
Abstract
Background
In gliomas molecular biomarkers are increasingly gaining diagnostic, prognostic and predictive significance. Determination of biomarker status after biopsy is important as not all patients are eligible for open tumor resection. We developed and validated prospectively (6/10–12/11) a protocol allowing for both reliable determination of multiple biomarkers and representative histological diagnoses from small-sized biopsies.
Methods
All molecular stereotactic biopsies were performed according to a detailed workflow. The selection of specimens best suited for molecular analyses was intra-operatively guided by the attending neuropathologist. Postoperative screening was done by methylation specific PCR using two distinct cryopreserved specimens to test for reproducibility of the findings and to rule out contamination. The DNA of a single best-suited specimen (1 mm3) was subjected to detailed molecular analysis (MGMT promoter methylation, IDH1/2 mutational status, LOH 1p and/or 19q).
Results
159 consecutively enrolled untreated gliomas were analyzed (94 glioblastomas, 2 gliosarcomas, 24 anaplastic astrocytomas, 10 oligo-tumors grade II/III, 20 grade II astrocytomas and 9 pilocytic astrocytomas). Transient morbidity was 2 %. Overall, the drop-out rate due to tissue contamination was 0.4 %. Median time from biopsy to histological and molecular genetic analyses was 3 and 5 days, respectively. Distributions of the respective biomarker status for tumor subgroups were consistent with the literature. The final histological diagnosis was changed/modified in 5/159 patients according to molecular findings. Treatment after molecular biopsy was highly personalized.
Conclusions
Molecular stereotactic biopsy is feasible and safe, can be implemented in daily clinical practice, improves diagnostic precision and enables personalized treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular biomarkers are differentially expressed among various types of gliomas with different prognoses. They gain an emerging role in tumor subclassification and prognostic/predictive modeling as well as treatment decisions and incorporation into study protocols for stratification of treatment results [1–4]. Mutations in both the TP53 gene and the IDH1 or IDH2 gene, for example, are frequently seen in low-grade astrocytomas, grade III astrocytomas, and secondary glioblastomas [5, 6]. A 1p19q codeletion occurs often in low- and high-grade oligodendrogliomas (∼75 %) and oligoastrocytomas (∼45 %), but seldom in fibrillary astrocytomas (∼8 %), and almost never in gemistocytic astrocytomas [7, 8]. IDH1/IDH2 mutations are significantly associated with other low-grade glioma-associated genetic abnormalities such as TP53 mutations, 1p/19q codeletions, but not with those seen in primary glioblastomas (i.e., EGFR amplifications and chromosome 10 loss) [9, 10].
Determination of the biomarker status can increase the diagnostic precision; the detection of a 1p/19q codeletion, for example, supports the diagnosis of an oligodendroglioma/oligoastrocytoma [11, 12]. Screening for IDH mutations is helpful to distinguish WHO grade I pilocytic astrocytomas and ependymomas (harbouring no IDH mutations) from diffuse astrocytomas and to differentiate diffuse tumor infiltration from reactive gliosis [6, 13–15]. There is a strong prognostic or predictive role of molecular biomarkers: MGMT promoter methylation has been established as a powerful favourable prognostic/predictive marker for malignant glioma patients receiving alkylating agents [16, 17]. Loss of heterozygosity on 1p/19q has been associated with favourable outcome measurements after chemotherapy and/or radiation therapy in tumors with an oligodendroglial component [1, 18, 19], and IDH1/2 mutations are associated with a better prognosis in anaplastic astrocytomas and glioblastomas but not in grade II astrocytomas [3, 6, 9, 20]. Notably, patients with anaplastic astrocytomas lacking an IDH1 mutation experienced a worse outcome than those with glioblastomas harbouring an IDH1 mutation [21].
Molecular genetic analyses are usually performed on tissue samples obtained from open tumor resection. Patients undergoing biopsy only are often left with uncertainties regarding their biomarker status and the prognosis of the disease [22]. Recent clinical trials have pointed out the impact of molecular biomarkers on treatment decisions [17, 23]. Particularly for the elderly subpopulation with glioblastomas; for example, determination of the MGMT promoter methylation status has been shown to be important in treatment decisions favoring radiotherapy alone or neo-adjuvant chemotherapy with temozolomide. Hence, in elderly patients with unresectable glioblastomas, a stereotactic biopsy with determination of molecular profiles is an important prerequisite for personalized treatment. Even though preliminary data on small study groups have already indicated that biomarker determination can be principally obtained from small sized biopsy specimens [10], biopsy-related methods and molecular genetic processing techniques suitable for implementation in daily clinical practise have not yet been defined [10]. In the present prospective study, we detail a workflow for the surgical procedure and the molecular genetic processing concerning the stereotactic glioma biopsy being termed as “molecular stereotactic biopsy”. Practicability and validity were tested in a large and consecutively treated study cohort. Using the presented workflow, we demonstrate that molecular stereotactic biopsy procedures can be done safely and successfully. We further show that the combining conventional histopathological and molecular genetic analyses improves the diagnostic precision of stereotactic biopsy procedures and enables personalized treatment.
Materials and methods
Patient selection
MoBxSt was considered to be indicated i) for tumors not completely resectable with no or only moderate spaceoccupying effect (i.e., not demanding decompressive surgery); ii) in tumors exhibiting an unfavourable risk/benefit estimation of microsurgery as compared to potentially alternative strategies (e.g., primary chemotherapy); iii) in tumors of unclear origin and identity, and iv) in patients with a significant comorbidity. All tumors were reviewed by the multidisciplinary tumor board with regard to tumor location, resectability, risk of surgical treatment, degree of spaceoccupying effects, and the spectrum, benefit, and risk of potentially suitable treatment strategies. Only those patients with histologically verified glioma were included in the current analysis, as the molecular genetic analysis protocol was applied in these tumors only. Patients with inflammatory lesions, metastases, lymphoma etc. (n = 119) were excluded. All enrolled patients gave informed consent; the study protocol was reviewed and approved by the institutional review board of the Ludwig-Maximilians-University, Munich, Germany (AZ 216/4).
Molecular stereotactic biopsy (MoStBx) – work flow
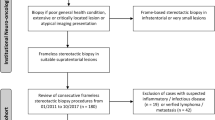
The term MoStBx refers to a procedure established at the Departments of Neurosurgery and Neuropathology of the Ludwig-Maximilians-University Munich. MoStBx aims to achieve a reliable pathological and molecular diagnosis on the basis of small stereotactic specimens (<1 mm3). As DNA yield from formalin-fixed and paraffin-embedded (FFPE) material is considerably lower, molecular genetic analyses of frozen material was generally preferred, which implies that histological and molecular analyses were intended to be performed on distinct samples. Given that the overall number of specimens available for histopathological and molecular genetic analyses is often limited, MoStBx should ideally enable detailed molecular genetic analyses on the basis of one single tissue specimen. It is therefore mandatory that tumor samples with no or only minimal contamination by non-neoplastic tissue, blood, or necrotic tissue are selected for molecular genetic analyses. Conversely, in cases of such contamination, MoStBx should enable the recognition of the contamination. It is important to note that the described selection procedure is only appropriate for the determination of the status of biomarkers, for which a homogeneous distribution throughout the tumor has been demonstrated (e.g., TP53-, IDH1/2-mutational status, MGMT promoter methylation status, LOH 1p/19q status), since a representative molecular diagnosis can be obtained from a single small tissue sample only for those markers [22, 24, 25]. The presented flow chart (Fig. 1) providing a comprehensive overview on the selection procedure includes links referring to the imaging details used for improved target determination in the planning phase (protocol A), the biopsy procedure itself (protocol B) and the applied histopathological and molecular genetic methods (protocols C-H).

The flowchart illustrates the MoStBx procedure in chronological order. Capital letters in brackets (A–J) refer to the respective detailed protocols in the materials and methods section. Step 1 Initial planning, surgical procedure and probe sampling. Step 2 Screening steps for selection of best suitable tumor tissue for molecular examination. The left box “MSP (methylation specific PCR) ambiguous” provides three commonly occurring combinations of histological and molecular findings: 1) no signal in MSP of both samples + viable tumor in all adjacent specimens → any sample suitable for molecular analysis; 2) “MSP signal at position −7, but not at −14 + viable tumor at −8 but non-tumor tissue at −15 → selection of −7 for molecular analysis; 3) Weak to moderate signals in MSP + viable tumor in all adjacent specimens → selection of the −2 for subsequent analysis due to best signal intensity at this level. The right box “MSP methylated” provides one example with clear signals in MSP of all three samples → any sample suitable for molecular analysis with preference of the −6 due to slightly better signal intensity. Step 3 Overview on the subsequent detailed analyses. Step 4 Diagnostic procedures including both initially independently-performed histopathological and molecular evaluation and, additionally, the option for re-evaluation and revision if needed.
-
Step 1 (guided-sampling) Selection of the tissue samples best suited for cryopreservation/molecular genetic analyses started in the operation room (OR) and was guided by the attending neuropathologist on site. Hereby, intraoperative evaluation of smear preparations (HE and methylene blue) guarantees immediate histological feedback without any delay (protocols B, C). For molecular analyses, it was generally intended to collect two tissue samples from different sites of the tumor in the direct vicinity (1 mm apart) of an intra-operatively identified solid and viable tumor (protocol B). The second sample was used in the screening phase of the procedure and represents a backup sample (in case of technical difficulties during the evaluation process).
-
Step 2 (screening) After DNA isolation (protocol D) a methylation specific PCR (MSP) for the MGMT promoter was used as a screening method for all (≥ 2) frozen samples (protocol E). When all samples homogeneously indicated methylation of the MGMT promoter, only one of the samples entered the subsequent detailed molecular genetic work flow. In cases with one or more samples exhibiting no or only weak methylation signals, histological sections of the adjacent stereotactic specimen were analyzed to ensure tumor content and to identify the specimen best suited for further molecular analyses. In cases of suspected contamination of all samples, molecular analysis from FFPE-material exhibiting a solid viable tumor was initiated.
-
Step 3 (detailed analysis) After MSP screening, DNA of one single sample was usually subjected to detailed analysis. MGMT promoter methylation status, 1p19q status and IDH1/2 mutation status were examined as described below (protocols E-H). Other analyses (e.g. BRAF V600E mutation analysis) could be performed if required
-
Step 4 (diagnosis) The histopathological and molecular diagnoses were expected to be made 3 days and 5–7 days, respectively, after MoBxSt. Both results were checked for congruency. In case of discordance, molecular pathological analysis and histological diagnosis were re-evaluated.
18FET-PET investigation (protocol A)
In selected patients with MRI-suspected low-grade gliomas, a dynamic 18FET-PET evaluation was performed, as described previously [25], for optimal target determination of the biopsy procedure. This approach was used, as pronounced inter- and intratumoral heterogeneity has recently been uncovered [25].
Molecular stereotactic biopsy technique (protocol B)
The multimodal image-guided surgical technique of MoStBx has been described: Co-registration of contrast enhanced computerized tomography (CT, slide thickness: 0.6 m); magnetic resonance imaging (MRI; axial T2-weigthed sequences, slide thickness: 2 mm, 3D T1-weighted sequences after administration of gadopentetate dimeglumine, slide thickness: 1 mm); and, 18FET-PET imaging data (if indicated) served for 3D visualization (i-plan stereotaxy, Brainlab) of the tumor and simulation of the “best” trajectory [10]. Whole biopsy specimens were consecutively collected in 1 mm steps towards the target point via a small skin incision (6–10 mm) and a 2 mm bore . Using micro forceps, the maximum amount of tissue per biopsy specimen was 1 mm3. The tissue sampling and selection procedure was guided by the attending neuropathologist. Tissue samples selected for molecular genetic analyses were stored at −20 °C immediately after surgery. Each of the tissue samples taken along the biopsy trajectory were consecutively numbered with regard to distance from the target point and documented in the biopsy protocol of each patient, thereby enabling corresponding analyses of the tissue, molecular, and imaging data. Another native CT-scan was done in all patients 1 day after biopsy to rule out surgery-related bleeding. Any adverse events within 30 days after surgery that may have been potentially attributable to the biopsy were considered as perioperative morbidity.
Histopathology (protocol C)
For the intraoperative diagnosis smear preparations were stained with methylene blue and/or hematoxylin and eosin. Stereotactic biopsy specimens chosen for final histopathological evaluation, were fixed in 4 % buffered formalin (Fisher Scientific GmbH, Schwerte, Germany) and embedded in paraffin. Tumor morphology in 2 μm paraffin sections was visualized using hematoxylin and eosin staining. Standardized immunohistochemistry was done on a benchmark staining machine with a 3, 3′-diaminobenzidine (DAB) detection system according to the manufacturer’s instructions (Ventana Medical Systems, Arizona, USA). Antibodies used were usually anti-human GFAP (monoclonal mouse, clone 6 F2, Dako, Glostrup, Denmark), anti-MAP2 (clone HM-2, Sigma, Saint Louis, Missouri, USA) and anti-human Ki67 antibody (mouse monoclonal, clone MIB-1, Dako, Glostrup, Denmark). Tumor classification and grading was done by members of the histopathological conference according to the 2007 WHO classification of tumors of the central nervous system [26].
Molecular genetic analyses
Isolation of DNA from a stereotactic biopsy sample (protocol D)
DNA extraction from frozen tissue and FFPE tissue was performed using a QIAmp DNA Micro Kit and a QIAmp DNA FFPE Tissue Kit, respectively (both: Qiagen, Hilden, Germany). Isolation from blood was performed using the QIAmp DNA Blood Kit (Qiagen, Hilden, Germany). The quantity and purity of DNA were assessed using a NanoDrop1 ND-1000 Spectrophotometer (NanoDrop, Wilmington, NC). DNA yield from one frozen sample was in the range of 15–40 ng/μl in a 70 μl elution volume; for one FFPE sample (10–25 ng/μl) the OD 260/280 ratio was ∼1.9. Alternatively, DNA was isolated on a Maxwell® 16 MDx instrument using the M16 tissue LEV DNA purification kit (frozen tissue), the M16 FFPE Tissue LEV DNA purification kit (FFPE tissue) or the M16 Blood DNA purification kit (blood) (all: Promega Deutschland GmbH, Mannheim, Germany). Due to up to 30 % higher DNA yield this was the method of choice for cases limited to FFPE material.
Determination of the MGMT promoter methylation status (protocol E)
Determination of the MGMT promoter methylation was done by methylation-specific PCR (MSP) and sequencing analysis. Bisulfite conversion of 200–400 ng DNA was performed with the EpiTect® Bisulfite Kit (Qiagen) as described previously [24]. For MSP as molecular screening (Fig. 1, step 2), two pairs of primers, each specific for either the methylated or the unmethylated MGMT promoter region, were used as originally described by Esteller and colleagues [27]. MSP and visualization of the amplicons by gel electrophoreses were performed according to standard protocols [24]. For detailed analysis with capillary sequencing (flowchart step 3), primers originally described by Moellemann and colleagues were used [28]. The sequencing reaction covers a 316 bp region of the MGMT promoter with 25 CpG sites, including those detected by MSP (corresponding to CpG positions 2–14). The MGMT promoter was considered “methylated” when more than half of the CpG sites (≥13 of the 25 CpG sites) were found to be “methylated” or “partially methylated”. A “partially methylated” CpG site was defined as the cytosine peaks being 50 % or more of the corresponding thymine peak. Positions with cytosine peaks as small as 10–50 % of the thymine peak were considered weakly methylated. When 9 – 12 of 25 CpG sites were “methylated/partially methylated” the MGMT promoter was considered “partially” methylated. When more than 9 of the 25 CpG sites were “methylated/partially methylated” the MGMT promoter was considered not methylated.
PCR amplification of microsatellite markers (protocol F)
For the LOH on 1p/19q status determination 5 tetranucleotide markers were used as described previously [11, 29]. Briefly, 10–20 ng of DNA (blood and tumor) was used for PCR (HotStarTaq® Master Mix Kit, QIAGEN®, Hilden, Germany) using microsatellite markers D1S1608, D1S548, D1S1592, D1S1184, D1S1161 for chromosome 1p and D19S433, D19S431, D19S718, D19S559, D19S601 for chromosome 19q with annealing temperatures as suggested by Hartmann et al. [11]. The distribution of the markers throughout the entire chromosomal arms also allowed the detection of partial deletions [30]. The PCR reaction was performed with an initial denaturation at 94 °C for 15 min, followed by 35 cycles (94 °C, 30s; annealing time 40s; 72°, 1 min) in an automated thermocycler (Eppendorf Mastercycler gradient) and final extension at 72 °C for 10 min. Given the minimal amounts of tumor DNA, special care was taken to avoid cross-contamination. The samples of each patient were analysed in separate experimental rounds with one no template control (NTC) for each microsatellite primer pair.
Microsatellite analysis by horizontal gel electrophoresis on synthetic high resolution gels and SYBR® Gold staining (protocol G)
From each PCR amplicon, 0.5 μl were analyzed on synthetic, highly resolving Spreadex® EL 800 wide mini gels (Elchrom Scientific®, Switzerland). Electrophoresis was performed at 120 V and 55 °C for 140 min by use of the Elchrom submerged gel electrophoresis system. This system controls for constant flow of the running buffer and provides a uniform electric field in the gel compartment which ensures reproducible experimental conditions. PCR products were visualized by SYBR® Gold (Molecular Probes®, Oregon, USA) which has a several fold higher sensitivity than ethidium bromide [31].
Detection of IDH1/2 mutations (protocol H)
IDH1 and IDH2 mutations were examined using pyrosequencing techniques. Primer design was performed with PSQ Assay design Software (Qiagen, Hilden, Germany) based on accession numbers NC_000002.11 (209100953–209119806), IDH1 and NC_000015.9 (90627210–90645708) and IDH2. For IDH1, a fragment of 88 bp length including codon 132 was amplified using the primer pair 5'-biotin-AAAAATATCCCCCGGCTTG-3' (forward) and 5'-TGCCAACATGACTTACTTGATCC-3' (reverse) [3]. For IDH2 a fragment of 83 bp length including codon 172 was amplified using the primer pair 5'-TCCCACGCCTAGTCCCTG -3' (forward) and 5'-biotin-CTCTCCACCCTGGCCTACC -3' (reverse). PCR was performed in a 200 μl tube with a final volume of 40 μl using the HotStarTaq DNA Polymerase Master Mix Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions with 20 ng of DNA. Each PCR was accompanied by a no template control (NTC). Initial denaturation (95 °C, 15 min) was followed by 40 cycles of 20 s at 94 °C, 20 s at 52 °C and 30 s at 72 °C and a final extension step at 72 °C for 5 min. The PCR product was checked by gel electrophoreses including no template controls (NTC). Next, 10 μl of the PCR product as well as the NTC were subjected to pyrosequencing on a PyroMark Q24 System using the Pyro Gold reagents kit (both: Qiagen, Hilden, Germany) and the sequencing primers 5'-TGATCCCCATAAGCA-3' for IDH1 or 5′-AGCCCATCACCATTG-3′ for IDH2, respectively. Subsequent purification and processing of the biotinylated single-strand DNA was performed according to the manufacturer’s instructions. Resulting data were analyzed and quantified with the PyroMark Q24 Software (Qiagen, Hilden, Germany).
Statistical analysis
The distribution of continuously scaled variables was analyzed with the Wilcoxon test. Associations between molecular alterations and histological subtypes were assessed with [chi]2 statistics or Fisher exact tests (in cases of small sample size). A value of p < 0.05 was considered significant.
Results
Patients
This prospective study was comprised of a series of 159 patients with a diagnosis of a de-novo glioma, who were consecutively treated during an 18 month period (06/2010–12/2011). In 47 (29 %) of the patients with MRI-suspected low grade gliomas a dynamic FET-PET guided biopsy was done. Overall, tumors were classified as glioblastoma (n = 94), anaplastic astrocytoma (n = 24), oligodendroglioma/oligoastrocytoma (Grade II + III; n = 10), astrocytomas WHO II (n = 20, incl. 4 brainstem gliomas), pilocytic astrocytomas (n = 9), and gliosarcoma (n = 2). Patients with low-grade gliomas (n = 26) were significantly younger (p < 0.01) than those with high-grade gliomas (ie., mean age: 37 vs. 61 years). Patient characteristic are summarized in Table 1. The mean number of biopsy specimens taken was 17.1 per patient (range 8–37). Transient morbidity rate was 2 %: two patients suffered from focal seizures after biopsy, another one from an erosion of the cornea as a consequence of inadequate protection during surgery. There was no permanent morbidity and no mortality. The median time from biopsy to histological and molecular genetic analyses was 3 and 5 days, respectively.
Molecular diagnosis
The drop-out rate due to insufficient amount of tumor tissue (contaminated specimens) was 0.4 %. Results of molecular genetic analyses of the respective glioma subgroups are summarized in Table 2. The MGMT promoter was methylated in 11 % of the astrocytomas grade I tumors, 65 % of the astrocytoma II tumors, 54 % of the astrocytoma WHO III tumors, 44 % of the glioblastoma multiforme WHO IV tumors and 100 % of oligodendroglioma/oligoastrocytoma grade II/III tumors. A 1p/19q codeletion was seen in 90 % of the oligodendroglioma/oligoastrocytoma grade II/III tumors, 15 % of astrocytoma grade II tumors, 8 % of astrocytoma grade III tumors, 3 % of glioblastoma multiforme WHO IV tumors, but not in the astrocytoma grade I tumors. 1p/19q codeleted tumors always exhibited both an IDH1/2 mutation and a methylated MGMT promoter. An IDH1/2 mutation could be detected in 70 % of the astrocytoma grade II tumors, in 90 % of oligo-tumors grade II/III but not in the grade I astrocytoma tumors. IDH1/2 mutations were seen in 29 % of the anaplastic astrocytomas and in 5 % of the glioblastomas; all these tumors exhibited a methylated MGMT promoter and were significantly younger than those harbouring wild type IDH1/2 (42 vs. 64 years, p < 0.001); they were classified as secondary malignant gliomas. Overall, the frequency of IDH1/2 mutations was significantly correlated with both the frequency of LOH 1p19q and/or MGMT promoter methylation (p < 0.001). Patients harbouring IDH1/2 mutations were younger (p < 0.001).
The final histological diagnosis was changed/modified in 5 of the 159 tumors according the molecular findings: i) the detection of a methylated MGMT promoter supported the diagnosis of grade II gliomas in two cases (exemplified in Fig. 2). Histological evaluation alone could not unequivocally distinguish between a reactive gliotic and a neoplastic lesion; ii) Lack of 1p19q codeletion supported the diagnosis of an anaplastic astrocytoma in another case. Histological evaluation alone was uncertain concerning an oligodendroglial component in this tumor; iii) The detection of LOH 1p19q in a glioblastoma lacking unequivocal histological signs of oligodendroglial differentiation led to the final diagnosis of a “glioblastoma with oligodendroglial component”; iv) The absence of an IDH1/2 mutation supported the diagnosis of a pilocytic astrocytoma in one case. Histological analysis of the small specimens could not rule out a diffuse astrocytoma grade II.

Example of a lesion histologically classified as “no tumor” (a–d) in which MoStBx (E, F) enabled the diagnosis of a glioma. a HE staining of a stereotactic specimen (−4), shows few conspicuous cells with irregular nuclei not sufficient for tumor diagnosis. b IHC analysis for GFAP shows a picture compatible with reactive astrocytosis. c IHC analysis for MAP2 also reveals no clear evidence for tumor cells. d IHC analysis for the proliferation marker Ki67 showed only one positive stained nucleus in all 17 samples. e MSP of the two specimens (at −3, −15) indicates methylation of the MGMT promoter. U: MSP product specific for unmethylated sequence, M: MSP product specific for the methylated sequence, NTC: no template control. f Capillary sequencing confirming methylation of the MGMT promotor led to a revision of the initial histological diagnosis in favor of a glioma. Arrowheads indicate “methylated/partially methylated” CpG sites, asterisks indicate weakly/unmethylated CpG sites. Same symbols in brackets indicate CpG positions which are not clearly seen in the forward sequencing reaction (shown) and were evaluated in the reverse sequencing reaction (not shown). g Pyrosequencing revealed that the case did not show an IDH1-mutation (picture). An histological tumor diagnosis using an antibody directed against the IDH1 (R132H) mutation was therefore not possible in this case
Reproducibility of molecular analyses
At the time of tumor progression, a second molecular analysis was done in 21 patients: Discordant findings (as compared to the initial molecular findings) were seen in 4 of the 21 patients and could be attributed to tissue contamination in two patients. The third case concerned an astrocytoma with borderline methylation, classified as “not methylated” in the initial analysis and as “at best partially methylated” in the follow-up analysis. The fourth case concerned an astrocytoma grade II (no 1p19q codeletion) that had progressed to GBM. Follow-up analysis indicated partial LOH on 1p (LOH for 2 of 5 informative markers) and 19q (LOH for 3 of 5 informative markers). Re-evaluation of the original gel electrophoresis with knowledge of the second analysis showed that 2 of the 10 markers showed a suspicious fainting of bands that was not considered however sufficient for the diagnosis of a partial 1p/19q codeletion. In the remaining 17 patients the biomarker pattern remained unchanged as compared to the initial analysis.
Diagnostic reliability of MSP for determination of MGMT promoter methylation
177 MGMT analyses (157 original and 20 follow-up at the time of tumor progression) were performed. Diagnostic results were obtained in 175 of the 177 of the cases (99 %). MGMT status was “unmethylated” in 89 of the 175 cases (51 %), “methylated” in 63 of the 175 cases (36 %) and “partial methylated” in 23 of the 175 cases (13 %). Concordant findings between the MSP and bisulfite-sequencing were seen in 151 of the 175 cases (86 %). In the remaining 24 cases (14 %) MSP showed false positive as well as false negative results as depicted in Fig. 3 (A). Positive predictive value (PPV), negative predictive value (NPV), sensitivity (SENS) and specificity (SPEC) for MSP were 92.1 %, 87.2 %, 88.2 % and 91.5 %, respectively, in cases showing “no” MSP signal (93/175); the corresponding values for those exhibiting a “strong” MSP signal (61/175) were 96.7 %, 96,5, 93.7 % and 98.2 %, respectively (Fig. 3B); PPV, NNV, SENS, and SPEC for tumors with “weak” MSP signal (21/175) were 47.6 %, 91.5 %, 43.5 % and 92.7 %, respectively.

MGMT methylation status: the diagnostic value of MSP and capillary sequencing. a Correlation of MSP screening results (left column) with final diagnosis determined by capillary sequencing (right column). b Corresponding positive predictive value (PPV), negative predictive value (NPV), sensitivity (SENS) and specificity (SPEC) for cases showing no (upper row), weak (middle row) and strong (lower row) signals in the MSP.
Treatment after biopsy
Treatment strategies after MoStBx are summarized in Table 3. Eleven patients underwent open tumor resection. MoStBx before resection was done because of differential diagnostic uncertainties in these cases. 89 % of the glioblastoma patients were treated according the EORTC protocol [32]. Those not receiving standard treatment (10 patients) were older (median age: 72 years vs. 65 years); these patients received radiotherapy (chemotherapy) alone in cases of an unmethylated (methylated) promoter status or palliative treatment only because of poor clinical conditions. Among grade III astrocytomas, those with MGMT promoter methylation were more likely to receive chemotherapy alone (p < 0.001). Four patients with anaplastic astrocytoma harbouring either wild type IDH (3 tumors) or an extremely high proliferation index (1 tumor; proliferation index 30%) underwent radiotherapy with concomitant and adjuvant temozolomide. A 1p/19q codeletion was significantly associated with the initiation of a primary chemotherapy (p < 0.001).
Discussion
During the last decade, genetic studies on gliomas have revealed molecular alterations that increasingly serve as diagnostic, prognostic and predictive markers and are used for stratification in clinical trials [1, 16, 33, 34]. It is therefore important to ensure these biomarkers can also be reliably analyzed from small sized stereotactic biopsies in daily clinical practice, as not all patients are eligible for open tumor resection or benefit from microsurgical treatment [35]. However, difficulties in the size adjusted molecular genetic analysis, doubts as to whether the molecular findings are representative, and concerns that additionally collected samples for genetic analysis might compromise the quality of conventional neuropathological tumor-diagnosis assessment, contribute uncertainty about the role of stereotactic biopsy in the era of molecular neuro-oncology.
We demonstrate for the first time that multiple molecular biomarkers can be reliably determined simultaneously from a single small sized cryopreserved biopsy sample (<1 mm3) containing a viable tumor. This could be achieved by a selection process of the collected samples as outlined in Fig. 1, which was guided by the attending neuropathologist in the OR. A prerequisite for such an approach is that the biomarker under investigation is homogeneously distributed throughout the tumor space. Exactly that has been shown for the described biomarkers of the current report; these biomarkers are considered early events in gliomagenesis [10, 24, 25]. We also showed, in accordance with this assumption, that the described biomarkers usually remained unchanged in cases of tumor recurrence or tumor progression. Similar results have also reported by Felsberg et al. [36]. We show that MoStBx is safe, can be applied in any location of the brain, and resulted in conclusive histological diagnoses and corresponding molecular profiles that are in line with biomarker profiles of WHO grade I-IV gliomas after open tumor resection. A histopathological and molecular diagnosis was typically available 3 days and 5 days after stereotactic biopsy, respectively. This enabled rapid personalized treatment planning. Our study shows that the status of the evaluated biomarkers in each tumor generally did not change in case of tumor recurrences after re-biopsy or open tumor resection, which further corroborates the reliability and validity of the analyses. One patient, however, was found with a partial 1p and 19q codeletion after malignant transformation, which was not seen in the initial biopsy of a grade II astrocytoma. Whether this finding can be interpreted as a new genetic alteration during the process of malignant transformation remains unclear.
MoStBx successfully contributes to the diagnostic classification of those tumors in which the growth pattern and composition of the tumor often cannot be totally captured by small sized biopsy specimens. For example, pilocytic astrocytoma grade I (usually harbouring wild type IDH), or gliomas with suspected oligodendroglial components (often harbouring 1p/19q codeletion); MoStBx has also been shown to be extremely useful in differentiating between reactive gliotic lesions and infiltrative low-grade gliomas (MGMT promoter often methylated). Interestingly, although molecular markers were diagnostically helpful, we also found some cases in which the histological picture seemed to be contradictory to the molecular findings (e.g., 3 tumors showing purely astrocytic differentiation exhibited a 1p19q codeletion, which is regarded as a hallmark of oligodendroglial/oligoastrocytic differentiation). It cannot be excluded that MoStBx had missed the oligodendroglial component in these cases. A recent study, however, cautions one to rely on molecular markers alone; two markers (IDH1 mutations and KIAA1549-BRAF fusions) that seemed to be mutually exclusive and were suggested to confidently distinguish diffuse astrocytomas (IDH1/2 mutations, no KIAA1549-BRAF fusion) and pilocytic astrocytomas (KIAA1549-BRAF fusion, no IDH1/2 mutations) were now found coexisting in some diffuse astrocytomas [37, 38]. In gliomas molecular biomarkers have the potential to refine and complement the current tumor classification and will provide additional prognostic and/or predictive data for personalized treatment strategies [39].
Selection of the analytical methods best suited for implementation of small size-adjusted molecular analyses in a clinical setting is challenging. Even though molecular genetic analyses of cryopreserved material have the disadvantage of histological and molecular genetic findings could not directly correlating, we could demonstrate that the approach presented herin using an alternating sampling technique for histological and molecular evaluation in 1-mm-steps was highly effective, This strongly contrasts to the experience of other groups who had applied FFPE based molecular analyses to small tissue specimens of glioblastoma patients [16].
LOH1p19q was determined by PCR based microsatellite analysis as this method also allows for the discrimination of isolated/partial deletions. Alternative methods (FISH, alpha-internexin immunohistochemistry) cannot detect these alterations or are not suited for the limited amount of tissue and DNA available [10, 11, 30]. As IDH1/2 mutations in gliomas cluster in only one codon, they are perfectly in the scope of pyrosequencing and thus were analyzed with this fast and reliable method. In contrast to immunohistochemical detection with an antibody directed against the most common IDH1 (R132H) mutation, pyrosequencing has the advantage of covering all potential IDH1 and IDH2 mutations in the sequence analyzed. The original publication describing this antibody revealed that the immunohistochemical results did not always correlate with the gold standard of sequencing [40]. Of note, in our own experience the number of cases in which IDH1 (R132H) immunohistochemistry does not correlate with the pyrosequencing results is even slightly higher. MSP is the most widely applied method for determination of the MGMT promoter methylation and has been shown to correlate well with clinical outcomes in several studies [16]. In the current study MSP was used as a general screening step before detailed molecular genetic analyses and was followed by capillary sequencing, which covers a larger number of CpG sites [24, 41]. Comparing both methods, MSP revealed (false positive and negative) results in 14 % of the analyzed tumors, mostly in the group of patients with partial methylation (Fig. 3). Failure to detect tumors with partial methylation indicates a natural limitation of MSP. A recent study systematically analyzed the impact of specific CpG sites within the MGMT promotor on the transcriptional regulation of MGMT using a luciferase reporter assay [42]. This study revealed that MSP is located within the optimal region for MGMT testing. However, the study also showed that substitution of a single CpG outside the MSP region almost completely inhibited the promotor activity. The authors observed high variability regarding the methylated positions, which is in line with our results. Given this, determination of a higher number of individual CpG sites by capillary sequencing might indicate sensitivity to alkylating substances more reliably than MSP. Additional clinical data are necessary to support our observations in favour of sequencing analysis.
While the MoBxSt approach presented here will currently be limited to larger centres having a neuropathologist on-site, emerging technologies may provide the basis for wider application in the near future. With the growing demand for personalized medicine, a new miniature endoscopic system has been developed to specifically identify vital tumor tissue by detection of protoporphyrin IX (PpIX) fluorescence which accumulates in highly proliferating tumor cells but not in necrosis upon administration of 5-aminolaevulinic acid (5-ALA) [43]. The endoscopic system could be easily integrated in the stereotactic setup of MoBxSt. In a glioblastoma patient, preliminary results indicated that endoscopic fluorescence-guided biopsy is able to detect viable tumor tissue suitable for both histological and molecular pathological examination independent of intraoperative evaluation. This technique needs further validation and might enable sending samples to centres experienced in molecular pathological analyses of small-sized stereotactic specimens. Personalized biomarker assisted diagnosis and treatment concepts could become more readily available in the near future (Fig. 4).

Future perspective – endoscopy based stereotactic biopsy. a MRI indicating a left side temporal ring enhanced lesion. Target level and stereotactic trajectory (green dotted line) are shown. The White scale [mm] depicted the stepwise biopsy procedure throughout the tumor. b Endoscopic image (left) on level at −33 did not show 5-ALA-induced fluorescence (blue). Histological analysis at this position revealed no tumor tissue, as demonstrated in HE stained sections in low (middle) and high (right) magnification. Scale bar: 50 μm. c Endoscopic image at level −31 shows 5-ALA-induced fluorescence (red), indicating tumor tissue. Corresponding histological examination revealed a glioblastoma (WHO grade IV) as demonstrated in HE stained sections in low (middle) and high (right) magnification. Scale bar: 50 μm. d Endoscopic image on consecutive level at −30 shows 5-ALA-induced fluorescence (red), also indicating tumor tissue. Biopsy sample −30 was selected for molecular analysis. MSP (middle) and capillary sequencing (right) demonstrates an unmethylated MGMT promotor sequence. (U and M): PCR specific for the unmethylated and methylated MGMT promotor sequence, respectively. Asterisks (*) indicate unmethylated CpG positions
Taken together, the current study demonstrated that MoStBx: i) is feasible and safe and can be implemented in daily clinical practise; ii) provides valid and reproducible molecular analyses of multiple biomarkers without comprising the quality of histological classification and grading; iii) improves diagnostic accuracy; iv) provides a platform for analyses of further prognostic/predictive biomarkers in the future (e.g., BRAF-V600E); and, v) enables personalized treatment planning.
References
Erdem-Eraslan L, Gravendeel LA, de Rooi J, Eilers PH, Idbaih A, Spliet WG, den Dunnen WF, Teepen JL, Wesseling P, Sillevis Smitt PA, Kros JM, Gorlia T, van den Bent MJ, French PJ (2013) Intrinsic molecular subtypes of glioma are prognostic and predict benefit from adjuvant procarbazine, lomustine, and vincristine chemotherapy in combination with other prognostic factors in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. J Clin Oncol 31:328–336
Tabatabai G, Hegi M, Stupp R, Weller M (2012) Clinical implications of molecular neuropathology and biomarkers for malignant glioma. Curr Neurol Neurosci Rep 12:302–307
Thon N, Eigenbrod S, Kreth S, Lutz J, Tonn JC, Kretzschmar H, Peraud A, Kreth FW (2012) IDH1 mutations in grade II astrocytomas are associated with unfavorable progression-free survival and prolonged postrecurrence survival. Cancer 118:452–460
Weiler M, Wick W (2012) Molecular predictors of outcome in low-grade glioma. Curr Opin Neurol 25:767–773
Stander M, Peraud A, Leroch B, Kreth FW (2004) Prognostic impact of TP53 mutation status for adult patients with supratentorial World Health Organization Grade II astrocytoma or oligoastrocytoma: a long-term analysis. Cancer 101:1028–1035
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Appin CL, Gao J, Chisolm C, Torian M, Alexis D, Vincentelli C, Schniederjan MJ, Hadjipanayis C, Olson JJ, Hunter S, Hao C, Brat DJ (2013) Glioblastoma with Oligodendroglioma Component (GBM-O): Molecular genetic and clinical characteristics. Brain Pathol 23(4):454–461
Okamoto Y, Di Patre PL, Burkhard C, Horstmann S, Jourde B, Fahey M, Schuler D, Probst-Hensch NM, Yasargil MG, Yonekawa Y, Lutolf UM, Kleihues P, Ohgaki H (2004) Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol 108:49–56
Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El HS, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154
Thon N, Eigenbrod S, Grasbon-Frodl EM, Ruiter M, Mehrkens JH, Kreth S, Tonn JC, Kretzschmar HA, Kreth FW (2009) Novel molecular stereotactic biopsy procedures reveal intratumoral homogeneity of loss of heterozygosity of 1p/19q and TP53 mutations in World Health Organization grade II gliomas. J Neuropathol Exp Neurol 68:1219–1228
Hartmann C, Mueller W, Lass U, Kamel-Reid S, von Deimling A (2005) Molecular genetic analysis of oligodendroglial tumors. J Neuropathol Exp Neurol 64:10–14
Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP (1994) Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 145:1175–1190
Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, Hartmann C, von Deimling A (2010) Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, Jeuken JW, Wesseling P, Reifenberger G, von Deimling A (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474
Rivera-Zengotita M, Yachnis AT (2012) Gliosis versus glioma?: don’t grade until you know. Adv Anat Pathol 19:239–249
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003
Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, Nikkhah G, Papsdorf K, Steinbach JP, Sabel M, Combs SE, Vesper J, Braun C, Meixensberger J, Ketter R, Mayer-Steinacker R, Reifenberger G, Weller M (2012) Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol 13:707–715
Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D, Buckner J, Fink K, Souhami L, Laperierre N, Mehta M, Curran W (2006) Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 24:2707–2714
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Enting RH, French PJ, Dinjens WN, Vecht CJ, Allgeier A, Lacombe D, Gorlia T, Hoang-Xuan K (2013) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31:344–350
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, Westphal M, Schackert G, Meyermann R, Pietsch T, Reifenberger G, Weller M, Loeffler M, von Deimling A (2010) Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120:707–718
Thon N, Eigenbrod S, Grasbon-Frodl EM, Lutz J, Kreth S, Popperl G, Belka C, Kretzschmar HA, Tonn JC, Kreth FW (2011) Predominant influence of MGMT methylation in non-resectable glioblastoma after radiotherapy plus temozolomide. J Neurol Neurosurg Psychiatry 82:441–446
Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, Abacioglu U, Tavelin B, Lhermitte B, Hegi ME, Rosell J, Henriksson R (2012) Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol 13:916–926
Grasbon-Frodl EM, Kreth FW, Ruiter M, Schnell O, Bise K, Felsberg J, Reifenberger G, Tonn JC, Kretzschmar HA (2007) Intratumoral homogeneity of MGMT promoter hypermethylation as demonstrated in serial stereotactic specimens from anaplastic astrocytomas and glioblastomas. Int J Cancer 121:2458–2464
Kunz M, Thon N, Eigenbrod S, Hartmann C, Egensperger R, Herms J, Geisler J, la Fougere C, Lutz J, Linn J, Kreth S, von Deimling A, Tonn JC, Kretzschmar HA, Popperl G, Kreth FW (2011) Hot spots in dynamic (18)FET-PET delineate malignant tumor parts within suspected WHO grade II gliomas. Neuro Oncol 13:307–316
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG (2000) Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 343:1350–1354
Mollemann M, Wolter M, Felsberg J, Collins VP, Reifenberger G (2005) Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer 113:379–385
Mueller W, Hartmann C, Hoffmann A, Lanksch W, Kiwit J, Tonn J, Veelken J, Schramm J, Weller M, Wiestler OD, Louis DN, von Deimling A (2002) Genetic signature of oligoastrocytomas correlates with tumor location and denotes distinct molecular subsets. Am J Pathol 161:313–319
Eigenbrod S, Roeber S, Thon N, Giese A, Krieger A, Grasbon-Frodl E, Egensperger R, Tonn JC, Kreth FW, Kretzschmar HA (2011) alpha-Internexin in the diagnosis of oligodendroglial tumors and association with 1p/19q status. J Neuropathol Exp Neurol 70:970–978
Tuma RS, Beaudet MP, Jin X, Jones LJ, Cheung CY, Yue S, Singer VL (1999) Characterization of SYBR Gold nucleic acid gel stain: a dye optimized for use with 300-nm ultraviolet transilluminators. Anal Biochem 268:278–288
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10:459–466
Weller M, Stupp R, Hegi ME, van den Bent M, Tonn JC, Sanson M, Wick W, Reifenberger G (2012) Personalized care in neuro-oncology coming of age: why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol 14(Suppl 4):iv100–iv108
Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, Sabel MC, Koeppen S, Ketter R, Meyermann R, Rapp M, Meisner C, Kortmann RD, Pietsch T, Wiestler OD, Ernemann U, Bamberg M, Reifenberger G, von Deimling A, Weller M (2009) NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880
Kreth FW, Thon N, Simon M, Westphal M, Schackert G, Nikkhah G, Hentschel B, Reifenberger G, Pietsch T, Weller M, Tonn JC (2013) Gross total but not incomplete resection of glioblastoma prolongs survival in the era of radiochemotherapy. Ann Oncol 24:3117–3123
Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, Schackert G, Kreth FW, Pietsch T, Loffler M, Weller M, Reifenberger G, Tonn JC (2011) Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer 129:659–670
Badiali M, Gleize V, Paris S, Moi L, Elhouadani S, Arcella A, Morace R, Antonelli M, Buttarelli FR, Figarella-Branger D, Kim YH, Ohgaki H, Mokhtari K, Sanson M, Giangaspero F (2012) KIAA1549-BRAF fusions and IDH mutations can coexist in diffuse gliomas of adults. Brain Pathol 22:841–847
Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, Pfister S, Von Deimling A, Hartmann C (2009) Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 118:401–405
Louis DN (2012) The next step in brain tumor classification: “Let us now praise famous men”… or molecules? Acta Neuropathol 124:761–762
Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601
Esteller M (2005) Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol 45:629–656
Malley DS, Hamoudi RA, Kocialkowski S, Pearson DM, Collins VP, Ichimura K (2011) A distinct region of the MGMT CpG island critical for transcriptional regulation is preferentially methylated in glioblastoma cells and xenografts. Acta Neuropathol 121:651–661
Gobel W, Brucker D, Kienast Y, Johansson A, Kniebuhler G, Ruhm A, Eigenbrod S, Fischer S, Goetz M, Kreth FW, Ehrhardt A, Stepp H, Irion KM, Herms J (2012) Optical needle endoscope for safe and precise stereotactically guided biopsy sampling in neurosurgery. Opt Express 20:26117–26126
Funding
No funding to declare.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Eigenbrod and R. Trabold contributed equally to this work
Rights and permissions
About this article

Cite this article
Eigenbrod, S., Trabold, R., Brucker, D. et al. Molecular stereotactic biopsy technique improves diagnostic accuracy and enables personalized treatment strategies in glioma patients. Acta Neurochir 156, 1427–1440 (2014). https://doi.org/10.1007/s00701-014-2073-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-014-2073-1