
Abstract
Methylphenidate (MPH) abuse damages brain cells. The neuroprotective effects of topiramate (TPM) have been reported previously, but its exact mechanism of action still remains unclear. This study investigated the in vivo role of various doses of TPM in the protection of rat amygdala cells against methylphenidate-induced oxidative stress and inflammation. Seventy adult male rats were divided into seven groups. Groups 1 and 2 received normal saline (0.7 ml/rat) and MPH (10 mg/kg), respectively, for 21 days. Groups 3, 4, 5, 6, and 7 were concurrently treated with MPH (10 mg/kg) and TPM (10, 30, 50, 70, and 100 mg/kg), respectively, for 21 days. elevated plus maze (EPM) was used to assess motor activity disturbances. In addition, oxidative, antioxidantand inflammatory factors and CREB, Ak1, CAMK4, MAPK3, PKA, BDNF, and c FOS gene levels were measured by RT-PCR, and also, CREB and BDNF protein levels were measured by WB in isolated amygdalae. MPH significantly disturbed motor activity and TPM (70 and 100 mg/kg) neutralized its effects. MPH significantly increased lipid peroxidation, mitochondrial GSSG levels and IL-1β and TNF-α level and CAMK4 gene expression in isolated amygdala cells. In contrast, superoxide dismutase, glutathione peroxidase, and glutathione reductase activities and CREB, BDNF Ak1, MAPK3, PKA, BDNF, and c FOS expression significantly decreased. The various doses of TPM attenuated these effects of MPH. It seems that TPM can be used as a neuroprotective agent and is a good candidate against MPH-induced neurodegeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Methylphenidate (MPH) is a neural stimulant approved for the management of hyperactive children (Challman and Lipsky 2000; Motaghinejad et al. 2015c; Gomes et al. 2010). MPH inhibits the dopamine and norepinephrine reuptake into presynaptic terminals (Motaghinejad et al. 2015c, d). MPH is structurally and functionally similar to methamphetamine and cocaine which creates high potential for abuse and dependency (Tagaya 2010; Huss and Lehmkuhl 2001; Motaghinejad et al. 2015d; Schwartz et al. 2006). Some previous studies have indicated an increase in MPH abuse frequency, particularly among college students (Klein-Schwartz 2002; Babcock and Byrne 2000). The neurochemical alterations caused by chronic MPH abuse in amygdaloidal nucleuses remain unclear, and just some of the effect of its abuse on the brain and behavior are clarified (Williams et al. 2004; Barrett and Pihl 2002; Trinh et al. 2013). Previous studies have confirmed that chronic abuse of MPH by doses of 2, 3, and 10 mg/kg induces apoptosis, oxidative damage and a rise in the expression of inflammatory cytokines, such as TNF-α and IL-1β levels. It decreases antioxidant enzymes in the central nervous system in human and animal models. These studies showed that mentioned alteration biomarkers correlated with animal behavioral changes. (Patrick and Markowitz 1997; Motaghinejad et al. 2015e; Jones and Dafny 2014).
TPM has successfully been used in the management of alcohol, methamphetamine, and cocaine addiction as an anticonvulsant (Garnett 2000; Arnone 2005). The neuroprotective effects of TPM have been reported in several studies, but its exact mechanism of action (Kudin et al. 2004; Motaghinejad et al. 2015e; Motaghinejad and Motevalian 2016; Mao et al. 2015) still remains unclear. Previous studies have suggested that the protective effects of TPM are mediated by a decrease in lipid peroxidation and increase in glutathione in the cell system (Motaghinejad and Motevalian 2016; Motaghinejad et al. 2015f). In addition, it has been shown that TPM has antioxidant and anti-inflammatory properties which can act as an excellent candidate for the management of oxidative stress and inflammation (Kutluhan et al. 2009). They have also demonstrated that TPM exhibits a synergistic effect on superoxide dismutase and catalase activities in striatum and mid-brain regions (Armaǧan et al. 2008; Kutluhan et al. 2009).
Recent findings also suggest that TPM can change mitochondrial respiratory enzymes and activate biogenesis of mitochondria. In addition, it has indicated that TPM has potent anti-inflammatory properties. TPM can reduce TNF-α and TGF-β1, and it also attenuates the inflammation and injury of brain cells (Koçer et al. 2009). The main anti-inflammatory properties of TPM were mediated by GABA enhancement, inhibition of glutamate Ca2+ channel influx, and blocking of Na+ channel (Armaǧan et al. 2008; Demirci et al. 2013). A similar study suggests that topiramate has high anti-inflammatory properties and a repositioning potential for the treatment of autoimmune diseases. The inhibition of inflammation and the decrease in inflammatory markers are responsible for these properties (Dudley et al. 2011; Pinheiro et al. 2015). Previous studies have shown that TPM can be proposed as a putative agent in the treatment of some neurodegenerative disorders (Shi et al. 2010; Shaldubina et al. 2002).
Some previous studies showed that neuroprotection of TPM was mediated by GABA activity enhancement on inhibition of AMPA/Kinate in glutamatergic system (Motaghinejad and Motevalian 2016; Yang and Shen 2009). On the other hand, previous studies have shown that cyclic AMP response element binding protein (CREB) acts as a major transcription factor in brain development and neurogenesis (Blendy 2006; Lee et al. 2005). CREB is activated in a phosphorylated form and some protein kinase, such as Ak1, CAMK4, MAPK3, PKA enzymes phosphorylate this transcription factor and convert CREB to its active form (Carlezon et al. 2005; Kitagawa 2007; Réus et al. 2011; Aguiar et al. 2011). CREB acts on DNA and prompts the production of BDNF and cFOS proteins, which are important in neurogenesis and the development of neurons (Réus et al. 2011; Aguiar et al. 2011). Keeping in mind the role of the amygdala in cognition, depression and anxiety-like behaviors, we aim to study the effects of TPM against MPH-induced oxidative stress and inflammation and clarify the role of the phosphorylated CREB signaling pathway in this type of neuroprotection in amygdala. We also attempt to indicate the possible role of Ak1, CaMK4, MAPK3, PKA, as an upstream of CREB signaling, gene expression in the phosphorylation and activation of CREB protein and c-FOS as well as BDNF as a downstream product of CREB effect on genes.
Materials and methods
Animals
Seventy male adult rats (mean weight 220 ± 10 g) and 8 weeks old obtained from the experimental research center of Iran University of Medical Sciences (IUMS, Tehran, Iran) and transferred to the laboratory. The animals were held for 2 weeks before the initiation of the experiment at room temperature with free access to standard food, tap water and a standard dark/light cycle. Our experimental protocol was approved by the research council of Iran University of Medical Sciences.
Drugs
TPM and MPH were purchased from Sigma-Aldrich (USA) and dissolved in normal saline for injection, freshly prepared just before administration. The volume of injection was 0.7 ml/rat.
Experimental design
Seventy adult male rats were randomly divided to six groups. Group 1 (as negative control) was treated with normal saline (0.7 ml/rat, i.p) for 21 days. Group 2 (as positive control) received MPH (10 mg/kg) for 21 days. Groups 3, 4, 5, 6, and 7 were treated concurrently for 21 days by MPH (10 mg/kg, ip) and TPM with doses of 10, 30, 50, 70, and 100 mg/kg by i.p. injection, respectively, administration of MPH in combination with TPM was done separately, and there was 1 h between TPM and MPH administration: first, TPM was injected, and then, MPH was administrated.
On day 22, after drug administration, elevated plus maze (EPM), a standard behavioral method used for study of anxiety and amygdaloidal nucleus degeneration, was used to evaluate the level of anxiety in the experimental animals. After behavioral assay in day 23, all animals were anesthetized by administration of 50 mg/kg of thiopental, and their brain tissue was removed. Keeping in mind the importance of the amygdala in anxiety-like behaviors, we evaluated the effects of administration of TPM on MPH-induced oxidative stress and inflammation in this region of brain. Considering the importance of CREB and BDNF and their upstream and downstream signaling pathways in neuroprotection, we evaluated the effects of TPM on MPH-induced disturbances in the CREB/BDNF signaling pathway using real-time reverse transcriptase-PCR (RT-PCR) and western Blotting. The isolation of amygdala was done according to guide by previous similar study (McCool and Botting 2000).
Elevated plus maze (EPM)
Another test applied to assess anxiety level in rodents is the elevated plus maze (EPM). The equipment consists of two opposite arms 55 × 15 cm, which are connected by a central square (10 × 10 cm), the whole apparatus shaped as a plus sign. One arm was kept open, while the other arm was enclosed with 40-cm elevated wall. The entire apparatus was elevated 50 cm above the ground. All animals were located individually in the center of the maze in front of an enclosed arm and the time which the animal spent in the open arms was recorded during 5 min for each rat. More time spent in open arms indicated non-depressive behavior. All behavioral changes and scoring of parameters were evaluated by a blind experimenter who did not know anything about drug treatment of groups and the protocol of experiment. In addition, each animal was examined three times, and the mean of the scores was reported as final data.
It should be mentioned that all the animals were tested in the first day of experiment by EPM (base line level of behavior), and also, this test was repeated in the last day after drug administration (the effect of drug administration on behavior), and this was done to be able to interpret the obtained results.
Mitochondrial preparation
By administrating 50 mg/kg of thiopental, all animals were anesthetized and euthanized, and amygdala was isolated according to the previous procedure explained already (McCool and Botting 2000). The amygdaloidal tissue was homogenized in a cold homogenization buffer, containing 25 mM 4-morpholinepropanesulfonic acid, 400 mM sucrose, 4 mM MgCl2, and 0.05 mM EGTA at pH 7.3. The homogenized cells were centrifuged at 4500g for 10 min, and after that, its supernatant was centrifuged at 12,000g for 10 min. The final sediment was resuspended in homogenization buffer and stored on ice. Total mitochondrial proteins in tissues were determined using protein Dc assay kit (Bio-Rad) briefly; Bradford reagent (1 part Bradford: 4 parts dH2O) was added to serial dilution series (0.1–1.0 mg/ml) of a known protein sample concentration such as BSA, dissolved in homogenization buffer. These serial dilutions were prepared for providing a standard curve. Then, in separate processes, 10, 15, 20, 25, and 30 μl of the mentioned protein extracts (homogenized cell solutions) were added to multiple wells, and Bradford reagent (100 µl) was added to each well. Color density of all wells was read by plate reader at 630 nm, and finally, using the mentioned standard curve, protein quantity of unknown protein extracts was obtained. These homogenized cell solutions were analyzed for the measurement of oxidative stress and inflammatory markers (Motaghinejad et al. 2015b, d).
Measurement of oxidative stress and inflammatory biomarkers
Study of lipid peroxidation
Lipid peroxides are unstable indicators of oxidative stress in cells that decompose to form more complex and reactive compounds, such as malondialdehyde (MDA), a natural byproduct of lipid peroxidation. To assess MDA, 100 μl of homogenized sample solution or MDA standard was added to separate micro centrifuge tubes, and then, 100 μl of SDS lysis solution was added to both sample and standard tubes, and mixed thoroughly. After 5 min incubation at room temperature, 250 μl of TBA reagent was added to each of the samples and standard tubes. The tubes were again incubated, but this time, at 95 °C for 45–60 min. Furthermore, the tubes were centrifuged at 10,000 rpm for 15 min and 300 μl of supernatant from each tube was transferred to new tubes. 300 μl of n-Butanol was added to each tube and after vortexing (1–2 min), and the tubes were centrifuged for 5 min at 10,000g. At the end, 200 μl of both the MDA standards and the samples were transferred to a 96-well microplate (compatible with a spectrophotometric plate reader), and absorbance was read at 532 nm. Results were expressed as nmol/mg of protein (Motaghinejad et al. 2015a, b; Shamoto-Nagai et al. 2007).
GSH and GSSG levels
Within the cells, glutathione exists in reduced (GSH) and oxidized (GSSG) forms. In healthy cells and tissues, more than 90 % of the total glutathione pool is in the reduced form, while less than 10 % exists in the disulfide form (GSSG). To measure GSH and GSSG levels, 25 μl of the 1X glutathione reductase solutions was added to each well of a 96-well plate, followed by the addition of 25 μl of the IX NADPH solution, and then, 100 μl of the prepared standard solution of glutathione or homogenized sample solution was added to each well and mixed thoroughly. In addition, 50 μl of the 1X Chromogen was also added to each well and mixed. Immediately after that, the absorbance was read at 405 nm for each GSSG/GSH standard and sample. Finally, using a standard curve, the amount of GSSG/GSH was determined and expressed as nmol/mg of protein (Motaghinejad et al. 2015a, b; Shamoto-Nagai et al. 2007).
Study of manganese superoxide dismutase activity
Superoxide dismutase (SOD), which catalyzes the dismutation of the superoxide anion (O2 −) into hydrogen peroxide and molecular oxygen, is one of the most important antioxidative enzymes that exist. To determine the SOD activity, 20 μl of the unknown sample solution was added to each well and 2nd blank wells, and 20 μl of ddH2O (double distilled water) was added to the 1st and 3rd blank wells, and then, 200 μl of WST working solution (1 ml of water-soluble tetrazolium salt; WST dissolved in 19 ml of buffer solution) was added to each well and mixed. 20 μl of dilution buffer was added to the 2nd and 3rd blank wells. Furthermore, 20 μl of enzyme working solution was added to each sample as well as the 1st blank well. After mixing thoroughly, the plates were incubated at 37 °C for 20 min, and absorbance was read at 450 nm using a microplate reader. As recommended by the manufacturer, SOD activity was calculated using the following equation: SOD activity = {[(A blank 1 − A blank 3) − (A sample − A blank 2)]/(A blank 1 − A blank 3)} × 100.
Data were reported as U/ml/mg protein (Motaghinejad et al. 2015a, b; Shamoto-Nagai et al. 2007).
Measurement of glutathione peroxidase (GPx) activity
Glutathione peroxidase (GPx) is one of the most important enzymes for the detoxification of peroxides in living cells. GPx plays a crucial role in protecting cells from damage by free radicals which are formed by peroxide decomposition. To assess GPx activity, 20 μl of the sample (which was diluted beforehand with the assay buffer) or assay buffer alone was added to the sample and its corresponding well. Then, 200 μl of the reaction solution was added to each well. After setting up the microtiter plate reader at 340 nm over a period of 8 min at 25 °C, 20 μl of peroxide substrate solution was added to each well and absorbance was measured. For data evaluation, delta OD was used between 2 and 8 min. As recommended by the manufacturer, change in absorbance [ΔA340/min] was calculated by the following equation: ΔA340/min = A340nm (Start) − A340nm (Stop)/Reaction time (min), any change in absorbance is directly proportional to GPx activity.
GPx activity: ΔA340/min × Reaction volume (ml) × Dilution factor of the original sample/Extinction coefficient for NADPH at 340 nm × Volume of the tested sample. Results were expressed as mU/mg protein (Motaghinejad et al. 2015a, b; Shamoto-Nagai et al. 2007).
Measurement of glutathione reductase (GR) activity
GR plays an important role in defending cells from injury by free radicals produced by peroxide decomposition. To assess GR activity, 25 μl of the sample (which was diluted beforehand with the assay buffer) or assay buffer alone was added to the sample and its corresponding well, and then, 250 μl of the special reaction solution was added to each well according to manufacturer instructions. After that, the microplates were read at 340 nm. The OD of sample wells was inserted in the standard curve which was drawn previously by manufacture kits, and the activity of GR in unknown sample solution was measured by insertion of OD and calculation of enzyme activity. Results were expressed as mU/mg protein (Motaghinejad et al. 2015a, b; Shamoto-Nagai et al. 2007).
Measurement of inflammatory parameters
IL-1β and TNF-α measurement
Concentrations of interleukin 1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α) in the supernatant of amygdaloidal cells were measured using a commercially available special ELISA kit (Genzyme Diagnostics, Cambridge, USA). Briefly, 96-well microtitre plates (Nunc) were coated with sheep anti-rat IL-1β and TNF-α polyclonal antibodies (2 mg/ml in bicarbonate coating buffer; 0.1 M NaHCO3, 0.1 M NaCl, pH 8.2, for 20 h at 48 °C), then washed three times with washing buffer (0.5 M NaCl, 2.5 mM NaH2PO4, 7.5 mM Na2HPO4, 0.1 % Tween 20, pH 7.2). 100 ml of a 1 % (w/v), after that ovalbumin (Sigma Chemical Co., Poole, Dorset, UK) solution in bicarbonate as coating buffer was added to each well and incubated at 37 °C for 1 h. After three washes, 100 ml of samples and standards were added and plates were incubated at 48 °C for 20 h. After three washes, 100 ml of the biotinylated sheep anti-rat IL-1β or TNF-α antibody (1:1000 dilutions in washing buffer containing 1 % sheep serum, Sigma Chemical Co., Poole, and Dorset, UK) was added to each well. The further incubation was carried out for 1 h at room temperature. After three washes, 100 ml avidin-HRP (Dako Ltd, UK) (1:5000 dilution in wash buffer) was added to each well, and plates were incubated at room temperature for 15 min. After three washes, 100 ml of TMB substrate solution (Dako Ltd., UK) was added to each well, and the plates were incubated for 10 min at room temperature. At the end of the incubation period, 100 ml of 1 M H2SO4 was added to each well to stop the reaction and to facilitate the color development. Absorbance was read at 450 nm on a microtitre plate reader. The detection limit of the assay was determined to be 4.3 pg/ml. Results were expressed as ng IL-1β/ml or TNF-α/ml (Motaghinejad et al. 2015a, b; Shamoto-Nagai et al. 2007).
Real-time reverse transcriptase-PCR (RT-PCR) studies
Total RNA was extracted from ~200 µg of amygdala using ONE STEP-RNA Reagent (Bio Basic, Canada inc.) according to the manufacturer’s instructions, and the quantity and quality of RNA were analyzed using a nanodrop (ND-1000, Thermo Scientific Fisher, US) and gel electrophoresis. To eliminate any genomic contamination, RNA was treated with DNase I (Qiagen, Hilden, Germany) as described by the manufacturer. Complementary DNA (cDNA) was synthesized using 1 µg of total RNA. The integrity and quality of cDNA were examined with GAPDH primers as housekeeping. Real-time reverse transcriptase-PCR (RT-PCR) was carried out to evaluate the differences in expression patterns of cAMP response element-binding protein (CREB), adenylate kinase 1 (Ak1), calcium/calmodulin-dependent protein kinase IV(CAMK4), mitogen-activated protein kinase 3 (MAPK3), protein kinase A (PKA), brain derived neurotrophic factor (BDNF), and c FOS genes among samples of each group. The specific primers corresponding to the coding sequence, including BDNF Forward: 5′-GGAGGCTAAGTGGAGCTGAC-3′; BDNF Reverse: 5′-GCTTCCGAGCCTTCCTTTAG-3′; Akt1 Forward: 5′-AAGGAGATCATGCAGCACCG-3′; Akt Reverse: 5′-GGTGGGCTCACCTTCTTCTC-3′, CAMK4 Forward: 5′-AGCAGCAGTCACACCAACAT-3′; Reverse: 5′-TCTGTCTTGTCCTTGCCGTC-3′, MAPK3 Forward: 5′-TATCAACACCACCTGCGACC-3′; Reverse: 5′-ATGATCTCTGGGGCTCGGTA-3′; cFOS Forward: 5′-GGGAGCTGACAGATACGCTC-3′; Reverse: 5′-TTGGCAATCTCGGTCTGCAA-3′. CREB1 Forward: 5′-CAGACAACCAGCAGAGTGGA-3′. Reverse: 5′-CTGGACTGTCTGCCCATTG-3′. GAPDH Forward: 5′-AGACAGCCGCATCTTCTTGT, Reverse: 5′-CCGTTCACACCGACCTTCA-3′. PKA Forward: 5′-GCAGGAGAGCGTGAAAGAGT-3′, Reverse: 5′-CTGAGAAGGGGTCTCCCATTT-3′, were designed by Primer 3 software version 0.4 (frodo.wi.mit.edu). Real-time RT-PCR was performed in 20 μl reactions containing 1 μl cDNA target, 100 nM forward and reverse primers and 1× SYBR® Premix Ex Taq™ II (Takara, Tokyo, Japan). Experiments were carried out in triplicate using a CFX96™ Real-Time System (C1000TM Thermal Cycler) (Bio-Rad, Hercules, CA, USA). Amplification conditions were as follow: initial denaturation at 95 °C for 10 min, followed by 40 cycles (denaturation at 95 °C for 15 s and annealing and extension at 60 °C for 60 s). The relative value of the mRNA expression level of CREB, Ak1, CAMK4, MAPK3, PKA, BDNF, and c FOS gene was calculated by comparing the cycle thresholds (CTs) of the target gene with that of housekeeping gene (GAPDH) using the 2−ΔΔct method and REST 2009 software. Serial dilutions of cDNAs were used to calculate the efficiencies of the primer sets on real-time PCR. In this regard, it was found that the efficiencies of the various primer sets were similar (Motaghinejad et al. 2015f; Peng et al. 2012).
Western blot
We studied the immunoreactivity, total and phosphorylated CREB, and BDNF contents of the isolated amygdala by Western blotting. Electrotransfer of the resolved bands from gel to polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, USA) was fulfilled in 90 min at 0.7 mA/cm2 using a semi-dry transfer apparatus (PeQlab). After transferring step, the membrane was weakly stained for about 3 min with Coomassie blue G-250 (Sigma Aldrich, UK) 1 µg/100 ml distil water without methanol. Then, the membrane was dried and cut into 2-mm wide stripes. After destaining with methanol, the strips washed and blocked with 2 % BSA overnight at 4 °C and then added with 1:100 diluted human or ovine sera at room temperature (RT) for 2 h on a shaker. The membranes then were washed with PBS-T (three washing steps in a total of 10 min) and incubated with following conjugated polyclonal anti-rabbit antibody: BDNF, CREB, total, and phosphorylated (1:500 dilutions in BSA, 360 min, RT; Sigma Aldrich, Germany), then all strips were exposed by secondary HRP conjugated polyclonal Rabbit anti-Sheep antibody (1:5000 dilution in BSA, 120 min, RT; Sina Biotech, Iran). The strips were washed and incubated with chemiluminescent substrate (Luminol and H2O2) for 2 min in RT. Finally, the reactive bands were detected on X-ray film within 10–20 s under safelight condition (Motaghinejad et al. 2015f; Woo et al. 2005).
Statistical analysis
All of the data obtained were statistically analyzed using version 6 of Graph Pad PRISM Software. The data were averaged in every experimental group and expressed as mean ± standard error of the means (SEM). Then, the differences between control and treatment groups were calculated by one way ANOVA. Differences between severities of behaviors in groups were evaluated by Tukey’s post hoc test. p < 0.05 was taken as statistically significant.
Results
The assessment of behavior in elevated plus maze (EPM)
The MPH treated animals spent less time in open arms and had fewer numbers of open arm entries in comparison to the animals in control group (p < 0.05). The treatment of animals with various doses of TPM increased time spent in open arms, as well as numbers of entry to open arms. This increase was statistically significant in groups treated with 70 and 100 mg/kg of TPM in comparison to MPH (10 mg/kg) only treated group (p < 0.05) (Table 1). Baseline measurements for all the animals did not show any significant difference between groups.
The effect of various doses of TPM on MPH-induced lipid peroxidation in mitochondria
MPH increased lipid peroxidation and MDA levels in isolated mitochondria from animals treated with MPH. Comparing MPH with negative control group regarding lipid peroxidation, it was showed that MPH-treated group has significantly increased in MDA level (p < 0.001). The various doses of TPM attenuated the decrease in MDA concentration in groups treated with 70 and 100 mg/kg of TPM, and the decrease was statistically significant in comparison to the group receiving MPH (10 mg/kg) only (p < 0.001) (Fig. 1).

The effects of various doses of TPM (10, 30, 50, 70, and 100 mg/kg) on MPH-induced lipid peroxidation in rat isolated amygdala mitochondria. All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
The effect of various doses of TPM on MPH-induced GSH/GSSG alterations in mitochondria
Treating animals with only MPH decreased the GSH content in isolated mitochondria in comparison to the negative control group (p < 0.001). The animals treated with 70 and 100 mg/kg of TPM showed noticeably higher amounts of GSH in comparison to the group receiving only MPH (10 mg/kg) (Table 2). In addition, GSSG levels in groups treated with MPH were notably higher than those of the negative control group (p < 0.001). TPM at doses of 70 and 100 mg/kg prevented an increase in GSSG levels induced by MPH, and this was significant comparing to the group treated with MPH (10 mg/kg) only (p < 0.001) (Table 2).
The effect of various doses of TPM on MPH-induced attenuation in superoxide dismutase (SOD) activity in mitochondria
Comparing MPH with negative control group regarding superoxide dismutase (SOD) activity, it was showed that MPH-treated group (10 mg/kg) has significant inhibition of SOD activity (p < 0.001). Administration of 70 and 100 mg/kg of TPM significantly increased the activity of manganese superoxide dismutase in comparison to the administration of MPH only (10 mg/kg) (p < 0.001) (Fig. 2).

The effect of TPM (10, 30, 50, 70, and 100 mg/kg) on MPH-induced attenuation in manganese superoxide dismutase activity in rat isolated amygdala mitochondria. All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
The effect of various doses of TPM on MPH- induced changes in glutathione peroxidase (GPx) activity in mitochondria
MPH alone caused attenuation of glutathione peroxidase (GPx) activity. This decrease was statistically significant in comparison to the negative control group (p < 0.001). In addition, the injection of TPM at doses of 70 and 100 mg/kg in MPH-treated animals increased the glutathione peroxidase activity (GPx), and these increases were statistically significant in comparison to the MPH (10 mg/kg) only group (p < 0.001) (Fig. 3).

The effect of TPM (10, 30, 50, 70, and 100 mg/kg) on MPH -induced attenuation in glutathione peroxidase (GPx) activity in rat isolated amygdala mitochondria. All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
The effect of various doses of TPM on MPH-induced changes in glutathione reductase (GR) activity in mitochondria
MPH (10 mg/kg) caused attenuation of glutathione reductase (GR) activity. This decrease was statistically significant in comparison to the negative control group (p < 0.001). In addition, the injection of TPM at doses of 70 and 100 mg/kg in MPH-treated animals increased glutathione reductase (GR) activity, which was statistically significant in comparison to the MPH (10 mg/kg) treated only group (p < 0.001) (Fig. 4).

The effect of TPM (10, 30, 50, 70, and 100 mg/kg) on MPH-induced attenuation in glutathione reductase (GR) activity in rat isolated amygdala mitochondria. All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
The effect of various doses of TPM on MPH-induced IL-1β and TNF-α level
MPH-induced increases in IL-1β and TNF-α level. These were significantly greater comparing to the negative control group (p < 0.001). TPM (70 and 100 mg/kg) decreased the MPH-induced inflammation, and this was statistically significant in comparison to the MPH (10 mg/kg) only group (p < 0.001) (Figs. 5, 6).

The effect of TPM (10, 30, 50, 70, and 100 mg/kg) on MPH-induced TNF-α alteration in rat isolated amygdala mitochondria. All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate

The effect of TPM (10, 30, 50, 70, and 100 mg/kg) on MPH-induced IL-β alteration in rat isolated amygdala mitochondria. All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
The effect of various doses of TPM on MPH-induced changes in Ak1, CAMK4, MAPK3, PKA, and c FOS by RT-PCR
MPH-induced decreases in expression of Ak1, MAPK3, PKA, and c FOS genes and increases in CAMK4 in the amygdala in comparison to the negative control group (p < 0.001). Correspondingly, TPM (100 mg/kg) increased Ak1, MAPK3, PKA and c FOS genes and decreased CAMK4 gene in comparison to the MPH (10 mg/kg) only (p < 0.001) (Figs. 7, 8, 9, 10, 11).

The alterations of expression (RT-PCR) of Akt1 in amygdala in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate

The alterations of expression (RT-PCR) of MAPK3 in amygdala in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate

The alterations of expression (RT-PCR) of PKA in amygdala in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate

The alterations of expression (RT-PCR) of cFOS in amygdala in control and groups under treatment with 10 mg/kg of MPH in absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate

The alterations of expression (RT-PCR) of CAMK4 in amygdale in control and groups under treatment with 10 mg/kg of MPH in absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
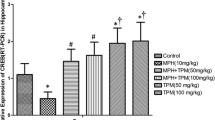
The effect of various doses of TPM on MPH-induced decreases in CREB by RT-PCR
MPH-induced decreases in expression of CREB genes in the amygdala in comparison to the negative control group (p < 0.001). Correspondingly, TPM (100 mg/kg) increased these genes in comparison to the MPH (10 mg/kg) only treated group (p < 0.001) (Fig. 12).

The alterations of expression (RT-PCR) of CREB in amygdala in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
The effect of various doses of TPM on MPH-induced decrease in BDNF by RT-PCR
MPH-induced decreases in expression of BDNF genes in the amygdala in comparison to the negative control group (p < 0.001). Correspondingly, TPM (100 mg/kg) increased these genes in comparison to the MPH (10 mg/kg) only treated group (p < 0.001) (Fig. 13).

The alterations of expression (RT-PCR) of BDNF in amygdala in control and groups under treatment with 10 mg/kg of MPH in absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data were expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate
The effect of various doses of TPM on MPH-induced decrease in total and phosphorylated-CREB by Western blot
The MPH-induced decreases in total and phosphorylated-CREB protein expression in the amygdala comparing to the negative control group (p < 0.001). There was a higher total and phosphorylated-CREB protein expression in amygdala of TPM (100 mg/kg) treated group in comparison to the MPH (10 mg/kg) only treated group (p < 0.001) (Figs. 14, 15).

The alterations of expression (WB) of total form of CREB in amygdala in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data are expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate, WB western blotting

The alterations of expression (WB) of phosphorylated form of CREB (P-CREB) in amygdala in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data are expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate, WB western blotting
The effect of various doses of TPM on MPH-induced decrease in BDNF by Western blot
The MPH-induced decreases in BDNF protein expression in the amygdala comparing to the negative control group (p < 0.001). There was a higher BDNF protein expression in amygdala of TPM (100 mg/kg) treated group in comparison to the MPH (10 mg/kg) only treated group (p < 0.001) (Fig. 16).

The alterations of expression (WB) of BDNF in amygdala in control and groups under treatment with 10 mg/kg of MPH in the absence and presence of TPM (10, 30, 50, 70, and 100 mg/kg). All data are expressed as Mean ± SEM (n = 8). *** Shows significant difference from MPH only treated group (p < 0.001). ### Shows significant difference from negative control group (p < 0.001). MPH methylphenidate, TPM topiramate, WB western blotting
Discussion
This study showed that treatments by various doses of TPM (10, 30, 50, 70, and 100 mg/kg) can reduce chronic MPH-induced oxidative stress and inflammation in the isolated amygdala of adult rats. Our study demonstrated that chronic MPH (10 mg/kg) abuse could induce anxiety like behavior in EPM and can alter biomarkers of oxidative stress in isolated amygdala. MPH can induce MDA, as marker of lipid peroxidation, increase GSSG content and TNF-α and IL-1β levels and could reduce GSH content, GPx, GR and SOD activities in the isolated amygdala of rats, While TPM in all mentioned doses in MPH-treated rats decreased anxiety like behavior in EPM and decreased MDA level, GSSG content, TNF-α and IL-1β levels, and also increased GSH levels, GPx, GR and SOD activities in the isolated amygdala of rats. MPH in 10 mg/kg can inhibit CREB and BDNF protein expression and alter their up and down stream signaling pathways, while TPM in all mentioned doses inhibit this effect of MPH and activate CREB and BDNF gene and protein expression and inhibit the effects of MPH on up and down stream signaling pathways of CREB. MPH is an amphetamine-like neural stimulant which inhibits the reuptake of dopamine and norepinephrine into presynaptic terminals, and it is structurally similar to amphetamines and cocaine, having high potential for abuse and addiction (Klein-Schwartz 2002; Huss and Lehmkuhl 2001). The results of our study showed that rats treated with MPH (10 mg/kg) spent less time in open arms and had fewer numbers of open arm entries in EPM in comparison to the control group; in addition, the treatment of animals with TPM especially by doses of 70 and 100 mg/kg increased time spent and numbers of entries to open arms in MPH (10 mg/kg) treated group. The baseline measurements of all the animals were not significantly different between groups and we can assume that there was no difference in behavioral parameters between animals before treatments. Administration of MPH or MPH in combination with TPM caused the behavioral changes in EPM, and we can conclude that these behavioral changes were due to drug treatments. Previous studies have demonstrated that amphetamine and MPH cause anxiety and motor activity disorder in juvenile rats (Vendruscolo et al. 2008; Davids et al. 2002). These studies showed that non-pharmacologic doses of MPH induce neurobehavioral alterations and anxiety-like behavior in the rat brain (Vendruscolo et al. 2008). Previous studies showed that TPM have anxiolytic and antidepressant effects and can diminish anxiety and depressive behavior in rodents (Khan and Liberzon 2004; Molina-Hernández et al. 2010; Mula et al. 2007; Cagetti et al. 2004). Some of these results showed that TPM can modulate both anxiety and depression of alcohol and amphetamine withdrawal syndrome (Cagetti et al. 2004). The results of our study showed that MPH (10 mg/kg) increased MDA in isolated amygdala. This was consistent with previous reports showing that MPH causes lipid peroxidation and protein damage in young rat brain (Martins et al. 2006). In addition, our results suggested that TPM in used doses could inhibit this effect of MPH and decrease the MDA level. Previous studies confirmed these results and showed that TPM can decrease lipid peroxidation in animal treated by pentylenetetetrazol (PTZ) or in cell treated by high glucose (Gibbs et al. 2006; Price et al. 2011).
In this study, chronic administration of 10 mg/kg of MPH decreased GSH and increased GSSG levels in the isolated amygdala. We could confirm our results referring to previous studies which show MPH and other compounds similar to amphetamines, induce neurotoxicity by converting glutathione from its reduced protective form (GSH) to its oxidized damaging form (GSSG), and by this effect, reduced the scavenging capacity of glutathione (Frey et al. 2006a, b; Motaghinejad et al. 2015b). However, our data showed that different doses of TPM could increase GSH content and decrease GSSG levels in MPH treated animals. In fact, TMP neutralizes the negative effects of MPH on the glutathione cycle. This results confirmed by previous findings which showed TPM positive effect on glutathione circle and inhibition of free radicals (Agarwal et al. 2011; Demirci et al. 2013).
In our study, the treatment of animals with MPH (10 mg/kg) decreased GPx, GR, and SOD activities in isolated amygdale. Recent studies have shown that MPH decreases antioxidant enzyme activity in some brain regions (Lau et al. 2000). Some previous studies have suggested that methylphenidate could decrease the production of antioxidant enzymes and cause DNA damage (Yano and Steiner 2007; Andreazza et al. 2007). In addition, previous studies have shown that chronic administration of MPH in adult and juvenile rats causes mitochondrial dysfunction, respiratory enzyme changes and decrease in antioxidant capacity in brain cells. They have also suggested that MPH can induce oxidative stress and start the beginning of programmed cell death processes in rat brain (Lau et al. 2000; Martins et al. 2006; Fagundes et al. 2007). Results of our study indicated that TPM in the mentioned doses could prevent the MPH-induced decrease in antioxidant enzymes and increase in GPx, GR, and SOD activities. Previous studies showed that TPM significantly increases SOD, CAT, and GPx activities. It also significantly decreases lipid peroxidation in experimental epilepsy models and are capable of modulating the oxidant-antioxidant system (Demirci et al. 2013; Yürekli and Nazıroğlu 2013). According to previous studies, GR is the main enzyme responsible for the conversion of glutathione from oxidized (GSH) to reduced form (GSSG), and TPM increases the conversion of GSSG to GSH by activating GR. Thus, in our studies, TPM protects the amygdale against MPH-induced oxidative stress by increase in GSH level (Nazıroğlu et al. 2009). In other words, topiramate can decrease oxidized form (harmful form) of glutathione and convert it to the reduced form (protective form) by activating GR and GPx (Cardenas-Rodriguez et al. 2013). Another study has shown that TPM has direct antioxidant activity, by increase of SOD, against H2O2, and some parts of TPM antioxidant activity are mediated by scavenging free radicals (Cardenas-Rodriguez et al. 2013; Armaǧan et al. 2008). The participation of oxidative stress in subjects with neuro-stimulant abuse has been established (Lau et al. 2000). Our study showed that MPH, as a neurostimulant with high abuse potential, could increase this type of oxidative damage and that TPM, as an antiepileptic drug with neuroprotective effects, can modulate these types of side effects of MPH.
Our study also showed that MPH at the dose of 10 mg/kg increased the inflammatory markers in the amygdala. Previous studies confirmed our findings and have shown that MPH induces the dopamine neuron loss and activation of microglia and cause increase of proinflammatory markers, such as TNF-α and IL-1β, and these results showed that activation of neuroinflammation is responsible for the neurodegenerative effects of long-term MPH administration (Kuczenski and Segal 2001; Yamamoto and Raudensky 2008; Sadasivan et al. 2012). Our study indicated that TPM at different doses significantly decreased the inflammatory markers in MPH treated rats. Some studies have shown that TPM has protective effects against inflammation by enhancement of GABA; these studies have suggested that TPM can attenuate TNF-α and TGF-β1 and also decrease inflammation and injury of the kidney and liver (Koçer et al. 2009; Armaǧan et al. 2008). Our study has also shown that MPH can inhibit gene and protein expression of CREB (total and phosphorylated) and its product, BDNF, in the amygdala, while TPM at doses of 70 and 100 mg/kg can activate phosphorylated form of CREB and BDNF expression in the amygdala. On the other hand, TPM at doses of 70 and 100 mg/kg increases CREB and BDNF expression at gene and protein levels and can probably cause neuroprotection by activating neurotrophic factors.
The protective role of CREB, as a transcription factor, has been established by many previous studies, and this transcription factor can affect DNA and cause production of BDNF and c-Fos and, by this mechanism, can initiate its protective role. CREB/BDNF protect brain cells against neurodegeneration (Dworkin and Mantamadiotis 2010; Almeida et al. 2005). It has been shown by many previous works that amphetamine abuse decreases CREB and BDNF expression and through this mechanism, disrupts cell survival and triggers neurodegeneration (Guo et al. 2011). Furthermore, the neuroprotective effect of TPM has been established in many previous studies, and based on our results, we believe that TPM can mediate its protective properties by activating the CERB/BDNF pathway (Pandey et al. 2005; Pinheiro et al. 2015).
Our data indicate that MPH can decrease Ak1, MAPK3, PKA, and cFOS levels while increasing CAMK4 at the gene level. On the other hand, our data showed that TPM (70 and 100 mg/kg) can increase Ak1, MAPK3, PKA, and cFOS levels while decreasing CAMK4 at the gene level. Based on these data, we can conclude that inhibition of CREB phosphorylation, by MPH administration, could be done by either of Ak1, MAPK3, and PKA. TPM can remove/prevent this inhibition. Many previous studies showed that influx of exorbitance Ca2+ in neural cells can be responsible for some neurodegenerative effects of methamphetamine type stimulants and in other neurodegenerative disorders (Arundine and Tymianski 2003; Mattson 2007). On the other hand, Ca2+-mediated events occur when the released Ca2+ binds to and activates the regulatory protein calmodulin and cause CAMK4 formation, our data showed that MPH can cause increase in expression of CAMK4, and it can be suggested that Ca2+ influx may be involved in increase of oxidative stress and inflammation. In addition, TPM (50 and 100 mg/kg) could decrease CAMK4 expression and possibly inhibit the degeneration caused by exorbitance Ca2+ influx. However, we have tried to show the involvement of the kinase enzymes (Ak1 or MAPK3, and PKA) in CREB phosphorylation to evaluate the role of CREB and its product, BDNF, in TPM neuroprotection against MPH-induced neurodegeneration. TPM, 50 and 100 mg/kg, in the absence of MPH, can also increase Ak1, MAPK3, and PKA, while decreasing CAMK4 at the gene level, and this mechanism increases phosphorylated CERB/BDNF expression at gene and protein levels and activate CERB/BDNF signaling. Based on our data, we can suggest that CERB/BDNF can be one of the important signaling pathways involved in TPM neuroprotection.
Conclusion
The results of this study supported the hypothesis that TPM might be beneficial against MPH-induced oxidative stress and inflammation in rat amygdala and could be applied for the treatment of patients abusing MPH and suffering from its neurodegenerative effects. We suggested that topiramate was useful in the management of problems associated with the use of MPH which was mediated by phosphorylated form of CREB and BDNF protein expression, and it is up and down stream pathways, but further studies were required with human subjects.
References
Agarwal NB, Agarwal NK, Mediratta PK, Sharma KK (2011) Effect of lamotrigine, oxcarbazepine and topiramate on cognitive functions and oxidative stress in PTZ-kindled mice. Seizure 20(3):257–262
Aguiar AS, Castro AA, Moreira EL, Glaser V, Santos AR, Tasca CI, Latini A, Prediger RD (2011) Short bouts of mild-intensity physical exercise improve spatial learning and memory in aging rats: involvement of hippocampal plasticity via AKT, CREB and BDNF signaling. Mech Ageing Dev 132(11):560–567
Almeida R, Manadas B, Melo C, Gomes J, Mendes C, Graos M, Carvalho R, Carvalho A, Duarte C (2005) Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ 12(10):1329–1343
Andreazza AC, Frey BN, Valvassori SS, Zanotto C, Gomes KM, Comim CM, Cassini C, Stertz L, Ribeiro LC, Quevedo J (2007) DNA damage in rats after treatment with methylphenidate. Prog Neuropsychopharmacol Biol Psychiatry 31(6):1282–1288
Armaǧan A, Kutluhan S, Yılmaz M, Yılmaz N, Bülbül M, Vural H, Soyupek S, Nazıroǧlu M (2008) Topiramate and vitamin E modulate antioxidant enzyme activities, nitric oxide and lipid peroxidation levels in pentylenetetrazol-induced nephrotoxicity in rats. Basic Clin Pharmacol Toxicol 103(2):166–170
Arnone D (2005) Review of the use of topiramate for treatment of psychiatric disorders. Ann Gen Psychiatry 4(1):5–15
Arundine M, Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34(4):325–337
Babcock Q, Byrne T (2000) Student perceptions of methylphenidate abuse at a public liberal arts college. J Am Coll Health 49(3):143–145
Barrett SP, Pihl RO (2002) Oral methylphenidate-alcohol co-abuse. J Clin Psychopharmacol 22(6):633–634
Blendy JA (2006) The role of CREB in depression and antidepressant treatment. Biol Psychiatry 59(12):1144–1150
Cagetti E, Baicy KJ, Olsen RW (2004) Topiramate attenuates withdrawal signs after chronic intermittent ethanol in rats. NeuroReport 15(1):207–210
Cardenas-Rodriguez N, Coballase-Urrutia E, Huerta-Gertrudis B, García-Cruz M, Pedraza-Chaverri J, Coria-Jiménez R, Bandala C, Ruíz-García M (2013) Antioxidant activity of topiramate: an antiepileptic agent. Neurological Sciences 34(5):741–747
Carlezon WA, Duman RS, Nestler EJ (2005) The many faces of CREB. Trends Neurosci 28(8):436–445
Challman TD, Lipsky JJ (2000) Methylphenidate: its pharmacology and uses. In: Mayo Clinic Proceedings, Elsevier, pp 711–721
Davids E, Zhang K, Tarazi FI, Baldessarini RJ (2002) Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning. Psychopharmacology 160(1):92–98
Demirci S, Kutluhan S, Nazıroğlu M, Uğuz AC, Yürekli VA, Demirci K (2013) Effects of selenium and topiramate on cytosolic Ca2 + influx and oxidative stress in neuronal PC12 cells. Neurochem Res 38(1):90–97
Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ (2011) Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Science translational medicine 3(96):96ra76
Dworkin S, Mantamadiotis T (2010) Targeting CREB signalling in neurogenesis. Expert Opin Ther Targets 14(8):869–879
Fagundes AO, Rezin GT, Zanette F, Grandi E, Assis LC, Dal-Pizzol F, Quevedo J, Streck EL (2007) Chronic administration of methylphenidate activates mitochondrial respiratory chain in brain of young rats. Int J Dev Neurosci 25(1):47–51
Frey BN, Martins MR, Petronilho FC, Dal-Pizzol F, Quevedo J, Kapczinski F (2006a) Increased oxidative stress after repeated amphetamine exposure: possible relevance as a model of mania. Bipolar Disord 8(3):275–280
Frey BN, Valvassori SS, Gomes KM, Martins MR, Dal-Pizzol F, Kapczinski F, Quevedo J (2006b) Increased oxidative stress in submitochondrial particles after chronic amphetamine exposure. Brain Res 1097(1):224–229
Garnett WR (2000) Clinical pharmacology of topiramate: a review. Epilepsia 41(s1):61–65
Gibbs JE, Walker MC, Cock HR (2006) Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia 47(3):469–478
Gomes KM, Comim CM, Valvassori SS, Réus GZ, Inácio CG, Martins MR, Souza RP, Quevedo J (2010) Diurnal differences in memory and learning in young and adult rats treated with methylphenidate. J Neural Transm 117(4):457–462
Guo W, Crossey EL, Zhang L, Zucca S, George OL, Valenzuela CF, Zhao X (2011) Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS One 6(5):e19351
Huss M, Lehmkuhl U (2001) Methylphenidate and substance abuse: a review of pharmacology, animal, and clinical studies. J Atten Disord 6:S65–S71
Jones Z, Dafny N (2014) Acute and chronic dose–response effect of methylphenidate on ventral tegmental area neurons correlated with animal behavior. J Neural Transm 121(3):327–345
Khan S, Liberzon I (2004) Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology 172(2):225–229
Kitagawa K (2007) CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J 274(13):3210–3217
Klein-Schwartz W (2002) Abuse and toxicity of methylphenidate. Curr Opin Pediatr 14(2):219–223
Koçer A, Memişoğullari R, Domaç FM, Ilhan A, Koçer E, Okuyucu Ş, Özdemir B, Yüksel H (2009) IL-6 levels in migraine patients receiving topiramate. Pain Pract 9(5):375–379
Kuczenski R, Segal DS (2001) Locomotor effects of acute and repeated threshold doses of amphetamine and methylphenidate: relative roles of dopamine and norepinephrine. J Pharmacol Exp Ther 296(3):876–883
Kudin AP, Debska-Vielhaber G, Vielhaber S, Elger CE, Kunz WS (2004) The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia 45(12):1478–1487
Kutluhan S, Nazıroğlu M, Çelik Ö, Yılmaz M (2009) Effects of selenium and topiramate on lipid peroxidation and antioxidant vitamin levels in blood of pentylentetrazol-induced epileptic rats. Biol Trace Elem Res 129(1–3):181–189
Lau JW, Senok S, Stadlin A (2000) Methamphetamine-induced oxidative stress in cultured mouse astrocytes. Ann N Y Acad Sci 914(1):146–156
Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K (2005) Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci 25(5):1137–1148
Mao X-Y, Cao Y-G, Ji Z, Zhou H-H, Liu Z-Q, Sun H-L (2015) Topiramate protects against glutamate excitotoxicity via activating BDNF/TrkB-dependent ERK pathway in rodent hippocampal neurons. Prog Neuropsychopharmacol Biol Psychiatry 60:11–17
Martins MR, Reinke A, Petronilho FC, Gomes KM, Dal-Pizzol F, Quevedo J (2006) Methylphenidate treatment induces oxidative stress in young rat brain. Brain Res 1078(1):189–197
Mattson MP (2007) Calcium and neurodegeneration. Aging Cell 6(3):337–350
McCool BA, Botting SK (2000) Characterization of strychnine-sensitive glycine receptors in acutely isolated adult rat basolateral amygdala neurons. Brain Res 859(2):341–351
Molina-Hernández M, Téllez-Alcántara NP, Olivera-Lopez JI, Jaramillo MT (2010) Antidepressant-like or anxiolytic-like actions of topiramate alone or co-administered with intra-lateral septal infusions of neuropeptide Y in male Wistar rats. Peptides 31(6):1184–1189
Motaghinejad M, Motevalian M (2016) Involvement of AMPA/kainate and GABA A receptors in topiramate neuroprotective effects against methylphenidate abuse sequels involving oxidative stress and inflammation in rat isolated hippocampus. Eur J Pharmacol 784:181–191
Motaghinejad M, Karimian M, Motaghinejad O, Shabab B, Yazdani I, Fatima S (2015a) Protective effects of various dosage of Curcumin against morphine induced apoptosis and oxidative stress in rat isolated hippocampus. Pharmacological Reports 67(2):230–235
Motaghinejad M, Karimian SM, Motaghinejad O, Shabab B, Asadighaleni M, Fatima S (2015b) The effect of various morphine weaning regimens on the sequelae of opioid tolerance involving physical dependency, anxiety and hippocampus cell neurodegeneration in rats. Fundam Clin Pharmacol 29(3):299–309
Motaghinejad M, Motevalian M, Ebrahimzadeh A (2015c) Reduction of methylphenidate induced anxiety, depression and cognition impairment by various doses of venlafaxine in rat. Int J Prev Med 4:52–58
Motaghinejad M, Motevalian M, Larijani SF, Khajehamedi Z (2015d) Protective effects of forced exercise against methylphenidate-induced anxiety, depression and cognition impairment in rat. Adv Biomed Res 21:134–137
Motaghinejad M, Motevalian M, Shabab B (2015e) Effects of chronic treatment with methylphenidate on oxidative stress and inflammation in hippocampus of adult rats. Neurosci Lett 619:106–113
Motaghinejad M, Motevalian M, Shabab B (2015f) Neuroprotective effects of various doses of topiramate against methylphenidate induced oxidative stress and inflammation in rat isolated hippocampus. Clin Exp Pharmacol Physiol 43:360–371
Mula M, Pini S, Cassano GB (2007) The role of anticonvulsant drugs in anxiety disorders: a critical review of the evidence. J Clin Psychopharmacol 27(3):263–272
Nazıroğlu M, Kutluhan S, Uğuz AC, Çelik Ö, Bal R, Butterworth PJ (2009) Topiramate and vitamin E modulate the electroencephalographic records, brain microsomal and blood antioxidant redox system in pentylentetrazol-induced seizure of rats. J Membr Biol 229(3):131–140
Pandey SC, Chartoff EH, Carlezon WA, Zou J, Zhang H, Kreibich AS, Blendy JA, Crews FT (2005) CREB gene transcription factors: role in molecular mechanisms of alcohol and drug addiction. Alcohol Clin Exp Res 29(2):176–184
Patrick KS, Markowitz JS (1997) Pharmacology of methylphenidate, amphetamine enantiomers and pemoline in attention-deficit hyperactivity disorder. Hum Psychopharmacol Clin Exp 12(6):527–546
Peng J, Sarkar S, Chang SL (2012) Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR. Drug Alcohol Depend 124(3):223–228
Pinheiro RMC, de Lima MNM, Portal BCD, Busato SB, Falavigna L, Ferreira RDP, Paz AC, de Aguiar BW, Kapczinski F, Schröder N (2015) Long-lasting recognition memory impairment and alterations in brain levels of cytokines and BDNF induced by maternal deprivation: effects of valproic acid and topiramate. J Neural Transm 122(5):709–719
Price TO, Eranki V, Banks WA, Ercal N, Shah GN (2011) Topiramate treatment protects blood-brain barrier pericytes from hyperglycemia-induced oxidative damage in diabetic mice. Endocrinology 153(1):362–372
Réus GZ, Stringari RB, Ribeiro KF, Ferraro AK, Vitto MF, Cesconetto P, Souza CT, Quevedo J (2011) Ketamine plus imipramine treatment induces antidepressant-like behavior and increases CREB and BDNF protein levels and PKA and PKC phosphorylation in rat brain. Behav Brain Res 221(1):166–171
Sadasivan S, Pond BB, Pani AK, Qu C, Jiao Y, Smeyne RJ (2012) Methylphenidate exposure induces dopamine neuron loss and activation of microglia in the basal ganglia of mice. PLoS One 7(3):e33693
Schwartz K, Weizman A, Rehavi M (2006) The effect of psychostimulants on [3H] dopamine uptake and release in rat brain synaptic vesicles. J Neural Transm 113(9):1347–1352
Shaldubina A, Einat H, Szechtman H, Shimon H, Belmaker R (2002) Preliminary evaluation of oral anticonvulsant treatment in the quinpirole model of bipolar disorder. J Neural Transm 109(3):433–440
Shamoto-Nagai M, Maruyama W, Hashizume Y, Yoshida M, Osawa T, Riederer P, Naoi M (2007) In parkinsonian substantia nigra, α-synuclein is modified by acrolein, a lipid-peroxidation product, and accumulates in the dopamine neurons with inhibition of proteasome activity. J Neural Transm 114(12):1559–1567
Shi X-Y, Wang J-W, Cui H, Li B-M, Lei G-F, Sun R-P (2010) Effects of antiepileptic drugs on mRNA levels of BDNF and NT-3 and cell neogenesis in the developing rat brain. Brain Dev 32(3):229–235
Tagaya H (2010) Methylphenidate: pharmacology, indication and potential of abuse. Nihon rinsho Jpn J Clin Med 68(8):1550–1555
Trinh T, Kohllepel S, Yang P, Burau K, Dafny N (2013) Adult female rats’ altered diurnal locomotor activity pattern following chronic methylphenidate treatment. J Neural Transm 120(12):1717–1731
Vendruscolo LF, Izídio GS, Takahashi RN, Ramos A (2008) Chronic methylphenidate treatment during adolescence increases anxiety-related behaviors and ethanol drinking in adult spontaneously hypertensive rats. Behav Pharmacol 19(1):21–27
Williams RJ, Goodale LA, Shay-Fiddler MA, Gloster SP, Chang SY (2004) Methylphenidate and dextroamphetamine abuse in substance-abusing adolescents. Am J Addict 13(4):381–389
Woo NH, Teng HK, Siao C-J, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B (2005) Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci 8(8):1069–1077
Yamamoto BK, Raudensky J (2008) The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J Neuroimmune Pharmacol 3(4):203–217
Yang J, Shen J (2009) Elevated endogenous GABA concentration attenuates glutamate–glutamine cycling between neurons and astroglia. J Neural Transm 116(3):291–300
Yano M, Steiner H (2007) Methylphenidate and cocaine: the same effects on gene regulation? Trends Pharmacol Sci 28(11):588–596
Yürekli VA, Nazıroğlu M (2013) Selenium and topiramate attenuates blood oxidative toxicity in patients with Epilepsy: a clinical pilot study. Biol Trace Elem Res 152(2):180–186
Acknowledgments
We thank head of Razi drug research center in Iran University of medical sciences for providing helpful facilities for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Motaghinejad, M., Motevalian, M., Falak, R. et al. Neuroprotective effects of various doses of topiramate against methylphenidate-induced oxidative stress and inflammation in isolated rat amygdala: the possible role of CREB/BDNF signaling pathway. J Neural Transm 123, 1463–1477 (2016). https://doi.org/10.1007/s00702-016-1619-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-016-1619-1