinical diagnosis and brain imaging in patients attending a memory clinic. J Neurol 253:477–482
Biessels GJ, Deary IJ, Ryan CM (2008) Cognition and diabetes: a lifespan perspective. Lancet Neurol 7(2):184–190
Bitel CL, Kasinathan C, Kaswala RH, Klein WL, Frederikse PH (2012) Amyloid-beta and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J Alzheimers Dis 32:291–305
Blázquez E, Velázquez E, Hurtado-Carneiro V, Ruiz-Albusac JM (2014) Insulin in the brain: its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front Endocrinol (Lausanne) 5:161
Blennow K, Wallin A, Fredman P, Karlsson I, Gottfries CG, Svennerholm L (1990) Blood–brain barrier disturbance in patients with Alzheimer’s diseases related to vascular factors. Acta Neurol Scand 81(4):323–326
Brands AM, Kessels RP, Hoogma RP, Henselmans JM, van der Beek Boter JW, Kappelle LJ, de Haan EH, Biessels GJ (2006) Cognitive performance, psychological well-being, and brain magnetic resonance imaging in older patients with type 1 diabetes. Diabetes 55(6):1800–1806
Breteler MM (2000) Vascular risk factors for Alzheimer’s disease: an epidemiologic perspective. Neurobiol Aging 21:153–160
Brundel M, van den Berg E, Reijmer YD, de Bresser J, Kappelle LJ, Biessels GJ (2012) Cerebral haemodynamics, cognition and brain volumes in patients with type 2 diabetes. J Diabetes Complicat 26(3):205–209
Burghardt NS, Park EH, Hen R, Fenton AA (2012) Adult-born hippocampal neurons promote cognitive flexibility in mice. Hippocampus 22(9):1795–1808
Carvalho C, Santos MS, Oliveira CR, Moreira PI (2015) Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim Biophys Acta 1852:1665–1675
Chamberlain JJ, Rhinehart AS, Shaefer CF Jr, Neuman A (2016) Diagnosis and management of diabetes: synopsis of the 2016 American Diabetes Association Standards of Medical Care in Diabetes. Ann Intern Med 164(8):542–552
Chatterjee S, Peters SA, Woodward M, Mejia Arango S, Batty GD, Beckett N, Beiser A, Borenstein AR, Crane PK, Haan M, Hassing LB, Hayden KM, Kiyohara Y, Larson EB, Li CY, Ninomiya T, Ohara T, Peters R, Russ TC, Seshadri S, Strand BH, Walker R, Xu W, Huxley RR (2016) Type 2 diabetes as a risk factor for dementia in women compared with men: a pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care 39(2):300–307
Chen Y, Liang Z, Blanchard J, Dai CL, Sun S, Lee MH, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX (2013) A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol Neurobiol 47:711–725
Chen Y, Deng Y, Zhang B, Gong CX (2014) Deregulation of brain insulin signaling in Alzheimer’s disease. Neurosci Bull 30:282–294
Chiu WC, Ho WC, Liao DL, Lin MH, Chiu CC, Su YP, Chen PC (2015) Health Data Analysis in Taiwan (hDATa) Research Group. Progress of diabetic severity and risk of dementia. J Clin Endocrinol Metab 100(8):2899–3190
Chmiel-Perzynska I, Perzynski A, Urbanska EM (2014) Experimental diabetes mellitus type 1 increases hippocampal content of kynurenic acid. Pharmacol Rep 66:1134–1139
Chmiel-Perzyńska I, Kloc R, Perzyński A, Rudzki S, Urbańska EM (2011) Novel aspect of ketone action: beta-hydroxybutyrate increases brain synthesis of kynurenic acid in vitro. Neurotox Res 20:40–50
Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, Callaghan M, Arbuckle M, Behl C, Craft S (2015) Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis 44:897–906
Correia SC, Santos RX, Santos MS, Casadesus G, Lamanna JC, Perry G, Smith MA, Moreira PI (2013) Mitochondrial abnormalities in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. Curr Alzheimer Res 10:406–419
Craft S, Christen Y (2010) Diabetes, insulin and Alzheimer’s disease XIII. Springer, New York, p 250
Craft S, Watson GS (2004) Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 3:169–178
Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M, Porte D Jr (1998) Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: relationship to severity of dementia and apolipoprotein e genotype. Neurology 50:164–168
Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B (2012) Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol 69(1):29–38
Craft S, Cholerton B, Baker LD (2013) Insulin and Alzheimer’s disease: untangling the web. J Alzheimers Dis 33(Suppl 1):263–275
Cukierman T, Gerstein HC, Williamson JD (2005) Cognitive decline and dementia in diabetes–systematic overview of prospective observational studies. Diabetologia 48(12):2460–2469
Cukierman-Yaffe T, Gerstein HC, Williamson JD et al (2009) Relationship between baseline glycemic control and cognitive function in individuals with type 2 diabetes and other cardiovascular risk factors: the action to control cardiovascular risk in diabetes-memory in diabetes (ACCORD-MIND) trial. Diabetes Care 32(2):221–226
Cukierman-Yaffe T, Bosch J, Diaz R, Dyal L, Hancu N, Hildebrandt P, Lanas F, Lewis BS, Marre M, Yale JF, Yusuf S, Gerstein HC, ORIGIN Investigators (2014) Effects of basal insulin glargine and omega-3 fatty acid on cognitive decline and probable cognitive impairment in people with dysglycaemia: a substudy of the ORIGIN trial. Lancet Diabetes Endocrinol 2(7):562–572
Darreh-Shori T, Forsberg A, Modiri N, Andreasen N, Blennow K, Kamil C, Ahmed H, Almkvist O, Långström B, Nordberg A (2011) Differential levels of apolipoprotein E and butyrylcholinesterase show strong association with pathological signs of Alzheimer’s disease in the brain in vivo. Neurobiol Aging 32:2320.e15–2320.e32
de Felice FG, Benedict C (2015) A key role of insulin receptors in memory. Diabetes 64:3653–3655
de Felice FG, Lourenco MV, Ferreira ST (2014) How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 10(1 Suppl):S26–S32
de la Monte SM (2009) Insulin resistance and Alzheimer’s disease. BMB Rep 42(8):475–481
de la Monte SM (2012) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 9(1):35–66
de la Monte SM, Tong M (2014) Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol 88:548–559
de la Monte SM, Tong M, Lester-Coll N, Plater M Jr, Wands JR (2006) Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis 10:89–109
de la Monte SM, Re E, Longato L, Tong M (2012) Dysfunctional pro-ceramide, ER stress, and insulin/IGF signaling networks with progression of Alzheimer’s disease. J Alzheimers Dis 30(Suppl 2):S217–S229
de la Torre JC (2010) Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res Rev 9(3):218–225. doi:10.1016/j.arr.2010.04.002 (epub Apr 10)
de Matos AM, de Macedo MP, Rauter AP (2017) Bridging type 2 diabetes and Alzheimer’s disease: assembling the puzzle pieces in the quest for the molecules with therapeutic and preventive potential. Med Res Rev. doi:10.1002/med.21440
de Oliveira Lanna ME, Pimentel MLV, Novis SAP (2014) Diabetes effects in Alzheimer disease: the interactive role of insulin and Aβ peptide. J Alzheimers Dis Parkinsonism 4:151. doi:10.4172/2161-0460.1000151
Deng W, Saxe MD, Gallina IS, Gage FH (2009a) Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci 29(43):13532–13542
Deng Y, Li B, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX (2009b) Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: implication for Alzheimer’s disease. Am J Pathol 175:2089–2098
Diamant S, Podoly E, Friedler A, Ligumsky H, Livnah O, Soreq H (2006) Butyrylcholinesterase attenuates amyloid fibril formation in vitro. PNAS 103(23):8628–8633
Doi Y, Ninomiya T, Hata J, Fukuhara M, Yonemoto K, Iwase M et al (2010) Impact of glucose tolerance status on development of ischemic stroke and coronary heart disease in a general Japanese population: the Hisayama Study. Stroke 41:203–209
Du LL, Xie JZ, Cheng XS, Li XH, Kong FL, Jiang X, Ma ZW, Wang JZ, Chen C, Zhou XW (2014) Activation of sirtuin 1 attenuates cerebral ventricular streptozotocin-induced tau hyperphosphorylation and cognitive injuries in rat hippocampi. Age 36:613–623
Duarte JMN, Oses JP, Rodrigues RJ, Cunha RA (2007) Modification of purinergic signaling in the hippocampus of streptozotocin-induced diabetic rats. Neuroscience 149(2):382–391
Ekonomou A, Ballard CG, Pathmanaban ON, Perry RH, Perry EK, Kalaria RN, Minger SL (2011) Increased neural progenitors in vascular dementia. Neurobiol Aging 32(12):2152–2161
Esmaeili M, Ghaedi K, Nejati AS, Nematollahi M, Shiralyian H, Nasr-Esfahani MH (2016) Pioglitazone significantly prevented decreased rate of neural differentiation of mouse embryonic stem cells which was reduced by Pex11beta knock-down. Neuroscience 312:35–47
Feinkohl I, Price JF, Strachan MW, Frier BM (2015) The impact of diabetes on cognitive decline: potential vascular, metabolic, and psychosocial risk factors. Alzheimers Res Ther 7:46
Fitzpatrick AL, Kuller LH, Lopez OL, Diehr P, O´Meara ES, Longstreth WT Jr, Luchsinger JA (2009) Midlife and late-life obesity and the risk of dementia: cardiovascular health study. Arch Neurol 66:336–342
Friedman JI, Tang CY, de Haas HJ, Changchien L, Goliasch G, Dabas P et al (2014) Brain imaging changes associated with risk factors for cardiovascular and cerebrovascular disease in asymptomatic patients JACC. Cardiovasc Imaging 7(10):1039–1053
Frisardi V, Solfrizzi V, Capurso C, Imbimbo BP, Vendemiale G, Seripa D, Pilotto A, Panza F (2010) Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J Alzheimers Dis 21:57–63
Garwood CJ, Ratcliffe LE, Morgan SV, Simpson JE, Owens H, Vazquez-Villasenor I, Heath PR, Romero IA, Ince PG, Wharton SB (2015) Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol Brain 8:51
Gasparini L, Xu H (2003) Potential roles of insulin and IGF-1 in Alzheimer’s disease. Trends Neurosci 26(8):404–406
Gengler PL, McClean R, McCurtin VA, Ischer H (2012) Val(8)GLP-1 rescures synaptic plasticity and reduces dense core plaqgues in APP/PS1 mice. Neurobiol Aging 33:265–276
Gold SM, Dziobek I, Sweat V, Tirsi A, Rogers K, Bruehl H, Tsui W, Richardson S, Javier E, Convit A (2007) Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 50(4):711–719
Goldin A, Beckman JA, Schmidt AM, Creager MA (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114:597–605
Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S, American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia (2011) Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke 42(9):2672–2713
Gröber U, Schmidt J, Kisters K (2015) Magnesium in prevention and therapy. Nutrients 7(9):8199–8226
Grünblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S (2007) Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem 101(3):757–770
Gsell W, Strein I, Riederer P (1996) The neurochemistry of Alzheimer type, vascular type and mixed type dementias compared. J Neural Transm Suppl 47:73–101
Gudala K, Bansal D, Schifano F, Bhansali A (2013) Diabetes mellitus and risk of dementia: a meta-analysis of prospective observational studies. J Diabetes Investig 4(6):640–650
Hallschmid M, Benedict C, Schultes B, Born J, Kern W (2008) Obese men respond to cognitive but not to catabolic brain insulin signaling. Int J Obes 32:275–282
Hamed SA (2017) Brain injury with diabetes mellitus: evidence, mechanisms and treatment implications. Expert Rev Clin Pharmacol 10(4):409–428
Hao K, Di Narzo AF, Ho L, Luo W, Li S, Chen R, Li T, Dubner L, Pasinetti GM (2015) Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol Aspects Med 43–44:66–76
Haroon NN, Austin PC, Shah BR, Wu J, Gill SS, Booth GL (2015) Risk of dementia in seniors with newly diagnosed diabetes: a population-based study. Diabetes Care 38(10):1868–1875
Hassanzadeh G, Hosseini A, Pasbakhsh P, Akbari M, Ghaffarpour M, Takzare N, Zahmatkesh M (2015) Trimetazidine prevents oxidative changes induced in a rat model of sporadic type of Alzheimer’s disease. Acta Med Iran 53:17–24
Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide A-M, Krebs N, Machicao F, Fritsche A, Häring H-U, Staiger H (2011) Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One 6(6):e21594
Herath PM, Cherbuin N, Eramudugolla R, Anstey KJ (2016) The effect of diabetes medication on cognitive function: evidence from the PATH Through Life study. Biomed Res Int 2016:7208429. doi:10.1155/2016/7208429
Hertz MM, Paulson OB, Barry DI, Christiansen JS, Svendsen PA (1981) Insulin increases glucose transfer across the blood–brain barrier in man. Clin Investig 67(3):597–604
Horváth Z, Vécsei L (2009) Current medical aspects of pantethine. Ideggyogy Sz. 62:220–229
Hoyer S (1998) Is sporadic Alzheimer disease the brain type of non-insulin dependent diabetes mellitus? A challenging hypothesis. J Neural Transm 105(4–5):415–422
Hoyer S (2004) Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol 490(1–3):115–125
Hoyer S, Lannert H (2007) Long-term abnormalities in brain glucose/energy metabolism after inhibition of the neuronal insulin receptor: implication of tau-protein. J Neural Transm Suppl 72:195–202
Hoyer S, Lannert H, Nöldner M, Chatterjee SS (1999) Damaged neuronal energy metabolism and behavior are improved by Ginkgo biloba extract (EGb 761). J Neural Transm 106:1171–1188
Huang CC, Chung CM, Leu HB, Lin LY, Chiu CC, Hsu CY, Chiang CH, Huang PH, Chen TJ, Lin SJ, Chen JW, Chan WL (2014) Diabetes mellitus and the risk of alzheimer’s disease: a Nationwide Population-Based Study. PLoS One 9(1):e87095
Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M et al (2013) Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Investig 123(3):1299–1309
Irie F, Fitzpatrick AL, Lopez OL et al (2008) Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE e4. The cardiovascular health study cognition study. Arch Neurol 65:89–93
Isik AT, Soysal P, Yay A, Usarel C (2017) The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabetes Res Clin Pract 123:192–198
Israili ZH (2011) Advances in the treatment of type 2 diabetes mellitus. Am J Ther 18(2):117–152
Jacobson AM, Musen G, Ryan CM, Silvers N, Cleary P, Waberski B et al (2007) Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Group. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med 356:1842–1852
Javed H, Khan MM, Ahmad A, Vaibhav K, Ahmad ME, Khan A, Ashafaq M, Islam F, Siddiqui MS, Safhi MM, Islam F (2012) Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience 210:340–352
Jellinger KA (2015a) Cognitive impairment and the diabetic brain. Int J Neurol Neurother 2:033
Jellinger KA (2015b) The diabetic brain and dementia. J Alzheimer’s Dis Parkinsonism 5:193
Ji H, Dai D, Wang Y, Jiang D, Zhou X, Lin P, Ji X, Li J, Zhang Y, Yin H, Chen R, Zhang L, Xu M, Duan S, Wang Q (2015) Association of BDNF and BCHE with Alzheimer’s disease: meta-analysis based on 56 genetic case-control studies of 12,563 cases and 12,622 controls. Exp Ther Med 9:1831–1840
Jia JP, Jia JM, Zhou WD, Xu M, Chu CB, Yan X, Sun YX (2004) Differential acetylcholine and choline concentrations in the cerebrospinal fluid of patients with Alzheimer’s disease and vascular dementia. Chin Med J 117(8):1161–1164
Jiang Q, Zhang L, Ding G, Davoodi-Bojd E, Li Q, Li L, Sadry N, Nedergaard M, Chopp M, Zhang ZG (2016) Impairment of the glymphatic system after diabetes. J Cereb Blood Flow Metab 37(4):1326–1337
Jiang Q, Zhang L, Ding G, Davoodi-Bojd E, Li Q, Li L, Sadry N, Nedergaard M, Chopp M, Zhang Z (2017) Impairment of the glymphatic system after diabetes. J Cereb Blood Flow Metab 37(4):1326–1337
Kagansky N, Levy S, Rimon E, Cojocaru L, Fridman A, Ozer Z et al (2003) Hypoglycemia as a predictor of mortality in hospitalized elderly patients. Arch Intern Med 163:1825–1829
Kalaria RN (2009) Linking cerebrovascular defense mechanisms in brain ageing and Alzheimer’s disease. Neurobiol Aging 30(9):1512–1514
Kang S, Lee YH, Lee JE (2017) Metabolism-centric overview of the pathogenesis of Alzheimer’s disease. Yonsei Med J 58(3):479–488. doi:10.3349/ymj.2017.58.3.479
Kasiukiewicz A, Wojszel ZB, Klimiuk K, Wojskowicz A (2015) Potential glycemic overtreatment in patients admitted to the geriatric ward. Eur Geriatr Med 6(Suppl I):S142
Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR (2005) Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 64:1152–1156
Kempermann G (2015) Activity dependency and aging in the regulation of adult neurogenesis. Cold Spring Harb Perspect Biol 7(11):a018929
Kim HA, Miller AA, Drummond GR, Thrift AG, Arumugam TV, Phan TG, Srikanth VK, Sobey CG (2012) Vascular cognitive impairment and Alzheimer’s disease: role of cerebral hypoperfusion and oxidative stress. Naunyn Schmiedebergs Arch Pharmacol 385:953–959
Kimura N (2016) Diabetes mellitus induces Alzheimer’s disease pathology: histopathological evidence from animal models. Int J Mol Sci 17(4):503
Kimura K, Tanida M, Nagata N, Inaba Y, Watanabe H, Nagashimada M, Ota T, Asahara S, Kido Y, Matsumoto M, Toshinai K, Nakazato M, Shibamoto T, Kaneko S, Kasuga M, Inoue H (2016) Central insulin action activates Kupffer cells by suppressing hepatic vagal activation via the nicotinic alpha 7 acetylcholine receptor. Cell Rep 14(10):2362–2374
Kirkman MS, Briscoe VJ, Clark N et al (2012) Diabetes in older adults. Diabetes Care 35(12):2650–2664
Knezovic A, Osmanovic-Barilar J, Curlin M, Hof PR, Simic G, Riederer P, Salkovic-Petrisic M (2015) Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J Neural Transm 122:577–592
Könner AC, Brüning JC (2012) Selective insulin and leptin resistance in metabolic disorders. Cell Metab 16:144–152
Kosaraju J, Gali CC, Khatwal RB, Dubala A, Chinni S, Holsinger RM, Madhunapantula VS, Nataraj SKM, Basavan D (2013) Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 72:291–300
Kraska A, Santin MD, Dorieux O, Joseph-Mathurin N, Bourrin E, Petit F, Jan C, Chaigneau M, Hantraye P, Lestage P, Dhenain M (2012) In vivo cross-sectional characterization of cerebral alterations induced by intracerebroventricular administration of streptozotocin. PLoS One 7(9):e46196
Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Häring HU (2016) Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol Rev 96:1169–1209
Kumar R, Jaggi AS, Singh N (2010) Effects of erythropoietin on memory deficits and brain oxidative stress in the mouse models of dementia. Korean J Physiol Pharmacol 14:345–352
Kuo SC, Lai SW, Hung HC, Muo CH, Hung SC, Liu LL, Chang CW, Hwu YJ, Chen SL, Sung FC (2015) Association between comorbidities and dementia in diabetes mellitus patients: population-based retrospective cohort study. J Diabetes Complicat 29(8):1071–1076
Last D, Alsop DC, Abduljalil AM, Marquis RP, de Bazelaire C, Hu K et al (2007) Global and regional effects of type 2 diabetes on brain tissue volumes and cerebral vasoreactivity. Diabetes Care 30(5):1193–1199
Lee S, Tong M, Hang S, Deochand C, de la Monte S (2013) CSF and brain indices of insulin resistance, oxidative stress and neuro-inflammation in early versus late Alzheimer’s disease. J Alzheimers Dis Parkinsonism 3:128
Lee Y, Kim YH, Park SJ, Huh JW, Kim SH, Kim SU, Kim JS, Jeong KJ, Lee KM, Hong Y, Lee SR, Chang KT (2014) Insulin/IGF signaling-related gene expression in the brain of a sporadic Alzheimer’s disease monkey model induced by intracerebroventricular injection of streptozotocin. J Alzheimers Dis 38:251–267
Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM (2006) Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis 9:13–33
Lin YL, Mettling C, Chou CK (2000) Complexes formation between insulin receptor and extracellular signal-regulated kinases ERKs. Mol Cell Biol Res Commun 4(4):234–238
Lin F, Jia J, Qin W (2014) Enhancement of β-amyloid oligomer accumulation after intracerebroventricular injection of streptozotocin, which involves central insulin signaling in a transgenic mouse model. Neuroreport 25:1289–1295. doi:10.1097/WNR.0000000000000261
Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2009) Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem 111:242–249
Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S, on behalf of the American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia-AHA/ASA Scientific Statement (2011) Vascular Contributions to cognitive impairment and dementia. A statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 42:2672–2713
Luchsinger JA (2012) Type 2 diabetes and cognitive impairment: linking mechanisms. J Alzheimers Dis 30(Suppl 2):S185–S198
Luitse MJA, Biessels GJ, Rutten GE, Kappelle LJ (2012) Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol 11(3):261–271
Lupien SJ, McEwen BS, Gunnar MR, Heim C (2009) Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 10:434–445
Lutski M, Weinstein G, Goldbourt U, Tanne D (2017) Insulin resistance and future cognitive performance and cognitive decline in elderly patients with cardiovascular disease. J Alzheimers Dis 57(2):633–643
Macauley SL, Stanley M, Caesar EE, Yamada SA, Raichie ME, Perez R, Mahan TE, Sutphen CL, Holtzman DM (2015) Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J Clin Investig 125:2463–2467
Malek-Ahmadi M, Beach T, Obradov A, Sue L, Belden C, Davis K, Walker DG, Lue L, Adem A, Sabbagh MN (2013) Increased Alzheimer’s disease neuropathology is associated with type 2 diabetes and ApoE epsilon4 carrier status. Curr Alzheimer Res 10:654–659
Manschot SM, Brands AMA, van der Grond J, Kessels RPC, Algra A, Kappelle LJ et al (2006) Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 55(4):1106–1113
Marseglia A, Fratiglioni L, Laukka EJ, Santoni G, Pedersen NL, Bäckman L, Xu W (2016) Early cognitive deficits in type 2 diabetes: a population-based study. J Alzheimers Dis 53(3):1069–1078
Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, Sekita A, Suzuki SO, Kanba S, Kiyohara Y, Iwaki T (2010) Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 75(9):764–770
McCrimmon RJ, Ryan CM, Frier BM (2012) Diabetes and cognitive dysfunction. Lancet 379:2291–2299
Mirza Z, Kamal MA, Buzenadah AM, Al-Qahtani MH, Karim S (2014) Establishing genomic/transcriptomic links between Alzheimer’s disease and type II diabetes mellitus by meta-analysis approach. CNS Neurol Disord Drug Targets 13(3):501–516
Moran C, Phan TG, Chen J, Blizzard L, Beare R, Venn A et al (2013) Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care 36(12):4036–4042
Moran C, Beare R, Phan TG, Bruce DG, Callisaya ML, Srikanth V (2015) Alzheimer’s disease neuroimaging initiative. Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology 85:1123–1130
Musen G, Jacobson AM, Bolo NR, Simonson DC, Shenton ME, McCartney RL et al (2012) Resting-state brain functional connectivity is altered in type 2 diabetes. Diabetes 61(9):2375–2379
Mushtaq G, Greig NH, Khan JA, Kamal MA (2014) Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets 13(8):1432–1439
Nelson PT, Smith CD, Abner EA, Schmitt FA, Scheff SW, Davis GJ, Keller JN, Jicha GA, Davis D, Wang-Xia W, Hartman A, Katz DG, Markesbery WR (2009) Human cerebral neuropathology of Type 2 diabetes mellitus. Biochim Biophys Acta 1792:454–469
Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB (2008) Insulin resistance and Alzheimer’s disease: molecular links and clinical implications. Curr Alzheimer Res 5:438–447
Ninomiya T (2014) Diabetes mellitus and dementia. Curr Diabetes Rep 14(5):487
Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C (2002) Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 9:61–68
Nizam Z, Hyer L (2007) Vascular cognitive impairment: perspective and review. J Psychiatry 35:325
Nunley KA, Ryan CM, Orchard TJ, Aizenstein HJ, Jennings JR, Ryan J, Zgibor JC, Boudreau RM, Costacou T, Maynard JD, Miller RG, Rosano C (2015) White matter hyperintensities in middle-aged adults with childhood-onset type 1 diabetes. Neurology 84(20):2062–2069
Ohara K, Kondo N, Xie D, Tanabe K, Yamamoto T, Kosaka K, Miyasato K, Ohara K (1994) Normal sequences of muscarinic acetylcholine receptors (m1 and m2) in patients with Alzheimer’s disease and vascular dementia. Neurosci Lett 178(1):23–26
Ouwens DM, van Duinkerken E, Schoonenboom SN, de Wiza DH, Klein M, van Golen L, Pouwels PJ, Barkhof F, Moll AC, Snoek FJ, Teunissen CE, Scheltens P, Diamant M (2014) Cerebrospinal fluid levels of Alzheimer’s disease biomarkers in middle-aged patients with type 1 diabetes. Diabetologia 57(10):2208–2214
Ozbabalık D, Arslantaş D, Tuncer Elmacı N (2012) The epidemiology of vascular dementia, geriatrics, Prof. Craig Atwood (Ed.), ISBN: 978-953-51-0080-5, InTech
Palmer TD, Willhoite AR, Gage FH (2000) Vascular niche for adult hippocampal neurogenesis. J Comp Neurol 425(4):479–494
Passafaro M, Piëch V, Sheng M (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4(9):917–926
Pearson J, Tarabulsy GM, Bussières EL (2015) Foetal programming and cortisol secretion in early childhood: a meta-analysis of different programming variables. Infant Behav Dev 40:204–215
Pedrós I, Petrov D, Allgaier M, Sureda F, Barroso E, Beas-Zarate C, Auladell C, Pallàs M, Vázquez-Carrera M, Casadesús G, Folch J, Camins A (2014) Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim Biophys Acta 1842:1556–1566
Penet MF, Abou-Hamdan M, Coltel N, Cornille E, Grau GE, de Reggi M, Gharib B (2008) Protection against cerebral malaria by the low-molecular-weight thiol pantethine. Proc Natl Acad Sci U S A 105(4):1321–1326
Pimlott SL, Piggott M, Owens J, Greally E, Court JA, Jaros E, Perry RH, Perry EK, Wyper D (2004) Nicotinic acetylcholine receptor distribution in Alzheimer’s disease, dementia with Lewy bodies, Parkinson’s disease, and vascular dementia: in vitro binding study using 5-[(125)i]-a-85380. Neuropsychopharmacology 29(1):108–116
Plagemann A (2008) A matter of insulin: developmental programming of body weight regulation. J Matern Fetal Neonatal Med 21:143–148
Plaschke K, Hoyer S (1993) Action of the diabetogenic drug streptozotocin on glycolytic and glycogenolytic metabolism in adult rat brain cortex and hippocampus. Int J Dev Neurosci 11:477–483
Plaschke K, Müller D, Hoyer S (2010a) Insulin-resistant brain state (IRBS) changes membrane composition of fatty acids in temporal and entorhinal brain cortices of rats: relevance to sporadic Alzheimer’s disease? J Neural Transm 117:1419–1422
Plaschke K, Kopitz J, Siegelin M, Schliebs R, Salkovic-Petrisic M, Riederer P, Hoyer S (2010b) Insulin-resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer-like changes in Tg2576 AbetaPP-overexpressing mice. J Alzheimers Dis 19:691–704. doi:10.3233/JAD-2010-1270
Plastino M, Fava A, Pirritano D, Cotronei P, Sacco N, Sperlì T et al (2010) Effects of insulinic therapy on cognitive impairment in patients with Alzheimer disease and diabetes mellitus type-2. J Neurol Sci 288:112–116
Powers MA, Bardsley J, Cypress M et al (2015) Diabetes self-management education and support in type 2 diabetes: a joint position Statement of the American Diabetes Association, the American Association of Diabetes Educators, and the Academy of Nutrition and Dietetics. Diabetes Care 38(7):1372–1382
Praticò D, Clark CM, Lee VM, Trojanowski JQ, Rokach J, FitzGerald GA (2000) Increased 8,12-iso-iPF2alpha-VI in Alzheimer’s disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol 48:809–812
Praticò D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ (2002) Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch Neurol 59:972–976
Punthakee Z, Miller ME, Launer LJ et al (2012) Poor cognitive function and risk of severe hypoglycemia in type 2 diabetes: post hoc epidemiologic analysis of the ACCORD trial. Diabetes Care 35(4):787–793
Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV (2015) Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front Aging Neurosci 7(July):1–12
Ramos-Rodriguez JJ, Jimenez-Palomares M, Murillo-Carretero MI, Infante-Garcia C, Berrocoso E, Hernandez-Pacho F, Lechuga-Sancho AM, Cozar-Castellano I, Garcia-Alloza M (2015) Central vascular disease and exacerbated pathology in a mixed model of type 2 diabetes and Alzheimer’s disease. Psychoneuroendocrinology 62:69–79
Randazzo J, Zhang P, Makita J, Blessing K, Kador PF (2011) Orally active multi-functional antioxidants delay cataract formation in streptozotocin (type 1) diabetic and gamma-irradiated rats. PLoS One 6(4):e18980
Rao AA, Sridhar GR, Das UN (2007) Elevated butyrylcholinesterase and acetylcholinesterase may predict the development of type 2 diabetes mellitus and Alzheimer’s disease. Med Hypotheses 69(6):1272–1276
Raygani AV, Zahrai M, Soltanzadeh A, Doosti M, Javadi E, Pourmotabbed T (2004) Analysis of association between butyrylcholinesterase K variant and apolipoprotein E genotypes in Alzheimer’s disease. Neurosci Lett 371(2–3):142–146
Reid GA, Darvesh S (2015) Butyrylcholinesterase-knockout reduces brain deposition of fibrillar β-amyloid in an Alzheimer mouse model. Neuroscience 298:424–435
Reijmer YD, van den Berg E, de Bresser J, Kessels RP, Kappelle LJ, Algra A, Biessels GJ, Utrecht Diabetic Encephalopathy Study Group (2011) Accelerated cognitive decline in patients with type 2 diabetes: MRI correlates and risk factors. Diabetes Metab Res Rev 27(2):195–202
Rizvi SM, Shakil S, Biswas D, Shakil S, Shaikh S, Bagga P, Kamal MA (2014) Invokana (Canagliflozin) as a dual inhibitor of AChE and SGLT2: advancement in Alzheimer’s disease-diabetes type 2 linkage via an enzoinformatics study. CNS Neurol Disord Drug Targets 13(3):447–451
Rizzo MR, Barbieri M, Boccardi V, Angellotti E, Marfella R, Paolisso G (2014) Dipeptidyl peptidase-4 inhibitors have protective effect on cognitive impairment in aged diabetic patients with mild cognitive impairment. J Gerontol Ser A Biol Sci Med Sci 69:1122–1131
Rodrigues L, Biasibetti R, Swarowsky A, Leite MC, Quincozes-Santos A, Quilfeldt JA, Achaval M, Gonçalves CA (2009) Hippocampal alterations in rats submitted to streptozotocin-induced dementia model are prevented by aminoguanidine. J Alzheimer’s Dis 17:193–202
Rönnemaa E, Zethelius B, Sundelöf J, Sundström J, Degerman-Gunnarsson M, Berne C, Lannfelt L, Kilander L (2008) Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 71:1065–1071
Roriz-Filho JS, Sá-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML, Moriguti JC, Roriz-Cruz M (2009) (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta 1792:432–443
Rosales-Corral S, Tan DX, Manchester L, Reiter RJ (2015) Diabetes and Alzheimer disease, two overlapping pathologies with the same background: oxidative stress. Oxid Med Cell Longev 2015:985845
Rudolph JD, de Graauw M, van de Water B, Geiger T, Sharan R (2016) Elucidation of signaling pathways from large-scale phosphoproteomic data using protein interaction networks. Cell Syst 3(6):585–593
Sagare AP, Bell RD, Zlokovic BV (2012) Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med 2(10):011452
Salkovic-Petrisic M, Osmanovic J, Grünblatt E, Riederer P, Hoyer S (2009) Modeling sporadic Alzheimer’s disease: the insulin resistant brain state generates multiple long-term morphobiological abnormalities including hyperphosphorylated tau protein and amyloid-beta. J Alzheimers Dis 18(4):729–750. doi:10.3233/JAD-2009-1184
Salkovic-Petrisic M, Osmanovic-Barilar J, Brückner MK, Hoyer S, Arendt T, Riederer P (2011) Cerebral amyloid angiopathy in streptozotocin rat model of sporadic Alzheimer’s disease: a long-term follow up study. J Neural Transm 118:765–772
Samy DM, Ismail CA, Nassra RA, Zeitoun TM, Nomair AM (2016) Downstream modulation of extrinsic apoptotic pathway in streptozotocin-induced Alzheimer’s dementia in rats: erythropoietin versus curcumin. Eur J Pharmacol 770:52–60
Sato N, Takeda S, Uchio-Yamada K, Ueda H, Fujisawa T, Rakugi H, Morishita R (2011) Role of insulin signaling in the interaction between Alzheimer disease and diabetes mellitus: a missing link to therapeutic potential. Curr Aging Sci 4:118–127
Schmeichel AM, Schmelzer JD, Low PA (2003) Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes 52(1):165–171
Seaquist E (2015) The impact of diabetes on cerebral structure and function. Psychosom Med 77:616–621
Serlin Y, Levy J, Shalev H (2011) Vascular pathology and blood–brain barrier disruption in cognitive and psychiatric complications of type 2 diabetes mellitus. Cardiovasc Psychiatry Neurol 2011:609202
Shaikh S, Ahmad SS, Ansari MA, Shakil S, Rizvi SM, Shakil S, Tabrez S, Akhtar S, Kamal MA (2014) Prediction of comparative inhibition efficiencies for a novel natural ligand, galangin against human brain acetylcholinesterase, butyrylcholinesterase and 5-lipoxygenase: a neuroinformatics study. CNS Neurol Disord Drug Targets 13(3):452–459
Sharma M, Gupta YK (2003) Effect of alpha lipoic acid on intracerebroventricular streptozotocin model of cognitive impairment in rats. Eur Neuropsychopharmacol 13:241–247
Sharma N, Deshmukh R, Bedi KL (2010) SP600125, a competitive inhibitor of JNK attenuates streptozotocin induced neurocognitive deficit and oxidative stress in rats. Pharmacol Biochem Behav 96:386–394
Shemesh E, Rudich A, Harman-Boehm I, Cukierman-Yaffe T (2012) Effect of intranasal insulin on cognitive function: a systematic review. J Clin Endocrinol Metab 2(97):366–376
Shingo AS, Kanabayashi T, Kito S, Murase T (2013) Intracerebroventricular administration of an insulin analogue recovers STZ-induced cognitive decline in rats. Behav Brain Res 241:105–111
Shoham S, Bejar C, Kovalev E, Schorer-Apelbaum D, Weinstock M (2007) Ladostigil prevents gliosis, oxidative-nitrative stress and memory deficits induced by intracerebroventricular injection of streptozotocin in rats. Neuropharmacology 52:836–843
Sinclair AJ, Paolisso G, Castro M, Bourdel-Marchasson I, Gadsby R, Rodriguez ML (2011) European diabetes working party for older people. Clinical guidelines for type 2 diabetes mellitus. Executive summary. Diabetes Metab 37:27–38
Sinclair A, Dunning T, Rodriguez-Mañas L (2015) Diabetes in older people: new insights and remaining challenges. Lancet Diabetes Endocrinol 3(4):275–285
Sobngwi E, Boudou P, Mauvais-Jarvis F, Leblanc H, Velho G, Vexiau P, Porcher R, Hadjadj S, Pratley R, Tataranni PA, Calvo F, Gautier JF (2003) Effect of a diabetic environ- ment in utero on predisposition to type 2 diabetes. Lancet 361:1861–1865
Sohrab SS, Karim S, Kamal MA, Abuzenadah AM, Chaudhary AG, Al-Qahtani MH, Mirza Z (2014) Bacteriophage-a common divergent therapeutic approach for Alzheimer’s disease and type II diabetes mellitus. CNS Neurol Disord Drug Targets 13(3):491–500
Sonnen JA, Larson EB, Brickell K, Crane PK, Woltjer R, Montine TJ, Craft S (2009) Different patterns of cerebral injury in dementia with or without diabetes. Arch Neurol 66:315–322
Sridhar GR, Thota H, Allam AR, Suresh Babu C, Siva Prasad A, Divakar Ch (2006) Alzheimer’s disease and type 2 diabetes mellitus: the cholinesterase connection? Lipids Health Dis 5:28. doi:10.1186/1476-511X-5-28
Sridhar GR, Rao AA, Srinivas K, Nirmala G, Lakshmi G, Suryanarayna D, Rao PV, Kaladhar DG, Kumar SV, Devi TU, Nitesh T, Hanuman T (2010) Butyrylcholinesterase in metabolic syndrome. Med Hypotheses 75:648–651
Su F, Shu H, Ye Q, Wang Z, Xie C, Yuan B, Zhang Z, Bai F (2017) Brain insulin resistance deteriorates cognition by altering the topological features of brain networks. Neuroimage Clin 13:280–287
Sun P, Knezovic A, Parlak M, Cuber J, Karabeg MM, Deckert J, Riederer P, Hua Q, Salkovic-Petrisic M, Schmitt AG (2015) Long-term effects of intracerebroventricular streptozotocin treatment on adult neurogenesis in the rat hippocampus. Curr Alzheimer Res 12(8):772–784
Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R (2010) Diabetes accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA 107:7036–7041
Takeda S, Sato N, Rakugi H, Morishita R (2011) Molecular mechanisms linking diabetes mellitus and Alzheimer disease: beta-amyloid peptide, insulin signaling, and neuronal function. Mol Biosyst 7:1822–1827
Talbot K (2014) Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs. Neurodegener Dis Manag 4:31–40
Talbot K, Wang HY (2014) The nature, significance, and glucagon-like peptide-1 analog treatment of brain insulin resistance in Alzheimer’s disease. Alzheimers Dement 10(1 Suppl):12–25
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE (2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Investig 122:1316–1338
Tamaki C, Ohtsuki S, Terasaki T (2007) Insulin facilitates the hepatic clearance of plasma amyloid beta-peptide (1–40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol Pharmacol 2(4):850–855
Taurino F, Stanca E, Siculella L, Trentadue R, Papa S, Zanotti F, Gnoni A (2012) Mitochondrial proteome analysis reveals depression of the Ndufs3 subunit and activity of complex I in diabetic rat brain. J Proteom 75(8):2331–2341
Teipel SJ, Kurth J, Krause B, Grothe MJ (2015) The relative importance of imaging markers for the prediction of Alzheimer’s disease dementia in mild cognitive impairment—beyond classical regression. Neuroimage Clin 8:583–593
Thacker EL, Psaty BM, McKnight B, Heckbert SR, Longstreth WT Jr, Mukamal KJ et al (2011) Fasting and post-glucose load measures of insulin resistance and risk of ischemic stroke in older adults. Stroke 42:3347–3351
Thambisetty M, Metter EJ, Yang A, Dolan H, Marano C, Zonderman AB, Troncoso JC, Zhou Y, Wong DF, Ferrucci L, Egan J, Resnick SM, O’Brien RJ (2013) Glucoseintolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol 70:1167–1172
Thomas T, Thomas G, McLendon C, Sutton T, Mullan M (1996) β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380(6570):168–171
Tohgi H, Abe T, Kimura M, Saheki M, Takahashi S (1996) Cerebrospinal fluid acetylcholine and choline in vascular dementia of Binswanger and multiple small infarct types as compared with Alzheimer-type dementia. J Neural Transm (Vienna) 103(10):1211–1220
Tota S, Kamat PK, Saxena G, Hanif K, Najmi AK, Nath C (2012) Central angiotensin converting enzyme facilitates memory impairment in intracerebroventricular streptozotocin treated rats. Behav Brain Res 226:317–330
Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA (2015) Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem 133:739–749
Tschritter O, Preissl H, Hennige AM, Stumvoll M, Porubska K, Frost R, Marx H, Klösel B, Lutzenberger W, Birbaumer N, Häring HU, Fritsche A (2006) The cerebrocortical response to hyperinsulinemia is reduced in overweight humans: a magnetoencephalographic study. Proc Natl Acad Sci 103:12103–12108
Tschritter O, Preissl H, Yokoyama Y, Machicao F, Häring HU, Fritsche A (2007) Variation in the FTO gene locus is associated with cerebrocortical insulin resistance in humans. Diabetologia 50:2602–2603
Tseng C-K, Soroka O, Maney M, Aron DC, Pogach LM (2014) Assessing potential glycemic overtreatment in persons at hypoglycemic risk. JAMA Intern Med 174:259–268
Umegaki H (2012) Neurodegeneration in diabetes mellitus. Adv Exp Med Biol 724:258–265
Unger JW, Livingston JN, Moss AM (1991) Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog Neurobiol 36(5):343–362
Vagelatos NT, Eslick GD (2013) Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev 35:152–160
Valente T, Gella A, Fernandez-Busquets X, Unzeta M, Durany N (2010) Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol Dis 37:67–76
van Bussel FCG, Backes WH, Hofman PAM, van Oostenbrugge RJ, van Boxtel MPJ, Verhey FRJ, Steinbusch HWM, Schram MT, Stehouwer CDA, Wildberger JE, Jansen JFA (2017) Cerebral pathology and cognition in diabetes: the merits of multiparametric neuroimaging. Front Neurosci 5(11):188
van den Berg E, Reijmer YD, de Bresser J, Kessels RP, Kappelle LJ, Biessels GJ, Utrecht Diabetic Encephalopathy Study Group (2010) A 4 year follow-up study of cognitive functioning in patients with type 2 diabetes mellitus. Diabetologia 53(1):58–65
van Duinkerken E, Schoonheim MM, Sanz-Arigita EJ, IJzerman RG, Moll AC, Snoek FJ, Ryan CM, Klein M, Diamant M, Barkhof F (2012a) Resting-state brain networks in type 1 diabetic patients with and without microangiopathy and their relation to cognitive functions and disease variables. Diabetes 61(7):1814–1821
van Duinkerken E, Schoonheim MM, Ijzerman RG, Klein M, Ryan CM, Moll AC, Snoek FJ, Barkhof F, Diamant M, Pouwels PJ (2012b) Diffusion tensor imaging in type 1 diabetes: decreased white matter integrity relates to cognitive functions. Diabetologia 55(4):1218–1220
van Elderen SGC, de Roos A, de Craen AJM, Westendorp RGJ, Blauw GJ, Jukema JW et al (2010) Progression of brain atrophy and cognitive decline in diabetes mellitus: a 3-year follow-up. Neurology 75(11):997–1002
van Harten B, de Leeuw FE, Weinstein HC et al (2006) Brain imaging in patients with diabetes: a systematic review. Diabetes Care 29(11):2539–2548
van Harten B, Oosterman JM, van Loon B-JP, Scheltens P, Weinstein HC (2007) Brain lesions on MRI in elderly patients with type 2 diabetes mellitus. Eur Neurol 57(2):70–74
Vecsei L, Szalárdy L, Fülöp F, Toldi J (2013) Kynurenine in the CNS: recent advances and new questions. Nat Rev Drug Disc 12:64–82
Vedhara K, Miles JN, Wetherell MA et al (2010) Coping style and depression influence the healing of diabetic foot ulcers: observational and mechanistic evidence. Diabetologia 53(8):1590–1598
Verdile G, Fuller SJ, Martins RN (2015) The role of type 2 diabetes in neurodegeneration. Neurobiol Dis 84:22–38
Vousden KH (2010) Alternative fuel-another role for p53 in the regulation of metabolism. Proc Natl Acad Sci 107:7117–7118
Want Z, Jiang Y, Wang X, Du Y, Xiao D, Deng Y, Wang J (2015) Butyrylcholinesterase K variant and Alzheimer’s disease risk: a meta-analysis. Med Sci Monit 21:1408–1413
White MF (2002) IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 283(3):E413–E422
Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP Jr, Selby JV (2009) Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 301(15):1565–1572
Willette AA, Bendlin BB, Starks EJ, Birdsill AC, Johnson SC, Christian BT, Okonkwo OC, La Rue A, Hermann BP, Koscik RL, Jonaitis EM, Sager MA, Asthana S (2015a) Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA Neurol 72:1013–1020
Willette AA, Modanlo N, Kapogiannis D (2015b) Alzheimer’s disease neuroimaging initiative. insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes 64:1933–1940
Winner B, Winkler J (2015) Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb Perspect Biol 7(4):a021287
Yang L, Kress BT, Weber HJ, Thiyagarajan M, Wang B, Deane R et al (2013) Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med 11:107
Yarchoan M, Toledo JB, Lee EB, Arvanitakis Z, Kazi H, Han LY, Louneva N, Lee VM, Kim SF, Trojanowski JQ, Arnold SE (2014) Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol 128:679–689
Yeo HG, Lee Y, Jeon CY, Jeong KJ, Jin YB, Kang P, Kim SU, Kim JS, Huh JW, Kim YH, Sim BW, Song BS, Park YH, Hong Y, Lee SR, Chang KT (2015) Characterization of cerebral damage in a monkey model of Alzheimer’s disease induced by intracerebroventricular injection of streptozotocin. J Alzheimers Dis 46:989–1005. doi:10.3233/JAD-143222
Zhao WQ, Alkon DL (2001) Role of insulin and insulin receptor inmlearning and memory. Mol Cell Endocrinol 177:125–134
Zhou Q, Xiao M, Nicoll RA (2001) Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci U S A 98(3):1261–1266
Zlokovic BV (2008) The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron 57(2):178–201
Zoungas S, Patel A, Chalmers J, de Galan BE, Li Q, Billot L, ADVANCE Collaborative Group et al (2010) Severe hypoglycemia and risks of vascular events and death. N Engl J Med 363:1410–1418
Acknowledgements
Hakan Yaman will be supported by the Akdeniz University Research Management Unit.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Riederer, P., Korczyn, A.D., Ali, S.S. et al. The diabetic brain and cognition. J Neural Transm 124, 1431–1454 (2017). https://doi.org/10.1007/s00702-017-1763-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-017-1763-2