
Abstract
Ketone bodies formed during ketogenic diet or non-treated diabetes mellitus may exert neuroprotective and antiepileptic effects. Here, we assessed the influence of ketone body, β-hydroxybutyrate (BHB) on the brain synthesis of kynurenic acid (KYNA), an endogenous antagonist of glutamatergic and α7-nicotinic receptors. In brain cortical slices and in primary glial cultures, BHB enhanced KYNA production. KT 5270, an inhibitor of protein kinase A, has prevented this action. At hypoglycemia, under pH 7.0 and 7.4, profound (15 mM BHB), but not mild (3 mM) ketosis increased synthesis of KYNA. In paradigm resembling diabetic ketoacidosis in vitro (30 mM glucose, pH 7.0), neither mild nor profound ketosis influenced the production of KYNA. At pH 7.4 and in 30 mM glucose though, both mild and severe ketonemia evoked an increase of KYNA production. The activity of KYNA biosynthetic enzymes, KAT I and KAT II, in cortical homogenate was not altered by BHB (0.05–10.0 mM). However, in cultured glial cells exposed to BHB (10 mM), the activity of KATs increased. This effect was reversed by the co-incubation of cells with KT 5270. Presented data reveal a novel mechanism of action of BHB. Increased synthesis of KYNA in the presence of BHB is most probably mediated by protein kinase A-dependent stimulation of KATs expression/activity leading to an increase of KYNA formation. Ensuing attenuation of the excessive excitatory glutamate-mediated neurotransmission may, at least in part, explain the neuroprotective actions of BHB.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ketone bodies, β-hydroxybutyrate (BHB), acetoacetate, and acetone, are produced during incomplete oxidization of fatty acids which takes place during inadequate supply of glucose. In the brain, BHB and acetoacetate are the primary source of energy at early development, in the course of fasting, non-treated diabetes or high-fat diet (Fukao et al. 2004; Hartman and Vining 2007). Synthesized in periphery ketones easily penetrate via blood–brain–barrier. Locally, astrocytes may generate ample amounts of ketone bodies (Cullingford 2004).
Low-glucose, high-fat food regimen increasing serum level of ketones (ketogenic diet) is recognized for a long time as a valuable alternative treatment of epilepsy (Wheless 1995; Gasior et al. 2006). Such diet is surprisingly effective among a substantial number of patients with pharmacoresistant and was suggested to exert long-lasting disease-modifying actions (Lefevre and Aronson 2000; Gasior et al. 2006; Hartman et al. 2007). Therapeutic effects of ketogenic diet were attributed to various mechanisms including alterations in brain energy or altered handling of glutamate (Bough et al. 2006; Yudkoff et al. 2008). Most of the theories, however, indicate the crucial role of the profound rise in ketones level in the antiepileptic action of ketogenic diet.
Increasing experimental evidence revealed that ketone bodies display also neuroprotective effects in various models of neuronal injury, including Alzheimer’s and Parkinson’s disease (Kashiwaya et al. 2000; Cheng et al. 2010). However, the precise mechanisms underlying their efficacy are far from understanding. Ketones were suggested to enhance GABA formation, reduce the oxidative stress, or exert anti-inflammatory effects (Gasior et al. 2006). There are also limited data indicating that ketones may attenuate the glutamate-mediated neuronal cell loss (Noh et al. 2006).
Kynurenic acid (KYNA) is an endogenous neuroactive product of tryptophan metabolism formed within the brain and in the periphery, along kynurenine pathway, in the process catalyzed by kynurenine aminotransferases (KAT I–III) (Guidetti et al. 1997; 2007). Brain KYNA is synthesized preferentially, but not exclusively, within astrocytes and its extracellular levels are within nanomolar range (Moroni et al. 1988; Turski et al. 1989; Németh et al. 2006). Neuroprotective and anticonvulsant actions of KYNA, demonstrated in different experimental settings, are linked mainly with the inhibition of strychnine-insensitive site of N-methyl-d-aspartate (NMDA) receptors (Urbanska et al. 1991; Stone et al. 2001; Schwarcz and Pellicciari 2002; Németh et al. 2006). Due to the fact that KYNA blocks NMDA receptors at higher concentrations than these found physiologically in the brain (Stone et al. 2001; Schwarcz and Pellicciari 2002), its role in the modulation of glutamate-mediated neurotransmission has been received skeptically for some years. However, recent discoveries have reinforced the hypothesis suggesting an important role of KYNA in brain physiology and pathology. It was demonstrated that high nanomolar levels of KYNA are sufficient to block noncompetitively α7 nicotinic receptors or to increase the expression of α4β2 nicotinic receptors, whereas low nanomolar amounts of compound reduce the presynaptic release of glutamate (Carpenedo et al. 2001; Hilmas et al. 2001; Luccini et al. 2007).
The regulation of KYNA formation was evaluated under in vitro and in vivo conditions. A number of endogenous and exogenous factors influencing the central production of KYNA have been recognized. For example, mitochondrial toxins compromising the status of mitochondrial respiration, endogenous sulfur-containing amino acids and inhibitors of protein kinase A activity reduce formation of KYNA, whereas rise of intracellular cAMP level stimulates KYNA production (Urbanska et al. 1997; Luchowski et al. 2002; Kocki et al. 2003; Luchowska et al. 2005, 2009; Kloc et al. 2008). It was also shown that hyperglycemia enhances the inhibitory effect of mitochondrial toxins and d,l-homocysteine on the central production of KYNA (Chmiel-Perzyńska et al. 2007).
Here, we aimed to assess the influence of ketone body, β-hydroxybutyrate (BHB) on KYNA synthesis in vitro, using paradigms partially resembling ketogenic diet and acute diabetic ketoacidosis.
Materials and Methods
Animals
Brain tissue was obtained from (A) adult male Wistar rats, weighing 220–250 g (studies ex vivo, in cortical slices) or from (B) 1–2 days old Wistar rats of both sexes (studies in glial cultures). Animals were housed under standard laboratory conditions, at 20°C environmental temperature, with food and water available ad libitum. The experimental procedures used herein were approved by the First Local Ethics Committee in Lublin and complied with the European Communities Council Directive on the use of animals in experimental studies.
Substances
l-Kynurenine sulfate salt, kynurenic acid, d-glucose and d,l(±) 3-hydroxybutyrate sodium salt (BHB) were obtained from Sigma-Aldrich (St Louis, USA). (9R,10S,12S)-2,3,9,10,11,12-Hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid hexyl ester (KT 5720) was purchased from Tocris (Bristol, UK). All the high pressure liquid chromatography (HPLC) reagents were supplied by J.T.Baker Laboratory Chemicals (Holland). Other reagents were obtained from POCH (Gliwice, Poland).
KYNA Synthesis Ex Vivo in Cortical Slices
Synthesis of KYNA in rat cortical slices was carried out as described before (Urbanska et al. 1997). Slices (1 × 1 mm) were freshly prepared from frontal cortices of naïve rats using McIlwain tissue chopper, placed at random into culture wells (8 slices per well), and incubated in Krebs-Ringer buffer (KRB) in a final volume of 1 ml. Standard KRB contained: 118.5 mM NaCl, 4.75 mM KCl, 1.77 mM CaCl2, 1.18 mM MgSO4, 12.9 mM NaH2PO4, 3 mM Na2HPO4, 5 mM glucose and was oxygenated for 30 min with gas mixture (95% O2–5% CO2). The following versions of KRB were used: (A) partially resembling ketogenic diet (0.5 mM glucose; pH 7.0 or 7.4, (B) partially similar to diabetic ketoacidosis (30 mM glucose; pH 7.0) or acute ketosis (30 mM glucose; pH 7.4). The pH 7.0 of KRB was obtained by titration with minimal quantity of 2 mM HCl. After the preincubation period (15 min), studied substances were added to each well. The incubation (37°C, 2 h) was started by addition of l-kynurenine (final concentration 10 μM). At least six wells were used for each concentration of studied substances. Blanks contained all components of the incubation buffer except for the brain tissue. Upon ending the incubation period, media were rapidly separated from the tissue, acidified with 0.1 ml of 1 N HCl and 14 μl of 50% trichloroacetic acid and centrifuged (8730 g, 5 min, 4°C). Supernatants were applied to the cation-exchange columns (Dowex 50W, hydrogen form), prewashed with 1 ml of water and 1 ml of 0.1 N HCl. Columns were subsequently washed with 1 ml of 0.1 N HCl and 1 ml of water. KYNA was eluted with 2.5 ml of water. Each experiment was repeated at least twice.
KYNA Synthesis in Mixed Glial Cultures
Mixed glial cell cultures were prepared from the brains of postnatal Wistar rats (days 1–2), as described previously (Luchowska et al. 2009). Briefly, forebrains were stripped of meninges, chopped into small chunks, trypsinized, and seeded (one brain/75 cm2) in culture flasks (Falcon, Switzerland) containing 10 ml of Basal Medium Eagle with Earle’s salts (BME, Biochrom AG, Berlin), supplemented with 2 mM l-glutamine, penicillin–streptomycin (500 IU/ml–500 UG/ml; Gibco-BRL) and containing 10% heat-inactivated fetal bovine serum (Gibco, InVitrogen). The medium was replaced twice a week. Cultures were maintained at 37°C in a 5% CO2/95% air atmosphere. After 14–15 days, cells were replated on poly-l-lysine-coated 24-microwell plates (Nunc), and medium was replaced every third day. After approx. 1 week, a confluent astrocytic monolayer developed with scattered oligodendrocytes and microglia on top. Approx. 75–80% of the cells were glial fibrillary acidic protein-positive (as revealed by immunostaining). The production of KYNA was assessed in tissue cultures after 20–22 days in vitro.
The influence of tested compounds on the synthesis of KYNA was evaluated using different exposure times. Experimental procedures involved 2, 4 or 24 h incubation. During 2 h incubation, the medium was replaced with freshly prepared KRB (composed as above). Substances were added to cultures 15 min prior to the addition of l-kynurenine (final concentration 10 μM). The incubation with KYNA substrate lasted always for 2 h. During longer exposure times, the medium was initially changed into the fresh one, and the compounds were added to cultures (start point determining the beginning of incubation). Prior to the addition of l-kynurenine, i.e., 2 h before the end of incubation, the medium was replaced again with freshly prepared KRB. Fresh solutions of tested compounds were added again to the incubation media. This procedure ensured that cells were exposed to studied substances throughout the entire incubation time. Blanks contained all of the incubation buffer components, except for the cultured cells. Controls received an appropriate amount of saline. Further analyses were performed as indicated above.
Kynurenine Aminotransferases (KAT I and KAT II) Activity
Semi-Purified Cortical Homogenate
The activities of KAT I and KAT II were assayed in naïve set of animals, as described before (Kocki et al. 2003). Briefly, freshly obtained cortical brain tissue (whole cortices) was homogenized (1:9; wt:vol) in 5 mM Tris–acetate buffer, pH 8.0, containing 50 μM pyridoxal-5′-phosphate and 10 mM 2-mercaptoethanol. The resulting homogenate was centrifuged (8730 g; 10 min), the supernatant was placed in cellulose membrane dialysis tubing (Sigma) and dialyzed overnight at 8°C, against 4 l of the Tris–acetate buffer, composed as above. The obtained semi-purified enzyme preparation was incubated in the reaction mixture containing 2 μM l-kynurenine, 1 mM pyruvate, 70 μM pyridoxal-5′-phosphate, 150 mM Tris–acetate buffer, and solutions of tested drugs. The reaction was carried out at pH of 7.0 (KAT II) or 9.5 (KAT I). Glutamine (final concentration 2 mM), the inhibitor of KAT I, was added to samples assaying KAT II activity. Six replicates were used for each concentration and each experiment was repeated twice. BHB stock solution was added (10 μl) to assayed samples to yield final concentrations of 0.05–10.0 mM. The appropriate amount of solvent was added to control samples. Blanks contained the enzyme preparation that was heat-deactivated at 100°C for 10 min. The incubation (37°C, 2 h) was terminated by a rapid transfer of samples to an ice-cold water bath. Next, 14 μl of 50% trichloroacetic acid (wt:vol) and 100 μl of 1 N HCl were added to each sample. Further procedures were performed as indicated above.
Mixed Glial Cultures
The activity of KATs was assessed in mixed glial cultures, prepared as above. After changing medium into the fresh one, the cells were incubated for 4 or 24 h in the presence of studied substances. During last 2 h of incubation, the cells were incubated in standard KRB (5.0 mM glucose and pH 7.4). Control cells were incubated with the addition of saline. Upon ending the incubation, media were removed and cells were detached from the surface using 100 μl of cold, distilled H2O. The liquid containing cells from 12 wells was harvested and combined with the same volume of 5 mM Tris–acetate buffer, pH 8.0, containing 50 μM pyridoxal-5′-phosphate and 10 mM 2-mercaptoethanol. The cells were sonicated and further procedures were carried on as described above, except for the incubation time which lasted for 4 h.
Quantification of KYNA
Eluted KYNA was subjected to the HPLC and quantified fluorimetrically (Varian HPLC system; ESA catecholamine HR-80, 3 μm, C18 reverse-phase column), as previously described (Urbanska et al. 1997).
Statistical Analyses
Data are presented as a percentage of control values ± SD. The statistical analyses were performed using one-way analysis of variance (ANOVA), with the adjustment of P value by the Bonferroni method.
Results
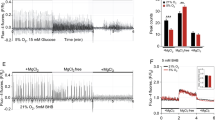
In brain cortical slices, BHB (10–20 mM) enhanced the synthesis of KYNA (Fig. 1a). BHB acted similarly in mixed astroglial cultures, but with higher potency. The effective concentrations were in the range of 5–10 mM (Fig. 1b). KT 5270 (0.25 μM), an inhibitor of protein kinase A, has prevented an increase of KYNA production induced by 10 mM BHB during 2 h incubation in tissue cultures (95.6 ± 4.2 vs 124.4 ± 3.8% of control; P < 0.05) (Fig. 1c).

The influence of β-hydroxybutyrate (BHB) (a, b, c) and KT5270 (c) on the kynurenic acid (KYNA) production in: a brain cortical slices, and b, c primary glial cultures. The data are presented as a percentage of control values ± SD. * P < 0.05 vs control (ANOVA). Control—standard KRB (5.0 mM glucose, pH 7.4)
Further, the influence of BHB on KYNA production was studied under conditions partially resembling ketogenic diet in vitro (cortical slices), i.e., in 0.5 mM glucose, at pH 7.0 or 7.4. KYNA synthesis in 0.5 mM glucose was reduced in both pH 7.0 and pH 7.4 (P < 0.05 vs control) (Fig. 2a–d). BHB at concentration not changing KYNA production per se (3.0 mM), has not affected the inhibitory effect of 0.5 mM glucose at both settings (Fig. 2a, b). In contrast, in the presence of 15 mM BHB, KYNA formation was not reduced by 0.5 mM glucose, at pH 7.0 (P < 0.05 vs 0.5 mM glucose). At pH 7.4 and in 0.5 mM glucose, KYNA synthesis was even enhanced (P < 0.05 vs control and vs 0.5 mM glucose) (Fig. 2c, d).

The influence of 3 and 15 mM β-hydroxybutyrate (BHB) on the kynurenic acid (KYNA) production in brain cortical slices under conditions partially resembling ketogenic diet, i.e., during hypoglycemia and pH 7.0 (a, c) or pH 7.4 (b, d). The data are presented as a percentage of control values ± SD. * P < 0.05 vs CTR (ANOVA), a P < 0.05 vs respective glucose concentration alone. CTR (control)—standard KRB (5.0 mM glucose, pH 7.4)
Next, the effects of BHB on KYNA synthesis were evaluated under conditions resembling diabetic ketoacidosis (30 mM glucose; pH 7.0) and acute diabetic ketosis (30 mM glucose; pH 7.4) in vitro (Fig. 3a–d). The production of KYNA in 30 mM glucose did not differ from control, either at pH 7.0 or pH 7.4 (Fig. 3a–d). BHB (3 mM) has not affected KYNA production in 30 mM glucose, at pH 7.0 (Fig. 3a). However, 3 mM BHB significantly enhanced synthesis of KYNA in 30 mM glucose, at pH 7.4 (P < 0.05 vs control) (Fig. 3b). In the presence of 15 mM BHB, there was no inhibition of KYNA production in 5 mM glucose, at pH 7.0 (P < 0.05 vs 5.0 mM glucose + pH 7.0) (Fig. 3c). 15 mM BHB significantly increased KYNA synthesis in 30 mM glucose, at pH 7.4 (P < 0.05 vs control and vs 30 mM glucose alone) (Fig. 3d).

The influence of 3 and 15 mM β-hydroxybutyrate (BHB) on the kynurenic acid (KYNA) production in brain cortical slices under conditions resembling acute diabetic ketoacidosis, i.e., during hyperglycemia (30.0 mM glucose) and pH 7.0 (a, c) and acute hyperglycemia with ketosis (b, d). The data are presented as a percentage of control values ± SD. * P < 0.05 vs CTR (ANOVA), a P < 0.05 vs respective glucose concentration alone. CTR (control)—standard KRB (5.0 mM glucose, pH 7.4)
In glial cultures, similarly as in cortical slices, at pH 7.0 and in 5.0 mM glucose KYNA production was inhibited (P < 0.05 vs control). In 0.5 mM glucose, KYNA synthesis was diminished at pH 7.0 and pH 7.4 (Fig. 4a, b). There was no change in KYNA synthesis at pH 7.0 or 7.4 and in 30 mM glucose vs control (Fig. 4c, d). In the presence of BHB (10 mM), in 0.5 mM glucose KYNA production was not lowered, neither at pH 7.0 nor at 7.4 pH (P < 0.05 vs 0.5 mM glucose at pH 7.0 or 7.4, respectively) (Fig. 4a, b). In 30 mM glucose, in the presence of 10 mM BHB there was no change in KYNA synthesis at pH 7.0 vs control, however, at pH 7.4 the production of KYNA slightly increased (P < 0.05 vs control) (Fig. 4c, d).

The influence of 10 mM β-hydroxybutyrate (BHB) on the kynurenic acid production in primary glial cultures under conditions partially resembling ketogenic diet (a, b), acute diabetic ketoacidosis (c), and hyperglycemia with ketosis (d). The data are presented as a percentage of control values ± SD. * P < 0.05 vs CTR (ANOVA), a P < 0.05 vs respective glucose concentration alone. CTR (control)—standard KRB (5.0 mM glucose, pH 7.4)
The enzymatic studies revealed that the activity of partially purified preparation of KAT I and KAT II is not altered by BHB (0.05–10.0 mM) (Table 1), what indicates that BHB does not modify the function of already formed KATs proteins. However, such analyses do not reveal any changes in the expression pattern of the enzymes. Therefore, we have estimated the effect of BHB (10 mM) on the activity of KAT I and II in the mixed glial cells, following their exposure to the compound for 4 or 24 h. In such paradigm, the activity of KAT II and KAT I was increased (Table 2). This effect was reversed by the co-incubation of cells with KT 5270 (0.25 μM) (Table 2).
Discussion
In our study, BHB stimulated brain synthesis of KYNA both in cortical slices and in primary glial cells, at 5–20 mM concentrations (racemic mixture of BHB). Considering the fact that only the d(−) enantiomer occurs physiologically, the effective concentrations were in fact two times lower (approx. 2.5–10 mM of D-BHB). In humans, plasma concentrations of circulating ketone bodies range from 0.1 mM in the postprandial state up to 6 mM during prolonged fasting and 25 mM in uncontrolled diabetes (Robinson and Williamson 1980; Fukao et al. 2004). In children treated with ketogenic diet, circulating BHB reaches 4–10 mM (Huttenlocher 1976; Gilbert et al. 2000), and its serum level correlates well with the seizure control, better even than do urine ketones (Gilbert et al. 2000; van Delft et al. 2010). In adults, depending on the type of given ketogenic diet, serum BHB is lower and in a range of 1–3 mM, whereas brain BHB reaches approx. 1 mM (Johnstone et al. 2008; Pan et al. 2000). In rats, physiological plasma level of BHB attains 0.1–0.75 mM (Thavendiranathan et al. 2000; Nehlig 2004), whereas during ketogenic diet reaches 0.8–7.5 mM (Thavendiranathan et al. 2000; Likhodii et al. 2002).
Thus, the effective here concentrations of BHB are comparable with serum BHB levels achieved during ketogenic diet and diabetic ketosis (Gilbert et al. 2000; Fukao et al. 2004), albeit higher than these described in human brain (Pan et al. 2000). Providing the fact that glial cells intensely produce BHB (Cullingford 2004), it cannot be excluded that in the synaptic vicinity, local levels of BHB are higher than detected in the whole brain. Moreover, levels of BHB increasing KYNA synthesis correspond well with other data showing that 4–10 mM BHB exerts neuroprotective activity (4–10 mM) in vitro (Izumi et al. 1998; Kashiwaya et al. 2000; Imamura et al. 2006).
Our data confirm the earlier results demonstrating that acidosis or hypoglycemia inhibit synthesis of KYNA in rat cortical slices (Turski et al. 1989). We show that the effect of acidosis is enhanced by the hypoglycemia, and conversely, that hyperglycemia may restore the acidosis-impaired production of KYNA to control levels. However, as found previously (Chmiel-Perzyńska et al. 2007), hyperglycemia does not enhance KYNA production per se.
The effect of BHB was studied in vitro using buffers partially resembling major serum changes in ketogenic diet and acute diabetic ketoacidosis. The first condition is characterized by high level of circulating ketones accompanied by mild to severe hypoglycemia (Cullingford 2004; Greene et al. 2001). Blood pH usually remains within physiological range (Huttenlocher 1976; Gilbert et al. 2000), but some authors suggest that acidosis can also occur and contributes in part to therapeutic effects of ketones (al-Mudallal et al. 1995; Schwartzkroin 1999). Thus, in our study, ketogenic diet-like KRB contained low-glucose (0.5 mM), and was of pH 7.0 or 7.4. Ketoacidosis is a complex of acute metabolic disturbances resulting from insulin deficiency and increased concentrations of antagonistic hormones. It typically includes triad of hyperglycemia, acidosis and increased total body ketones (Kitabchi et al. 2009). In here, BHB used in KRB containing 30 mM glucose and of pH 7.0 was considered to create milieu resembling diabetic ketoacidosis.
We found that profound (15 mM BHB), but not mild (3 mM) ketosis enhanced the synthesis of KYNA in 5.0 and 0.5 mM glucose, at physiological pH. At pH 7.0, equivalent to extracellular acidosis, in 0.5 and 5.0 mM glucose, KYNA production was reduced to approx. 50 and 80% of control. This reduction did not occur in the presence of 15 but not 3 mM BHB. In 5 mM glucose, 15 mM BHB actually enhanced KYNA synthesis to approx. 130% of control. These data suggest that during fasting or ketogenic diet, high level of BHB may not only protect brain from the reduction of KYNA elicited by hypoglycemia and/or acidosis but actually enhance its synthesis above control values.
Further, we have investigated the conditions resembling acute uncontrolled ketoacidosis (30 mM glucose; pH 7.0). As shown before (Chmiel-Perzyńska et al. 2007), nonketotic hyperglycemia (30 mM glucose) did not change the brain synthesis of KYNA at pH 7.4. Interestingly, at pH 7.0 there was no reduction of KYNA synthesis in 30 mM glucose in contrast to lowered synthesis of KYNA when glucose was of 5 mM concentration. Neither 3 nor 15 mM BHB influenced the production of KYNA at pH 7.0 and in 30 mM glucose, i.e., KYNA level remained within control values. At pH 7.4 and in 30 mM glucose though, both mild and severe ketonemia evoked an increase of KYNA production vs control. These data suggest that brain KYNA levels stay unaltered in diabetic ketoacidosis. However, when extracellular pH returns to physiological values, the synthesis of KYNA may actually rise above control. Thus, correction of pH might be an important factor elevating brain KYNA levels during acute diabetic ketoacidosis.
Ample amounts of BHB are produced during ketogenic diet, recognized nowadays as valuable alternative therapy in pharmacoresistant epilepsy (Lefevre and Aronson 2000; Gasior et al. 2007; Hartman and Vining 2007; Hartman et al. 2007). The antiepileptic role of BHB was questioned in the view of data showing that in some animal models only acetone and acetoacetate, but not BHB, are effective (Rho et al. 2002). However, recent in vitro results demonstrate that pre-treatment with BHB diminishes the occurrence of hypoglycemia-induced seizures in the immature intact hippocampus, and reduces post-hypoxic hyperexcitability in organotypic hippocampal slice culture (Samoilova et al. 2010; Abdelmalik et al. 2007). In the view of our data, it is not clear whether reported ~150% increases in KYNA levels after the exposure to BHB are relevant as regards antiepileptic potential of ketogenic diet. We have previously reported that significant increases in KYNA levels (~150–200%) are produced by a number of antiepileptic drugs including: phenobarbital, felbamate, phenytoin, and lamotrigine but not by vigabatrin, gabapentin, and tiagabine (Kocki et al. 2006). If increases in KYNA had been indeed involved in the anticonvulsant effects produced by the KD, then the antiepileptic drugs that produce similar or greater increases in KYNA should have been effective in refractory epilepsy. Conversely, if increases in KYNA had been indeed involved in the anticonvulsant effect produced by the KD, then the antiepileptic drugs that reduce levels of KYNA should have been expected to worsen the refractory epilepsy, which was not observed clinically. Nevertheless, the described here increases of KYNA after administration of BHB, despite not having most probably anticonvulsant effects, could exert neuroprotective action, considered nowadays an important factor in preventing progressive worsening of epilepsy (Klitgaard and Pitkänen 2003; Noh et al. 2008).
The increase of KYNA reported here is comparable with the enhancement of KYNA synthesis (150–250%) observed also in other in vitro studies, e.g., following application of memantine, low concentrations of d,l-homocysteine, some β-adrenergic agonists or cAMP analogues, as well as after administration of β-adrenergic agonists in vivo (Luchowska et al. 2005, 2009; Kloc et al. 2008). It is lower though, than after in vivo application of KYNA precursor, l-kynurenine together with probenecid, reported to increase brain KYNA several-fold (Miranda et al. 1997; Shepard et al. 2003). Yet, having in mind that already nanomolar levels of KYNA block α7 nicotinic receptors and reduce the presynaptic release of glutamate (Carpenedo et al. 2001; Hilmas et al. 2001; Luccini et al. 2007), found enhancement of KYNA synthesis evoked by BHB seems to be clinically relevant, especially as regards potential neuroprotective effects of ketone bodies.
The mechanisms of central action of ketones such as acetoacetate or BHB, apart from their influence on the energy metabolism, are not fully clear. Experimental evidence suggests that ketones may augment the GABA-mediated inhibitory neurotransmission, reduce the formation of free radical or exert direct anti-inflammatory and neuroprotective action (Gasior et al. 2006). BHB and acetoacetate do not seem to affect directly the excitatory synaptic transmission (Thio et al. 2000; Leite et al. 2004). In astrocytic cultures, 2–20 mM BHB caused transient activation of astrocytes and increased S100B protein concentration in extracellular space (Leite et al. 2004). Neurotoxic effects induced by mitochondrial toxins, 3-NPA and 1-methyl-4-phenylpyridinium (MPP+), were prevented by 4–8 mM BHB (Kashiwaya et al. 2000; Imamura et al. 2006). BHB, at 0.5–10.0 mM concentration, protected neurons from structural changes evoked by limited glucose availability or inhibition of glycolysis (Izumi et al. 1998). Similarly, BHB was neuroprotective against hypoxia and ischemia both in vitro and in vivo (Masuda et al. 2005; Suzuki et al. 2002). Thus, BHB is an endogenous compound displaying reliable neuroprotective effects, what makes it an interesting candidate for preventing neuronal loss, not only related to seizures or metabolic insults, but also in the course of neurodegenerative disorders (Kashiwaya et al. 2000; Noh et al. 2006; Cheng et al. 2010).
Recently, it was demonstrated that BHB may change the mRNA expression of enzymatic proteins. In cultured astrocytes, BHB suppressed the expression of GABA-T, thus reducing astrocytic GABA degradation what possibly may increase GABA concentration in the epileptic brain (Suzuki et al. 2009).
Here, enzymatic analyses have not revealed any direct influence of BHB on the activity of semi-purified preparation of KYNA biosynthetic enzymes. However, studies in glial cultures demonstrate that BHB stimulates the expression of KAT I and II, what could explain the observed enhancement of de novo KYNA synthesis. Further insight into the underlying mechanisms comes from the experiments with KT 5720, an inhibitor of protein kinase A. KT 5720 has prevented an increase of KYNA production induced by 10 mM BHB in glial cultures and abolished the BHB-evoked increase of KATs expression in this paradigm. We have previously shown that the stimulatory effect of β-adrenergic agonists and memantine on KYNA production is mediated via cAMP/protein kinase A-related signal (Kloc et al. 2008; Luchowska et al. 2009). Possibly, similar mechanism underlies the action of BHB.
In summary, demonstrated here results reveal a novel mechanism of action of ketone body, BHB, associated with an enhancement of brain KYNA synthesis. It seems that BHB may indirectly attenuate the excessive excitatory glutamate-mediated neurotransmission through an increased availability of synaptic KYNA mediated by protein kinase A-related pathway. Increased levels of KYNA, via interaction with presynaptic NMDA receptors, can lead to reduced glutamate release. Moreover, it cannot be excluded that the local increases of brain KYNA evoked by BHB may be sufficient to reduce the activity of postsynaptic glutamate receptors and contribute to its neuroprotective effects.
Abbreviations
- BHB:
-
β-Hydroxybutyrate
- HPLC:
-
High pressure liquid chromatography
- KT 5270:
-
(9R,10S,12S)-2,3,9,10,11,12-Hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid hexyl ester
- KAT:
-
Kynurenine aminotransferase
- KRB:
-
Krebs–Ringer buffer
- KYNA:
-
Kynurenic acid
References
Abdelmalik PA, Shannon P, Yiu A, Liang P, Adamchik Y, Weisspapir M, Samoilova M, Burnham WM, Carlen PL (2007) Hypoglycemic seizures during transient hypoglycemia exacerbate hippocampal dysfunction. Neurobiol Dis 26:646–660
al-Mudallal AS, Levin BE, Lust WD, Harik SI (1995) Effects of unbalanced diets on cerebral glucose metabolism in the adult rat. Neurology 45:2261–2265
Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ (2006) Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol 60:223–235
Carpenedo R, Pittaluga A, Cozzi A, Attucci S, Galli A, Raiteri M, Moroni F (2001) Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur J Neurosci 13:2141–2147
Cheng B, Yang X, Chen C, Cheng D, Xu X, Zhang X (2010) D-beta-hydroxybutyrate prevents MPP+-induced neurotoxicity in PC12 cells. Neurochem Res 35:444–451
Chmiel-Perzyńska I, Perzyński A, Wielosz M, Urbańska EM (2007) Hyperglycemia enhances the inhibitory effect of mitochondrial toxins and D,L-homocysteine on the brain production of kynurenic acid. Pharmacol Rep 59:268–273
Cullingford TE (2004) The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot Essent Fatty Acids 70:253–264
Fukao T, Lopaschuk GD, Mitchell GA (2004) Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids 70:243–251
Gasior M, Rogawski MA, Hartman AL (2006) Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol 17:431–439
Gasior M, French A, Joy MT, Tang RS, Hartman AL, Rogawski MA (2007) The anticonvulsant activity of acetone, the major ketone body in the ketogenic diet, is not dependent on its metabolites acetol, 1,2-propanediol, methylglyoxal, or pyruvic acid. Epilepsia 48:793–800
Gilbert DL, Pyzik PL, Freeman JM (2000) The ketogenic diet: seizure control correlates better with serum β-hydroxybutyrate than with urine ketones. J Child Neurol 15:787–790
Greene AE, Todorova MT, McGowan R, Seyfried TN (2001) Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia 42:1371–1378
Guidetti P, Okuno E, Schwarcz R (1997) Characterization of rat brain kynurenine aminotransferases I and II. J Neurosci Res 50:457–465
Guidetti P, Amori L, Sapko MT, Okuno E, Schwarcz R (2007) Mitochondrial aspartate aminotransferase: a third kynurenate-producing enzyme in the mammalian brain. Neurochem 102:103–111
Hartman AL, Vining EP (2007) Clinical aspects of the ketogenic diet. Epilepsia 48:31–42
Hartman AL, Gasior M, Vining EP, Rogawski MA (2007) The neuropharmacology of the ketogenic diet. Pediatr Neurol 36:281–292
Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX (2001) The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increase non-alpha 7 nicotinic receptor expression; physiopathological implications. J Neurosci 21:7463–7473
Huttenlocher PR (1976) Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr Res 10:536–540
Imamura K, Takeshima T, Kashiwaya Y, Nakaso K, Nakashima K (2006) D-beta-hydroxybutyrate protects dopaminergic SH-SY5Y cells in a rotenone model of Parkinson’s disease. J Neurosci Res 84:1376–1384
Izumi Y, Ishii K, Katsuki H, Benz AM, Zorumski CF (1998) beta-Hydroxybutyrate fuels synaptic function during development. Histological and physiological evidence in rat hippocampal slices. J Clin Invest 101:1121–1132
Johnstone AM, Horgan GW, Murison SD, Bremner DM, Lobley GE (2008) Effects of a high-protein ketogenic diet on hunger, appetite, and weight loss in obese men feeding ad libitum. Am J Clin Nutr 87:44–55
Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL (2000) D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc Natl Acad Sci USA 97:5440–5444
Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN (2009) Hyperglycemic crises in adult patients with diabetes. Diabetes Care 32:1335–1343
Klitgaard H, Pitkänen A (2003) Antiepileptogenesis, neuroprotection, and disease modification in the treatment of epilepsy: focus on levetiracetam. Epileptic Disord 1:S9–S16
Kloc R, Luchowska E, Wielosz M, Owe-Larsson B, Urbanska EM (2008) Memantine increases brain production of kynurenic acid via protein kinase A-dependent mechanism. Neurosci Lett 435:169–173
Kocki T, Luchowski P, Luchowska E, Wielosz M, Turski WA, Urbanska EM (2003) L-cysteine sulphinate, endogenous sulphur-containing amino acid, inhibits rat brain kynurenic acid production via selective interference with kynurenine aminotransferase II. Neurosci Lett 346:97–100
Kocki T, Wielosz M, Turski WA, Urbanska EM (2006) Enhancement of brain kynurenic acid production by anticonvulsants – novel mechanism of antiepileptic activity? Eur J Pharmacol 541:147–151
Lefevre F, Aronson N (2000) Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics 105:E46
Leite M, Frizzo JK, Nardin P, de Almeida LM, Tramontina F, Gottfried C, Gonçalves CA (2004) Beta-hydroxy-butyrate alters the extracellular content of S100B in astrocyte cultures. Brain Res Bull 64:139–143
Likhodii SS, Musa K, Cunnane SC (2002) Breath acetone as a measure of systemic ketosis assessed in a rat model of the ketogenic diet. Clin Chem 48:115–120
Luccini E, Musante V, Neri E, Raiteri M, Pittaluga A (2007) N-methyl-D-aspartate autoreceptors respond to low and high agonist concentrations by facilitating, respectively, exocytosis and carrier-mediated release of glutamate in rat hippocampus. J Neurosci Res 85:3657–3665
Luchowska E, Luchowski P, Paczek R, Ziembowicz A, Kocki T, Turski WA, Wielosz M, Lazarewicz J, Urbanska EM (2005) Dual effect of DL-homocysteine and S-adenosylhomocysteine on brain synthesis of the glutamate receptor antagonist, kynurenic acid. J Neurosci Res 79:375–382
Luchowska E, Kloc R, Olajossy B, Wnuk S, Wielosz M, Owe-Larsson B, Urbanska EM (2009) β-Adrenergic enhancement of brain kynurenic acid production mediated via cAMP-related protein kinase A signaling. Prog Neuropsychopharmacol Biol Psychiatry 33:519–529
Luchowski P, Luchowska E, Turski WA, Urbanska EM (2002) 1-Methyl-4-phenylpyridinium and 3-nitropropionic acid diminish cortical synthesis of kynurenic acid via interference with kynurenine aminotransferases in rats. Neurosci Lett 330:49–52
Masuda R, Monahan JW, Kashiwaya Y (2005) D-beta-hydroxybutyrate is neuroprotective against hypoxia in serum-free hippocampal primary cultures. J Neurosci Res 80:501–509
Miranda AF, Boegman RJ, Beninger RJ, Jhamandas K (1997) Protection against quinolinic acid-mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience 78:967–975
Moroni F, Russi P, Lombardi G, Beni M, Carlà V (1988) Presence of kynurenic acid in the mammalian brain. J Neurochem 51:177–180
Nehlig A (2004) Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot Essent Fatty Acids 70:265–275
Németh H, Toldi J, Vécsei L (2006) Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm Suppl 70:285–304
Noh HS, Hah YS, Nilufar R, Han J, Bong JH, Kang SS, Cho GJ, Choi WS (2006) Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J Neurosci Res 83:702–709
Noh HS, Kim YS, Choi WS (2008) Neuroprotective effects of the ketogenic diet. Epilepsia 49(Suppl 8):120–123
Pan JW, Rothman DL, Behar KL, Stein DT, Hetherington HP (2000) Human brain β-hydroxybutyrate and lactate increase in fasting induced ketosis. J Cereb Blood Flow Metab 20:1502–1507
Rho JM, Anderson GD, Donevan SD, White HS (2002) Acetoacetate, acetone, and dibenzylamine (a contaminant in L-(+)-β-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia 43:358–361
Robinson AM, Williamson DH (1980) Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 60:143–187
Samoilova M, Weisspapir M, Abdelmalik P, Velumian AA, Carlen PL (2010) Chronic in vitro ketosis is neuroprotective but not anti-convulsant. J Neurochem 113:826–835
Schwarcz R, Pellicciari R (2002) Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther 303:1–10
Schwartzkroin PA (1999) Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res 37:171–180
Shepard PD, Joy B, Clerkin L, Schwarcz R (2003) Micromolar brain levels of kynurenic acid are associated with a disruption of auditory sensory gating in the rat. Neuropsychopharmacology 28:1454–1462
Stone TW, Behan WM, Jones PA, Darlington LG, Smith RA (2001) The role of kynurenines in the production of neuronal death, and the neuroprotective effect of purines. J Alzheimers Dis 3:355–366
Suzuki M, Suzuki M, Kitamura Y, Mori S, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A (2002) Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn J Pharmacol 89:36–43
Suzuki Y, Takahashi H, Fukuda M, Hino H, Kobayashi K, Tanaka J, Ishii E (2009) Beta-hydroxybutyrate alters GABA-transaminase activity in cultured astrocytes. Brain Res 1268:17–23
Thavendiranathan P, Mendonca A, Dell C, Likhodii SS, Musa K, Iracleous C, Cunnane SC, Burnham WM (2000) The MCT ketogenic diet: effects on animal seizure models. Exp Neurol 161:696–703
Thio LL, Wong M, Yamada KA (2000) Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology 54:325–331
Turski WA, Gramsbergen JBP, Traitler H, Schwarcz R (1989) Rat brain slices produce and liberate kynurenic acid upon exposure to L-kynurenine. J Neurochem 52:1629–1636
Urbanska E, Ikonomidou C, Sieklucka M, Turski WA (1991) Aminooxyacetic acid produces excitotoxic lesions in the rat striatum. Synapse 9:129–135
Urbanska EM, Kocki T, Saran T, Kleinrok Z, Turski WA (1997) Impairment of brain kynurenic acid production by glutamate metabotropic receptor agonists. Neuroreport 8:3501–3505
van Delft R, Lambrechts D, Verschuure P, Hulsman J, Majoie M (2010) Blood beta-hydroxybutyrate correlates better with seizure reduction due to ketogenic diet than do ketones in the urine. Seizure 19:36–39
Wheless JW (1995) The ketogenic diet: fa(c)t or fiction. J Child Neurol 10:419–423
Yudkoff M, Daikhin Y, Horyn O, Nissim I, Nissim I (2008) Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia 49:73–75
Acknowledgment
This study was supported by the grants from Medical University in Lublin, DS 450/06, 450/07, 450/08, 450/09.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chmiel-Perzyńska, I., Kloc, R., Perzyński, A. et al. Novel Aspect of Ketone Action: β-Hydroxybutyrate Increases Brain Synthesis of Kynurenic Acid In Vitro. Neurotox Res 20, 40–50 (2011). https://doi.org/10.1007/s12640-010-9220-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-010-9220-0