
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative brain disorder that leads to a progressive decline in a person’s memory and ability to communicate and carry out daily activities. The brain pathology in AD is characterized by extensive neuronal loss, particularly of cholinergic neurons, intracellular neurofibrillary tangles composed of the tau protein (NFTs) and extracellular deposition of plaques composed of β-amyloid (Aβ), a cleavage product of the amyloid precursor protein (APP). These two insoluble protein aggregates are accompanied by a chronic inflammatory response and extensive oxidative damage. Whereas dys-regulation of APP expression or processing appears to be important for the familial, early-onset form of AD, controversy exists between the “Baptists” (in favour of Aβ) and the “Tauists” (in favour of tau) as to which of these two protein dysfunctions occur at the earliest stages or are the most important contributors to the disease process in sporadic AD. However, more and more “non-amyloid” and “non-tau” causes have been proposed, including, glycation, inflammation, oxidative stress and dys-regulation of the cell cycle. However, to get an insight into the ultimate cause of AD, and to prove that any drug target is valuable in AD, disease-relevant models giving insight into the pathogenic processes in AD are urgently needed. In the absence of a good animal model for sporadic AD, we propose in this review that induced pluripotent stem cells, derived from dermal fibroblasts of AD patients, and differentiated into cholinergic neurons, might be a promising novel tool for disease modelling and drug discovery for the sporadic form of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Alzheimer’s disease
Alzheimer’s disease and the urgent need for early diagnosis and specific treatments
Alzheimer’s disease (AD) is a progressive neurodegenerative brain disorder that leads to progressive decline in a person’s memory and ability to learn, make judgments, communicate and carry out daily activities. In the course of the disease, episodic memory is affected early, caused by neuronal dysfunction and cell death in the hippocampus and other medial temporal structures. As the disease progresses further, neurons also die in other cortical regions of the brain (Arendt 2009). At that stage, sufferers develop abnormalities in a range of cognitive domains as well as neuropsychiatric symptoms such as apathy, agitation or psychotic symptoms (Aalten et al. 2008). Patients with AD not only suffer emotionally and physically, but also represent a significant financial and emotional burden for caregivers, society and the community. Thus, it is becoming increasingly important to find the initial cause(s) of AD.
Induced pluripotent stem cell (iPSC) technology, whereby a patient’s somatic cells can be reprogrammed to a pluripotent state by the forced expression of a defined set of transcription factors, may offer a way forward to the development of novel personalized neuroprotective therapies that prevent AD (Huber et al. 2006; Holmquist et al. 2007; Maczurek et al. 2008). In addition, a growing focus is on biomarkers which enable detection of the disease in its early stages and will allow for preventative treatment (Song et al. 2009).
Histopathology of Alzheimer’s disease: key to pathogenesis and therapy?
The brain pathology in AD that is associated with cognitive decline and profound dementia is characterized by extensive neuronal loss, particularly of cholinergic neurons, intracellular neurofibrillary tangles (NFTs) and extracellular deposition of β-amyloid (Aβ) plaques (Braak and Del Tredici 2004; Thal et al. 2006). These two insoluble protein aggregates accumulate in susceptible regions of the brain and are accompanied by a chronic inflammatory response and extensive oxidative damage (Sastre et al. 2006; Weisman et al. 2006; Fuller et al. 2010).
Senile plaques, composed of crosslinked (e.g. by glycation or oxidation) β-amyloid (Aβ) peptide, are present in specific brain regions of AD patients (Loske et al. 2000; Thal et al. 2002). Aβ has been proposed to have a variety of toxic properties such as the ability to block communication between neurons, to cause degeneration of neurites, to contribute to oxidative stress and ultimately to lead to neuronal cell death (Kuhla et al. 2004). In addition, Aβ causes inflammation, as evidenced by the activation of the inflammatory cells of the brain, microglia and astroglia, mainly around the amyloid plaques (Wong et al. 2001).
The second protein aggregation problem in AD is largely intracellular, and results from the deposition of neurofibrillary tangles in neurons. These tangles are mainly composed of the cytoskeletal protein tau, and it has been suggested that hyper-phosphorylation and glycation contribute to their insolubility (Chen et al. 2004). In the CNS, tau is found in greatest abundance in neurons, where it stabilizes microtubules and is therefore key to maintenance of axonal integrity. Hyper-phosphorylation leads to “neurofibrillary tangles”, destabilization of the cytoskeleton, axonal degeneration and eventually neuronal cell death (Goedert et al. 1995a, b; Gotz et al. 2010). Recent studies suggest, that tauopathy in sporadic Alzheimer’s disease may begin in the third decade and possibly starts in the lower brainstem rather than in the transentorhinal region (Braak et al. 2011).
Considerable controversy still continues amongst the “Baptists”, and the “Tauists” (favouring either amyloid or tau as the major contributor to the disease, respectively) as to which of these two protein dysfunctions occurs at the earliest stages or are the most important contributors to the disease process. In addition, more and more “non-amyloid” and “non-tau” causes have been proposed, including disturbances in the insulin/insulin receptor glycation, inflammation, oxidative stress and dys-regulation of the cell cycle. Detailed descriptions of these “non-amyloid” and “non-tau” causes have been published in extensive reviews by the group of Peter Riederer and his collaborators (Thome et al. 1996; Münch et al. 1997, 1998; Retz et al. 1998; Riederer and Hoyer 2006; Arendt et al. 2010; Rahmadi et al. 2011; Srikanth et al. 2011).
Anti-amyloid drugs
The most favoured hypothesis about the cause of AD is the ‘amyloid cascade hypothesis’. This hypothesis states that the aberrant production, aggregation and deposition of Aβ is the causative process in the pathogenesis of both familial and sporadic (late-onset) AD (FAD and LOAD) (Iwatsubo et al. 1994; Karran et al. 2011).
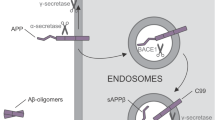
Aβ is a proteolytic fragment of the amyloid precursor protein (APP). In the amyloidogenic pathway, APP is first cleaved by β-secretase (BACE1) to generate a slightly shorter N-terminal ectodomain, APPs-β (Borchelt et al. 1996; Scheuner et al. 1996; Tomita et al. 1997). This is then cleaved by γ-secretase within the transmembrane domain to generate Aβ peptides of 40- and 42 amino acids in length (Gotz et al. 2010) .
The view that Aβ plays a central role in AD pathogenesis has developed from observations that patients with mutations in the APP and presenilin (PSEN) genes show both accelerated plaque deposition and the onset of dementia at an early age, and that all these patients demonstrate an increase in the production of Aβ, particularly the longer and more aggregation prone Aβ 1-42 (Butterfield et al. 2002).
Based on the “amyloid cascade hypothesis”, there was great hope to find a cure for AD by lowering the concentration of Aβ using a variety of different therapeutic approaches. Various “anti-amyloid drugs” targeting different pathways of Aβ42 production and/or aggregation have been developed and tested in clinical trials with AD patients. Their mechanisms of action include:
-
Inhibiting the enzymatic actions of the secretases with β- and γ-secretase inhibitors, thereby lowering the production of Aβ (Imbimbo and Giardina 2011).
-
Changing the action of the γ-secretase and changing Aβ production from β1-42 to shorter amyloid chain lengths using amyloid modulators (Czirr and Weggen 2006).
-
Eliciting an anti-Aβ antibody response (active immunization) or providing recombinant Aβ antibodies (passive immunization), both leading to amyloid removal by the immune system (Münch and Robinson 2002; Robinson et al. 2004; Dasilva et al. 2006; Panza et al. 2010).
-
Metal chelators which dissipate Aβ plaque deposits by chelating divalent metal ions (Fe2+, Cu2+ and Zn2+). A current example is clioquinol, with impressive effects in transgenic, APP overexpressing animal models and interesting results in phase I and II clinical trials (Bush 2002; Faux et al. 2010; Adlard et al. 2011; Bareggi and Cornelli 2012).
Unfortunately, all “anti-amyloid drug” candidates have so far failed to produce the expected therapeutic breakthroughs. They did, however, succeed in their effort to lower amyloid production and/or to remove amyloid plaques, but the cognitive decline in the treated patients did not slow down (Wan et al. 2009; Smith 2010; Castellani and Smith 2011).
These results suggest that Aβ might not be the dominant cause of sporadic AD (at least in some patients), and the “anti-amyloid approach” may not be an effective treatment for AD by itself, or may need to be given prophylactically (Golde et al. 2011).
Anti-tangle drugs
Tau, the protein component of the neurofibrillary tangles, is a microtubule-associated protein. It is proposed that the neurofibrillary tangles are formed as a result of abnormal hyper-phosphorylation, caused by an imbalance of kinase and phosphatase activities (Goedert et al. 1995a, b). A variety of kinases involved in the hyper-phosphorylation of tau have been described. These include glycogen synthase kinase-3 (GSK-3), mitogen-activated protein kinase (MAP)-kinase and microtubule affinity-regulating kinase 1 (MARK1). In addition, an insufficient activity of protein phosphatases, especially protein phosphatase 2A (PP2A), has also been suggested to be responsible for hyper-phosphorylation of tau. Another approach to the inhibition of tangle formation might be the inhibition of tau crosslinking, e.g. by advanced glycation endproducts by the use of anti-glycation agents (Kuhla et al. 2007; Krautwald and Münch 2010; Rahmadi et al. 2011). Furthermore, it has been suggested that microtubule (MT)-stabilizing drugs such as epothilone D (EpoD), which improve in existing tau pathology and related behavioural deficits in aged PS19 mice, might hold promise for the treatment of AD and related tauopathies.
Inhibitors of kinases and activators of phosphatases are the main classes of “anti-tangle” drugs (Navarrete et al. 2011). Two “anti-tangle” drugs, the GSK-3 inhibitors Tideglusib (Noscira, Spain) and the tau aggregation inhibitor methylthioninium chloride, Rember (TauRx Therapeutics, Singapore), have been tested in phase II clinical trials with some positive results, but the results of large phase II trials are still outstanding.
However, with growing uncertainty of the therapeutic potential of drugs targeting amyloid and tau, other novel therapies have recently been proposed, including those targeting glycation, oxidative stress and inflammation (Retz et al. 1998; Maczurek et al. 2008; Rahmadi et al. 2011; Srikanth et al. 2011).
However, to get an insight into the ultimate cause of AD, and to prove that any drug target is valuable in AD, disease-relevant models giving insight into the pathogenic processes in AD are urgently needed. In the absence of a good animal model for sporadic AD, we propose in this review that induced pluripotent stem cells (iPSCs), derived from dermal fibroblasts of AD patients, and differentiated into cholinergic neurons, might be a promising novel tool for disease modelling and drug discovery particularly for the sporadic form of AD. In the following sections, we will introduce the concept of iPSCs and review recently published studies in which these cells were used for AD disease modelling, and yielded interesting, and sometimes unexpected results.
Induced pluripotent stem cells (iPSCs) as a key to disease pathogenesis and drug discovery
Introduction to induced pluripotent stem cells (iPSCs)
The discovery of induced pluripotent stem cells (iPSCs), whereby a patient’s somatic cells can be reprogrammed to a pluripotent state by the forced expression of a defined set of transcription factors, has the potential to enable in vitro disease modelling and be used for drug discovery programs. In 2006, it was demonstrated that retroviral-mediated introduction of four transcription factors into mouse fibroblasts could convert them into cells closely resembling pluripotent embryonic stem cells (ESCs) (Takahashi and Yamanaka 2006). In that study, Yamanaka and his group found that the introduction of a combination of four transcription factors—octamer-binding protein 4 (also known as Pou5f1), Sox2, Krüppel-like factor 4 (Klf4) and c-Myc—into mouse fibroblasts was sufficient to induce the expression of endogenous pluripotency genes and thus reprogram the somatic cells to a new state with colony morphology, cell morphology, growth characteristics, gene expression and antigen expression similar to mouse ESCs. This stunning discovery was quickly replicated using human somatic cells, with the Yamanaka and Daley groups employing essentially the same gene cocktail (Takahashi et al. 2007; Park et al. 2008) and the Thomson group using a slightly different one (i.e. OCT4, NANOG, SOX2 and LIN28) (Yu et al. 2007). Further evidence that these reprogrammed cells are pluripotent was achieved by demonstrating they are capable of germ-line transmission in chimeric mouse assays (Okita et al. 2007; Wernig et al. 2007). Importantly, this work also showed that use of c-Myc should be avoided in reprogramming gene cocktails, as reactivation of the exogenous c-Myc transgene can lead to tumour formation.
The ability to reprogram human somatic cells to a pluripotent state provides a means to generate large numbers of patient-specific differentiated cells for both research and transplantation. Equally important, this reprogramming technology also enables the production of disease-specific cells from confirmed patients with disorders without a clear pattern of inheritance (“sporadic” cases). Accordingly, our group and others have generated hiPSCs from patients with sporadic AD. These new hiPSC lines will enable investigation of the development and maintenance of cholinergic neurons in a context uniquely related to AD, with the potential for high-throughput chemical screening to identify lead compounds for AD treatment (Fig. 1). In our hands, colonies of AD-iPSCs were morphologically indistinguishable from control (Co) iPSC and hESCs (Fig. 1a). Further characterization revealed that AD-iPSCs were genetically and phenotypically indistinguishable from control hESC/hiPSCs. Hypomethylated OCT4 promoter regions indicate successful reprogramming of AD-Fibs (24.4 vs. 66.7 %, Fig. 1b). Using immunofluorescence staining, feeder-free cultures of AD-iPSCs typically expressed undifferentiated pluripotent markers OCT4, NANOG, SSEA4 and TRA160 (Fig. 1c). Quantitative gene expression analyses of pluripotency-associated genes were not significantly different across all pluripotent cell lines. In contrast, parental fibroblasts (AD-Fib) expressed extremely low levels of NANOG, OCT4, SOX2 and GDF3, but had similar levels of CMYC and KLF4 expression (Fig. 1d). Furthermore, extended feeder-free culture showed no chromosomal abnormalities in ALZ1/ALZ7 as determined by standard G-banding karyotypic analysis (Fig. 1e).

Characterization of Alzheimer’s disease specific hiPSCs (AD-iPSCs). a Colony morphologies of hiPSCs from a non-demented control (Co-iPSC) and a sporadic AD patient (AD-IPSC) under feeder-free conditions. Scale 500 μm. b OCT4 promoter DNA methylation analysis using bisulfite sequencing. Open squares unmethylated, closed squares methylated. c Immunofluorescence staining of typical undifferentiated Co-hiPSCs; nuclear/surface markers, OCT4, NANOG, TRA160, SSEA4. Scale 200 μm. d Gene expression analyses of pluripotency-related gene of a non-demented control (Co-iPSC), two sporadic AD patients (ALZ1 and ALZ 27), and the original AD fibroblast cultures (AD-Fib) using quantitative PCR. ***p < 0.0005. e Standard G-banding karyotypic analysis of AD-iPSCs after extended propagation under feeder-free conditions. Experiments were approved by the UNSW HREC, approval number 08021
Although hiPSCs share key morphological and molecular characteristics with human embryonic stem cells (O’Connor et al. 2011), genetic and epigenetic differences have been identified (Bock et al. 2011; Nishino et al. 2011; Wang et al. 2011).
For example, the reprogramming process and/or subsequent culture can result in random DNA alterations not present within the genome of the parent cell (Hayden 2011). Since random integration of the transgene might lead to clonal heterogeneity and possible functional diversity, it is recommended to validate findings from one hiPSC clone with multiple independently derived hiPSC clones from the same patient (Sidhu 2011).
Drug discovery for AD using iPSCs
Another potential challenge for both high-throughput drug screening and developmental investigations of Alzheimer’s-specific hiPSC cell lines is the production of large numbers of highly purified, mature cholinergic neurons. Encouragingly, methods for differentiating and purifying cholinergic neurons from hiPSC cultures have been published though it is presently unclear whether these neurons are, or can be induced to become, fully mature (Israel et al. 2012).
A further challenge for hiPSC-based investigation of neurological disorders is that these disorders manifest themselves within the complex 3-dimensional architecture of the brain. This architecture is difficult to reproduce in vitro, leading to some limitations with in vitro drug screening for novel neurological pharmaceuticals. However, careful design of the drug screening assay parameters and detection methods should enable identification and pre-clinical validation of new lead compounds that have the potential to at least delay, if not cure, progression of diseases such as Alzheimer’s disease (Grskovic et al. 2011).
Although iPSC technology seems promising, several safety obstacles need to be addressed before iPSCs transits to the clinic; in particular, the risk of insertional mutagenesis when using integrative viruses and the transmission of pathogens when in media and/or feeder layers containing animal products. While transgene/viral-free methods have been developed, a majority of iPSCs are still derived on animal feeder layers, which offsets the benefits of a xeno-free autologous transplantation. The use of animal feeder layers also introduces inconsistent and variable reprogramming outcomes, making the screening of suitable, fully reprogrammed iPSC colonies labour intensive (Chan et al. 2009). This is reflected in iPSC lines generated under different experimental conditions and show varying degrees of differentiation into hematopoietic and neural lineages (Feng et al. 2010; Hu et al. 2010), which also limits its therapeutic potential. The use of defined media, however, minimizes variable and/or inhibitory components present in serum and growth-factor secreting feeder cells. Consequently, some laboratories have generated iPSCs under feeder-free conditions by using extracellular matrices and serum-free media (Sun et al. 2009; Vallier et al. 2009a, b). While the phenotypic outcomes have been tested, transcriptomic characterization of feeder-free derived iPSCs has not been fully explored. The advent of whole transcript gene expression microarrays was capitalized on to identify underlying molecular events that may underpin the differences between feeder-derived, feeder-free derived iPSCs and human embryonic stem cells (hESCs) propagated under identical conditions. We for the first time reported that feeder-free iPSCs (ff-iPSCs) resemble hESCs more than feeder-derived iPSCs (f-iPSCs) in terms of overall gene expression patterns governing pluripotency and other biological functions. The processes related to pluripotent signature in hESCs (i.e. DNA replication and cell cycle) were substantially enriched in ff-iPSCs and expression of bivalent genes was lower (Chung et al. 2012). The advantages of a feeder-free defined system are such that homogenous populations of patient-specific pluripotent stem cells can be generated, batch-to-batch differences created by serum and feeder-cells can be eliminated and scale-up cultures can be easily carried out. This is of particular interest in regenerative medicine.
AD disease modelling with iPSCs: identification of differences in cell phenotype and specific, AD-related cellular processes
The key to modelling any human disease is the identification of a disease-specific unique cellular phenotype. The most successful examples of this strategy have used diseases that have strong genetic components and affect a highly defined cell type leading to a characteristic difference to an unaffected cell, e.g. for diseases with a known molecular mechanism, such as in spinal muscular atrophy, Hutchinson–Gilford Progeria syndrome, familial Parkinson’s disease or Down syndrome (Ebben et al. 2011; Malpass 2011; Jung et al. 2012; Shi et al. 2012).
Similarly, iPSCs have been created from patients with familial AD (FAD) characterized by mutations in APP, PSEN1 and PSEN2. For example, Yagi et al. (2011) generated iPSCs from fibroblasts of AD patients with mutations in PSEN1 (A246E) and PSEN2 (N141I), and characterized their subsequent differentiation into neurons. They found that FAD-iPSC-derived neurons showed increased Aβ42 secretion. Furthermore, secretion of Aβ42 from these neurons sharply responds to γ-secretase inhibitors and modulators, indicating the potential for identification and validation of amyloid-lowering drugs (Yagi et al. 2011; Yahata et al. 2011).
In a further study, it was shown that iPSC-derived neuronal cells express functional proteins involved in Aβ production, including amyloid precursor protein, β-secretase, and γ-secretase, and were capable of secreting Aβ into the conditioned media (Yahata et al. 2011). Although Aβ production was inhibited by β- and γ-secretase inhibitors and an NSAID, there were different susceptibilities to all three drugs between early and late differentiation stages (Yahata et al. 2011).
In another study, Israel et al. created iPSCs from two patients with familial AD, both caused by a duplication of the amyloid-precursor protein gene (APP; termed APPDp), two with sporadic AD (termed sAD1, sAD2) and two non-demented controls (Israel et al. 2012). They showed that relative to controls, iPSC-derived, purified neurons from the two APPDp patients and patient sAD2 exhibited significantly higher levels of the pathological markers Aβ(1–40), phospho-tau (Thr 231) and active glycogen synthase kinase-3β. Neurons from these patients also accumulated large RAB5-positive early endosomes compared to controls, indicating an impairment of autophagy. Interestingly, they also showed that treatment of purified neurons with β-secretase inhibitors, but not γ-secretase inhibitors, caused significant reductions in phospho-Tau (Thr 231) and GSK-3β levels (Israel et al. 2012).
Encouragingly, methods for differentiation and purifying cholinergic neurons from hiPSC cultures have been published though it is presently unclear whether these neurons are, or can be induced to become, fully mature. Israel et al. (2012) described in their manuscript that differentiated and purified neurons contained glutamatergic, GABAergic and cholinergic neuronal subtypes. However, the two manuscripts about AD-iPSCs disease modelling did not contain descriptions about validated differentiation methods for pure cultures of cholinergic neurons (Yagi et al. 2011; Israel et al. 2012). However, protocols published describing the differentiation of ESCs to cholinergic neurons might prove useful for the cholinergic differentiation of iPSCs (Bissonnette et al. 2011).
These two studies demonstrate that iPSC technology can be used to observe patient-specific phenotypes in vitro, which reflect both the familial and the sporadic forms of the disease in a remarkable manner.
AD disease modelling with iPSCs: mapping differences in gene and protein expression
Both Yagi et al. (2011) and Israel et al. (2012) used protocols that induce differentiation to multiple neuronal subtypes. However, in the early stages of AD there is a preferential loss of cholinergic neurons and their innervation of the hippocampus and neocortex (Schliebs and Arendt 2011).
One of the most interesting questions is whether the neurons derived from iPSCs of AD patients can be differentiated into cholinergic neurons, by activating specific intracellular signalling pathways including repressor element 1-silencing transcription factor (REST) and its corepressor (CoREST) (Ooi and Wood 2007). Furthermore, it will be interesting to find out if AD iPSCs are distinctly different from those from age and gender-matched healthy controls in terms of global expression of mRNA and protein.
The induction of pluripotency largely revert somatic cells to their embryonic or ‘ground’ state. Using a developmental approach and disease-related perturbations or stressors, the life history of the disease can be recapitulated in vitro from iPSCs creating differentiable phenotypes. The alternative approach of culturing adult stem cells without a pluripotent stage would certainly maintain epigenetic cellular memory but it would be subsequently difficult to separate disease ‘cause’ from ‘effect’ (Murrell et al. 2008; Valenzuela et al. 2008). Similarly, the direct reprogramming of fibroblasts to neurons is likely to retain this ‘cell memory’ (Vierbuchen et al. 2010; Qiang et al. 2011).
However, there are also limitations and drawbacks on the use of iPSCs for disease modelling and drug discovery. One of the limiting factors in the utility of iPSC lines for drug discovery and safety is the considerable technical ‘noise’ obscuring the disease-related ‘signal’. A major contributor to this noise is the lack of consistency and poor target cell enrichment during iPSC differentiation. However, our feeder-free system for generating iPSCs offers a robust system for obtaining a homogeneous population of these cells that follow pluripotent signature patterns (Chung et al. 2012). The other key paradigm to circumvent the poor signal problem, which may then be tailored to the disease could be stratification of samples based on genetics (monogenic vs. polygenic/sporadic) and clinical history (responders vs. non-responders). A further problem is the large clonal variation of iPSCs, which requires the generation of a couple of clones from each patient and comparison of properties among these different clones to prove a general biological characteristic of the patient.
Outlook: personalized medicine for AD patients
The use of iPSC cellular models is therefore likely to lead to novel insights into the pathogenesis of AD, and to help discover new drugs for its treatment and/or prevention. Since these models are derived from individual patients, the cellular characteristics can be related to clinical features of the disease in that patient. An individual’s variations in the disease process and their cellular response to drugs may be reflected in the cellular model. If this is shown to be the case, a patient’s treatment regimen in the future could conceivably be individualized, based on the behaviour of the cellular model. For example, in the study by Israel et al. (2012) only 1 out of the 2 sporadic AD patients showed similarities to familial AD cell lines in terms of amyloid production, phospho-tau and active glycogen synthase kinase-3β levels, suggesting aetiological heterogeneity in the sporadic cases with a potential for differential treatment.
Defining AD subgroups with iPSC technology presents an excellent opportunity for a truly personalized approach to the treatment of AD. However, using current reprogramming and differentiation technology, it is unlikely that generating individual neurons from hiPSC for every patient for treatment will be economically viable. It is more likely that use of newly derived AD-hiPSCs will enable in vitro disease modelling that then enables patient-specific therapies based upon appropriate characterisation of AD patient groups by genetics and biomarker profile, and subsequent appropriate, targeted drug treatment.
References
Aalten P, Verhey FR, Boziki M, Brugnolo A, Bullock R, Byrne EJ, Camus V, Caputo M et al (2008) Consistency of neuropsychiatric syndromes across dementias: results from the European Alzheimer Disease Consortium. Part II. Dement Geriatr Cogn Disord 25:1–8
Adlard PA, Bica L, White AR, Nurjono M, Filiz G, Crouch PJ, Donnelly PS, Cappai R et al (2011) Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer’s disease. PLoS ONE 6:17669
Arendt T (2009) Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol 118:167–179
Arendt T, Bruckner MK, Mosch B, Losche A (2010) Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol 177:15–20
Bareggi SR, Cornelli U (2012) Clioquinol: review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci Ther 18:41–46
Bissonnette CJ, Lyass L, Bhattacharyya BJ, Belmadani A, Miller RJ, Kessler JA (2011) The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 29:802–811
Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF et al (2011) Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144:439–452
Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G et al (1996) Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron 17:1005–1013
Braak H, Del Tredici K (2004) Alzheimer’s disease: intraneuronal alterations precede insoluble amyloid-beta formation. Neurobiol Aging 25:713–718 (discussion 743–716)
Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70:960–969
Bush AI (2002) Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol Aging 23:1031–1038
Butterfield DA, Griffin S, Münch G, Pasinetti GM (2002) Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer’s disease brain exists. J Alzheimers Dis 4:193–201
Castellani RJ, Smith MA (2011) Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is ‘too big to fail’. J Pathol 224:147–152
Chan EM, Ratanasirintrawoot S, Park IH, Manos PD, Loh YH, Huo H, Miller JD, Hartung O et al (2009) Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol 27:1033–1037
Chen F, Wollmer MA, Hoerndli F, Münch G, Kuhla B, Rogaev EI, Tsolaki M, Papassotiropoulos A et al (2004) Role for glyoxalase I in Alzheimer’s disease. Proc Natl Acad Sci USA 101:7687–7692
Chung HC, Lin RC, Logan GJ, Alexander IE, Sachdev PS, Sidhu KS (2012) Human induced pluripotent stem cells derived under feeder-free conditions display unique cell cycle and DNA replication gene profiles. Stem Cells Dev 21:206–216
Czirr E, Weggen S (2006) Gamma-secretase modulation with Aβ42-lowering nonsteroidal anti-inflammatory drugs and derived compounds. Neuro-degenerative Dis 3:298–304
Dasilva KA, Aubert I, McLaurin J (2006) Vaccine development for Alzheimer’s disease. Curr Pharm Des 12:4283–4293
Ebben JD, Zorniak M, Clark PA, Kuo JS (2011) Introduction to induced pluripotent stem cells: advancing the potential for personalized medicine. World Neurosurg 76:270–275
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L et al (2010) PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J Alzheimers Dis 20:509–516
Feng Q, Lu SJ, Klimanskaya I, Gomes I, Kim D, Chung Y, Honig GR, Kim KS et al (2010) Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells 28:704–712
Fuller S, Steele M, Munch G (2010) Activated astroglia during chronic inflammation in Alzheimer’s disease—do they neglect their neurosupportive roles? Mutat Res 690:40–49
Goedert M, Jakes R, Spillantini MG, Crowther RA, Cohen P, Vanmechelen E, Probst A, Gotz J et al (1995a) Tau protein in Alzheimer’s disease. Biochem Soc Trans 23:80–85
Goedert M, Spillantini MG, Jakes R, Crowther RA, Vanmechelen E, Probst A, Gotz J, Burki K et al (1995b) Molecular dissection of the paired helical filament. Neurobiol Aging 16:325–334
Golde TE, Schneider LS, Koo EH (2011) Anti-Aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron 69:203–213
Gotz J, Lim YA, Ke YD, Eckert A, Ittner LM (2010) Dissecting toxicity of tau and beta-amyloid. Neurodegener Dis 7:10–12
Grskovic M, Javaherian A, Strulovici B, Daley GQ (2011) Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat Rev Drug Discov 10:915–929
Hayden EC (2011) Stem cells: the growing pains of pluripotency. Nature 473:272–274
Holmquist L, Stuchbury G, Berbaum K, Muscat S, Young S, Hager K, Engel J, Münch G (2007) Lipoic acid as a novel treatment for Alzheimer’s disease and related dementias. Pharmacol Ther 113:154–164
Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA, Zhang SC (2010) Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci USA 107:4335–4340
Huber A, Stuchbury G, Burkle A, Burnell J, Münch G (2006) Neuroprotective therapies for Alzheimer’s disease. Curr Pharm Des 12:705–717
Imbimbo BP, Giardina GA (2011) γ-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: disappointments and hopes. Curr Top Med Chem 11:1555–1570
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S et al (2012) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482:216–220
Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13:45–53
Jung YW, Hysolli E, Kim KY, Tanaka Y, Park IH (2012) Human induced pluripotent stem cells and neurodegenerative disease: prospects for novel therapies. Curr Opin Neurol 25:125–130
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10:698–712
Krautwald M, Münch G (2010) Advanced glycation end products as biomarkers and gerontotoxins—a basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp Gerontol 45:744–751
Kuhla B, Loske C, Garcia De Arriba S, Schinzel R, Huber J, Münch G (2004) Differential effects of “Advanced glycation endproducts” and beta-amyloid peptide on glucose utilization and ATP levels in the neuronal cell line SH-SY5Y. J Neural Transm 111:427–439
Kuhla B, Haase C, Flach K, Luth HJ, Arendt T, Münch G (2007) Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. J Biol Chem 282:6984–6991
Loske C, Gerdemann A, Schepl W, Wycislo M, Schinzel R, Palm D, Riederer P, Münch G (2000) Transition metal-mediated glycoxidation accelerates cross-linking of beta-amyloid peptide. Eur J Biochem 267:4171–4178
Maczurek A, Shanmugam K, Munch G (2008) Inflammation and the redox-sensitive AGE-RAGE pathway as a therapeutic target in Alzheimer’s disease. Ann NY Acad Sci 1126:147–151
Malpass K (2011) Parkinson disease: induced pluripotent stem cells—a new in vitro model to investigate alpha-synuclein dysfunction in Parkinson disease. Nat Rev Neurol 7:536
Münch G, Robinson SR (2002) Potential neurotoxic inflammatory responses to Aβ vaccination in humans. J Neural Transm 109:1081–1087
Münch G, Thome J, Foley P, Schinzel R, Riederer P (1997) Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res Brain Res Rev 23:134–143
Münch G, Schinzel R, Loske C, Wong A, Durany N, Li JJ, Vlassara H, Smith MA et al (1998) Alzheimer’s disease—synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm 105:439–461
Murrell W, Wetzig A, Donnellan M, Feron F, Burne T, Meedeniya A, Kesby J, Bianco J et al (2008) Olfactory mucosa is a potential source for autologous stem cell therapy for Parkinson’s disease. Stem Cells 26:2183–2192
Navarrete LP, Perez P, Morales I, Maccioni RB (2011) Novel drugs affecting tau behavior in the treatment of Alzheimer’s disease and tauopathies. Curr Alzheimer Res 8:678–685
Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A (2011) DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet 7:e1002085
O’Connor MD, Kardel MD, Eaves CJ (2011) Functional assays for human embryonic stem cell pluripotency. Methods Mol Biol 690:67–80
Okita K, Ichisaka T, Yamanaka S (2007) Generation of germline-competent induced pluripotent stem cells. Nature 448:313–317
Ooi L, Wood IC (2007) Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet 8:544–554
Panza F, Frisardi V, Imbimbo BP, D’Onofrio G, Pietrarossa G, Seripa D, Pilotto A, Solfrizzi V (2010) Bapineuzumab: anti-beta-amyloid monoclonal antibodies for the treatment of Alzheimer’s disease. Immunotherapy 2:767–782
Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C et al (2008) Disease-specific induced pluripotent stem cells. Cell 134:877–886
Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, Rhee D, Doege C, Chau L et al (2011) Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell 146:359–371
Rahmadi A, Steiner N, Münch G (2011) Advanced glycation endproducts as gerontotoxins and biomarkers for carbonyl-based degenerative processes in Alzheimer’s disease. Clin Chem Lab Med 49:385–391
Retz W, Gsell W, Münch G, Rosler M, Riederer P (1998) Free radicals in Alzheimer’s disease. J Neural Transm Suppl 54:221–236
Riederer P, Hoyer S (2006) From benefit to damage. Glutamate and advanced glycation end products in Alzheimer brain. J Neural Transm 113:1671–1677
Robinson SR, Bishop GM, Lee HG, Münch G (2004) Lessons from the AN 1792 Alzheimer vaccine: lest we forget. Neurobiol Aging 25:609–615
Sastre M, Klockgether T, Heneka MT (2006) Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int J Dev Neurosci 24:167–176
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J et al (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2:864–870
Schliebs R, Arendt T (2011) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221:555–563
Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ (2012) A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med 4:124ra129.
Sidhu KS (2011) New approaches for the generation of induced pluripotent stem cells. Expert Opin Biol Ther 11:569–579
Smith AD (2010) Why are drug trials in Alzheimer’s disease failing? Lancet 376:1466
Song F, Poljak A, Smythe GA, Sachdev P (2009) Plasma biomarkers for mild cognitive impairment and Alzheimer’s disease. Brain Res Rev 61:69–80
Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Münch G (2011) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32:763–777
Sun N, Panetta NJ, Gupta DM, Wilson KD, Lee A, Jia F, Hu S, Cherry AM et al (2009) Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci USA 106:15720–15725
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H (2006) The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ 2006:re1.
Thome J, Kornhuber J, Münch G, Schinzel R, Taneli Y, Zielke B, Rosler M, Riederer P (1996) New hypothesis on etiopathogenesis of Alzheimer syndrome. Advanced glycation end products (AGEs). Nervenarzt 67:924–929
Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J et al (1997) The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid beta protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci USA 94:2025–2030
Valenzuela MJ, Dean SK, Sachdev P, Tuch BE, Sidhu KS (2008) Neural precursors from canine skin: a new direction for testing autologous cell replacement in the brain. Stem Cells Dev 17:1087–1094
Vallier L, Touboul T, Brown S, Cho C, Bilican B, Alexander M, Cedervall J, Chandran S et al (2009a) Signaling pathways controlling pluripotency and early cell fate decisions of human induced pluripotent stem cells. Stem Cells 27:2655–2666
Vallier L, Touboul T, Chng Z, Brimpari M, Hannan N, Millan E, Smithers LE, Trotter M et al (2009b) Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS ONE 4:6082
Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463:1035–1041
Wan HI, Jacobsen JS, Rutkowski JL, Feuerstein GZ (2009) Translational medicine lessons from flurizan’s failure in Alzheimer’s disease (AD) trial: implication for future drug discovery and development for AD. Clin Transl Sci 2:242–247
Wang A, Huang K, Shen Y, Xue Z, Cai C, Horvath S, Fan G (2011) Functional modules distinguish human induced pluripotent stem cells from embryonic stem cells. Stem Cells Dev 20:1937–1950
Weisman D, Hakimian E, Ho GJ (2006) Interleukins, inflammation, and mechanisms of Alzheimer’s disease. Vitam Horm 74:505–530
Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R (2007) In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448:318–324
Wong A, Luth HJ, Deuther-Conrad W, Dukic-Stefanovic S, Gasic-Milenkovic J, Arendt T, Münch G (2001) Advanced glycation endproducts co-localize with inducible nitric oxide synthase in Alzheimer’s disease. Brain Res 920:32–40
Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H et al (2011) Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet 20:4530–4539
Yahata N, Asai M, Kitaoka S, Takahashi K, Asaka I, Hioki H, Kaneko T, Maruyama K et al (2011) Anti-Aβ drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS ONE 6:e25788
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920
Acknowledgments
We thank Peter Riederer for his brilliant scientific ideas and valuable mentorship in his long and distinguished scientific career. We gratefully acknowledge the grant support of the National Health and Medical Research Council (Grant IDs: 436797, 606543, 1046227) and Alzheimer’s Australia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ooi, L., Sidhu, K., Poljak, A. et al. Induced pluripotent stem cells as tools for disease modelling and drug discovery in Alzheimer’s disease. J Neural Transm 120, 103–111 (2013). https://doi.org/10.1007/s00702-012-0839-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-012-0839-2